

Abstract
In the course of catalysis or signaling, large multimodular proteins often undergo conformational changes that reposition the modules with respect to one another. The mechanisms that direct the reorganization of modules in these proteins are of considerable importance, but distinguishing alternate conformations is a challenge. Cobalamin-dependent methionine synthase (MetH) is a 136-kDa multimodular enzyme with a cobalamin chromophore; the color of the cobalamin reflects the conformation of the protein. The enzyme contains four modules and catalyzes three different methyl transfer reactions that require different arrangements of these modules. Two of these methyl transfer reactions occur during turnover, when homocysteine is converted to methionine by using a methyl group derived from methyltetrahydrofolate. The third reaction is occasionally required for reactivation of the enzyme and uses S-adenosyl-l-methionine as the methyl donor. The absorbance properties of the cobalamin cofactor have been exploited to assign conformations of the protein and to probe the effect of ligands and mutations on the distribution of conformers. The results imply that the methylcobalamin form of MetH exists as an ensemble of interconverting conformational states. Differential binding of substrates or products alters the distribution of conformers. Furthermore, steric conflicts disfavor conformers that juxtapose a methyl group on substrate with one on methylcobalamin. These results suggest that the methylation state of the cobalamin will influence the distribution of conformers during turnover.
In the course of catalysis or signaling large multimodular proteins often undergo conformational changes that reposition the modules with respect to one another. Various scenarios can be envisioned for these rearrangements. At one extreme, the protein might sample accessible conformations stochastically, searching a series of states until the required conformation is reached. At the other extreme, the transitions between discrete states during a catalytic cycle may be strictly regulated by interactions between the protein and its ligands. Rigorous evaluation of these options has proved extremely challenging, even in cases where alternate conformations have been structurally characterized, because it is difficult to distinguish different conformations experimentally. We have chosen to examine the conformational ensemble of the large modular enzyme, cobalamin-dependent methionine synthase (MetH), because the presence of the colored cobalamin cofactor at the active site provides an embedded reporter of the conformation of the protein.
MetH of Escherichia coli catalyzes the methylation of homocysteine (Hcy) to form methionine (Fig. 1A). The methyl group donated by methyltetrahydrofolate (CH3–H4folate) is transferred to an enzyme-bound cobalamin cofactor and then captured by Hcy. The cobalamin remains bound to its carrier module during the methyl transfer reactions and each substrate is presented to it in turn. Thus the enzyme must undergo conformational changes that permit the bound substrates to access the cobalamin. During catalysis the cobalamin cofactor cycles between cob(I)alamin, in which the cobalt is in the +1 oxidation state, and methylcobalamin, in which the cobalt is formally in the +3 oxidation state. Cob(I)alamin is readily oxidized by oxygen, and during turnover in an aerobic environment, the cofactor is occasionally oxidized to cob(II)alamin, producing an inactive form of the enzyme. MetH has evolved a self-repair mechanism that returns the cob(II)alamin form to the catalytically competent methylcobalamin form by a reductive methylation in which reducing equivalents are supplied by flavodoxin (1) and a methyl group is donated by S-adenosyl-l-methionine (AdoMet) (2). Thus three different substrates, CH3–H4folate, Hcy and AdoMet, are presented to the cobalamin at different points in the reaction cycles. Furthermore, AdoMet must be excluded from reaction with cob(I)alamin during turnover to prevent futile cycling.
Fig. 1.

The reaction cycle for methionine synthase (A) and definitions of the catalytic roles and conformational states of MetH (649–1227) and MetH (2–1227) (B). (A) During catalysis (the reactions shown in red) the cobalamin cofactor is alternately demethylated by Hcy and remethylated by CH3–H4folate. Transfer of the methyl group of methylcobalamin (formally the cobalt is in the +3 oxidation state) as a methylcarbocation leaves an electron pair on the cobalt, forming cob(I)alamin. Cob(I)alamin reacts with molecular oxygen during aerobic turnover (≈1/2,000 turnovers) and forms an inactive cob(II)alamin enzyme species. Return of this species to the catalytic cycle requires reduction to cob(I)alamin with electrons from reduced flavodoxin and then methylation to form methylcobalamin with a methyl group derived from AdoMet. The deactivation/reactivation cycle is shown in green. (B) The modules in MetH are represented schematically, from the N to the C terminus of the protein: H (green), Hcy-binding region; F (gold), methyltetrahydrofolate-binding region; B (red), cobalamin-binding region; A (blue), AdoMet binding region. The Rossmann domain of the cobalamin-binding region (B) is represented by an oval, the four-helix bundle is represented by four boxes, and the cobalamin is represented by a parallelogram. The structure shown for state 1 is the x-ray crystal structure of the cobalamin-bearing fragment isolated from the wild-type protein (7). The structure shown for state 4 is the x-ray crystal structure of the His759Gly variant of MetH (649–1227) (11).
An x-ray crystal structure of the full-length MetH polypeptide is not available, but limited proteolysis and expression of fragments have proven useful in delineating the functional and structural organization of MetH (3–5). The enzyme is composed of four regions: the N-terminal half of the protein comprises modules for binding Hcy and CH3–H4folate, whereas the C-terminal half houses the cobalamin- and AdoMet-binding regions. Studies of the N-terminal half of the protein have shown it to be competent in methyl transfers between Hcy and exogenously supplied methylcobalamin and between CH3–H4folate and exogenously supplied cob(I)alamin (5). Furthermore, methionine synthase activity can be reconstituted when protein fragments that correspond to the N- and C-terminal halves are combined (6).
Structural studies of MetH fragments and of related proteins that catalyze analogous methyl transfer reactions provide detailed pictures of the structural features that are responsible for binding of substrates and the B12 cofactor (7–11) by the isolated modules. The Hcy- and folate-binding regions of MetH are related to betaine–Hcy methyl transferase (12) and CH3–H4folate-corrinoid iron–sulfur protein methyl transferase (13), respectively. Betaine–Hcy methyl transferase is a zinc metalloprotein that catalyzes the methylation of Hcy by betaine (10). CH3–H4folate-corrinoid iron-sulfur protein methyltransferase catalyzes the methylation of cob(I)alamin by CH3–H4folate (13). Each of these proteins is a (βα)8 barrel with an active site that is located at the C-terminal end of the barrel strands (9, 10). The x-ray crystal structure of the B12-binding module shows that this module consists of an N-terminal four-helix bundle and a C-terminal β/α Rossmann domain, with the cobalamin cofactor sandwiched between them (7). In free cobalamins, a nitrogen of the dimethylbenzimidazole nucleotide appended to the corrin macrocycle is coordinated to the cobalt of cobalamin. However, in MetH, the dimethylbenzimidazole is displaced and inserted into a pocket in the Rossmann domain, and the imidazole side-chain of His-759 is coordinated to the cobalt. In the structure of the tryptic fragment, the upper face of the corrin macrocycle, where methyl transfer reactions are expected to occur, is masked by a four-helix bundle, “the cap.” The C-terminal module that binds AdoMet is an unusual helmet-shaped domain with the AdoMet binding site located on the inner surface of the helmet (8). This domain also contains determinants for binding of flavodoxin (14, 15).
The structures of the individual modules demonstrate that only one domain at a time can access the cobalamin, requiring the minimal ensemble of conformational states designated in Fig. 1B. During catalysis, the four-helix bundle that caps B12 (state 1) must be displaced and the Hcy-binding domain positioned vis-à-vis the methylcobalamin to allow methyl transfer to form methionine (state 3). This module must be displaced from the upper face of the corrin to allow access of the CH3–H4folate-binding module (state 2). When cob(I)alamin is oxidized, the AdoMet-binding domain must associate with cobalamin for reductive reactivation (state 4). Therefore, major conformational changes are necessary to facilitate both catalysis and reductive methylation. Primary catalysis proceeds with a turnover number of 27 s-1, so the conformational changes that are essential for catalysis must occur in the millisecond time range.
However, indiscriminate access of cob(I)alamin, generated by methyl transfer to Hcy in state 3, to AdoMet would result in futile cycling. The observation that MetH discriminates against methyl groups derived from AdoMet during catalysis, and against methyl groups derived from CH3–H4folate during reductive methylation (2, 16), argues for tight control of access to the reactivation conformation (state 4).
The crystal structure of the His759Gly variant of the C-terminal half of MetH (residues 649-1227) was solved recently (11). His-759 supplies the imidazole ligand to cobalt in the wild-type protein, and in its absence methylcobalamin lacks a coordinated imidazole (it is base-off) and exhibits a shift in absorbance maximum from 525 nm (red) to 450 nm (yellow). The structure revealed a dramatic rearrangement of the cobalamin-binding module to allow the AdoMet-binding domain to access the upper face of the corrin. The four-helix bundle or cap is rotated 65° and translated 25 Å from the position it occupies in the structure of the isolated cobalamin-binding domain (7), and the corrin macrocycle has moved away from the Rossmann domain and toward the AdoMet-binding domain; the cobalt is 2.3 Å farther away from the Cα of residue 759 than in the cap-on structure. The increased distance between the cobalt and the Cα of residue 759 (11) is consistent with spectral evidence for the formation of base-off cob(II)alamin in the course of reductive methylation of the wild-type protein (16); in the conformation required for reductive methylation (state 4, the reactivation conformation), a 2.3-Å displacement of the cobalt away from residue 759 would be sufficient to sever the coordination to the imidazole side-chain in the wild-type enzyme and would lead to the formation of base-off cob(II)alamin. The movement of the helical bundle to permit access to the cobalamin was expected, but the extensive rearrangement of the cobalamin and the specific interactions between the cobalamin and the backbone of the AdoMet binding domain were unforeseen. In light of this crystal structure, which shows that the His759Gly variant adopts a conformation more suitable for reactivation (state 4) than for catalysis, one can now rationalize the substantial decrease in the rate of turnover (105-fold) that is observed with this variant relative to the wild-type protein (17).
The imidazole side chain of His-759 is linked by a hydrogen bond to the carboxylate of Asp-757 and thence to the hydroxyl group of Ser-810. In contrast to the severe loss of activity that is associated with the His759Gly mutation, mutations of Asp-757 or Ser-810 lead to substantially less diminution of enzymatic activity, 27-fold and 1.5-fold, respectively (17, 18). Mutations of ligand triad residues decrease the rate of demethylation of methylcobalamin by Hcy and increase the rate of reactivation of the cob(II)alamin cofactor, suggesting that the changes in activity correspond to a shift in the ensemble of conformers to favor state 4 (17, 19). Consistent with this hypothesis, mutation of Asp-757 to Glu increases the propensity for cob(II)alamin to assume the base-off form (17).
In this communication, the effects of ligands on the conformation of MetH in the methylcobalamin form are probed, relying on the dramatic change in color of methylcobalamin as it undergoes conversion between base-on and base-off forms to infer the fraction of enzyme in the reactivation conformation (state 4). Both MetH (residues 2–1227) and the C-terminal fragment (residues 649-1227) are used in these studies and the effect of the Asp757Glu mutation is examined in both contexts. The observations suggest that interconversions between the base-on and off states of the cobalamin are strongly modulated by substrates and products, and that MetH in the methylcobalamin form can be described as an ensemble of the four conformational states shown in Fig. 1B.
Materials and Methods
Materials. (6-R,S)-5-Methyltetrahydrofolate (calcium salt) was purchased from Schircks Laboratories (Jona, Switzerland); AdoMet, aquocobalamin, DEAE-Sepharose fast flow, and l-homocysteine thiolactone were purchased from Sigma.
l-Homocysteine thiolactone was converted to l-homocysteine and quantitated as described (20). Q-Sepharose (fast flow) and phenyl Sepharose CL-4B were purchased from Amersham Pharmacia. The E. coli XL1-Blue strain was purchased from Stratagene.
Expression and Purification of the Wild-Type and Asp757Glu Variants of MetH (2–1227). The wild-type and Asp757Glu variants of MetH (2–1227) were overexpressed in XL1-Blue E. coli strains bearing either the pMMA-07 (wild-type) or the pMMA-21 (Asp757Glu) plasmid (18). The proteins were purified by using DEAE-Sepharose and MonoQ anion exchange chromatographies as described (20). Both proteins were reductively methylated by AdoMet in an electrochemical cell (20).
Construction of an Expression Plasmid for Asp757Glu MetH (649–1227). The Asp757Glu mutation was introduced into MetH (649–1227) by ligation of a fragment of DNA from the pMMA-21 vector (18) containing the mutation. Briefly, pMMA-21 was digested with BglII at 37°C, and then with BsiWI at 55°C. The ≈800-bp fragment was purified from an agarose gel and ligated into the similarly digested pMMA-11 vector (6) to yield pVB-6. The presence of the Asp757Glu mutation in MetH (649–1227) was verified by sequencing at the DNA Sequencing Core Facility of the University of Michigan Biomedical Research Core Facility.
Expression, Purification, and Assay of Wild-Type and Asp757Glu MetH (649–1227). Both proteins were overepxressed in E. coli and purified as described (6) with minor modifications. Assays of Asp757Glu MetH (649–1227) were performed as described for wild-type MetH (649–1227) (6).
Results
For the initial studies of conformational equilibria in MetH, we chose to examine the properties of the C-terminal fragments. In contrast to the full-length MetH, where a minimal ensemble of four conformational states can be envisioned, the minimal ensemble for the C-terminal fragment of MetH comprises only two states. Both the intact protein and the fragment share a conformation where the cobalamin is capped by a 4-helix bundle (state 1) and a reactivation conformation in which the AdoMet binding domain and the cobalamin are juxtaposed (state 4). Because the primary effect of ligand triad mutations was proposed to be a shift in the conformational equilibria toward the reactivation conformation (17), which is signaled by a base-off conformation of the cobalamin, we first compared the properties of the wild-type and Asp757Glu variant fragments.
Temperature-Dependent Changes in the UV-Visible Spectra of MetH Fragments. The effect of temperature on the absorbance spectrum of wild-type MetH (649–1227) is minimal (Fig. 2A). The spectra were deconvoluted, by using reference spectra for base-on and base-off methylcobalamin (see Fig. 9, which is published as supporting information on the PNAS web site, www.pnas.org), to obtain the contributions of each coordination variant to the observed spectrum. The spectral deconvolutions yield the concentrations of base-on and base-off methylcobalamin in the samples and allow calculation of the equilibrium constant for the interconversion of the two ligation forms of cobalamin (Keq = [base-off]/[base-on]). Deconvolution of the base-on and base-off components of the spectrum at 37°C indicates that the base-on form predominates in wild-type MetH (649–1227), with a Keq ≈ 0.20.
Fig. 2.

Effects of temperature on the UV/visible spectra of methylated wild type (A) and Asp757Glu MetH (649–1227) (B). The MetH fragments (≈3 μM) were diluted into 0.1 M KPi buffer (pH 7.2), at 15°C (red) and 40°C (orange) for the wild-type protein (A), or 15°C (red), 20°C (green), 25°C (black), 30°C (blue), or 40°C (orange) for the Asp757Glu variant (B) and allowed to equilibrate for 2 min before spectral acquisition.
In contrast, the UV-visible spectrum of Asp757Glu MetH (649–1227) changes with temperature from red (532 nm maximum) at 15°C to yellow-orange (450 nm maximum) at 40°C (Fig. 2B). The spectral changes indicate a reversible interconversion of enzyme-bound methylcobalamin cofactor between base-on (532 nm peak) and base-off (450 nm peak) forms. Spectral deconvolutions yield the concentrations of base-on and base-off methylcobalamin in the samples. The equilibrium constant is estimated to be ≈1 at 37°C for the Asp757Glu MetH (649–1227) variant, establishing that the Asp757Glu mutation does indeed shift the ensemble to favor the reactivation conformation.
The UV/visible spectrum of wild-type full-length MetH is not perturbed when the temperature of the sample is raised over the same range as the C-terminal fragment, and resembles fully base-on methylcobalamin (data not shown). The optical spectrum of the Asp757Glu MetH (2–1227) variant suggests that this variant of the full length protein also remains almost fully in the base-on conformation (Keq ≈ 0.14, data not shown).
The Asp757Glu Variant of MetH (649–1227) Supports Methionine Formation in the Presence of MetH (2–649). Despite the differences in the temperature dependence of the coordination environment of the Asp757Glu variant of MetH (649–1227), this variant supports methionine synthesis, albeit at a reduced rate. Previous studies had shown that wild type MetH (649–1227), when combined with MetH (2–649), can catalyze the formation of methionine at a rate (47,000 M-1·s-1) that is first order in both protein components (6). The second order rate constant determined for the reaction with the Asp757Glu variant is ≈600 M-1·s-1 (see Fig. 10, which is published as supporting information on the PNAS web site).
A Working Model. The observation of temperature-dependent changes in the composition of MetH (649–1227) suggests that the protein in solution is an ensemble of states that are in equilbrium with one another. It leads to a working model in which the distribution of these states will depend on the presence of ligands and on the oxidation and alkylation state of the cobalamin. Ligands may bias the stability of these states because they are likely to have differential binding affinities, depending on whether or not their respective modules are juxtaposed with the cobalamin. Our strategy was to employ substrates and products to alter the composition of the conformational ensemble, using the UV-visible spectra of the protein as a signal of the ligand environment of the cobalamin.
S-Adenosylhomocysteine Stabilizes Base-Off Methylcobalamin. AdoHcy is a product of the reductive methylation of the cobalamin cofactor, and by virtue of its position at the interface between the cobalamin and the C-terminal AdoMet binding modules (ref. 11, D. P. Huddler and M.L.L., unpublished results) may selectively stabilize the reactivation conformation. Assuming that the concentration of the base-off species corresponds to the concentration of the reactivation conformation, the population of this species should be enhanced by addition of AdoHcy to form an AdoHcy·methylcobalamin product complex, as shown in Fig. 3.
Fig. 3.

Thermodynamic cycles associated with the binding of AdoHcy to MetH. (Left) Binding of AdoHcy to MetH (649–1227) in the methylcobalamin form is presumed to selectively stabilize the product complex (state 4) in which the AdoMet-binding domain is positioned above the cobalamin. The dissociation constant KAdoHcy for binding of AdoHcy to state 4 will therefore be smaller than the dissociation constant K′AdoHcy for AdoHcy binding to state 1. The binding of AdoHcy will shift the equilbrium between states 1 and 4 to favor state 4, so that K′conf = [state 4·AdoHcy]/[state 1·AdoHcy] will be larger than Kconf = [state 4]/[state 1]. The standard free energy difference ΔΔG between RTlnK′AdoHcy and RTlnKAdoHcy will be equivalent to the free energy difference between the RTlnK′conf and RTlnKconf. (Right) The binding of AdoHcy to states 1, 2, and 3 of full-length MetH (2–1227) is represented by an composite dissociation constant K′AdoHcy. The standard free energy difference ΔΔG for binding of AdoHcy to state 4 vs. the other states is 0.9 kcal·mol-1 as determined from the ratio of Kconf to K′conf. It is likely that the Kd values for binding of AdoHcy to states 1, 2, and 3 are very similar.
Addition of AdoHcy to Asp757Glu MetH (649–1227) (Fig. 4A) and to Asp757Glu MetH (2–1227) (Fig. 4B) increases the fraction of base-off cobalamin. As indicated in Fig. 4 Insets, the effect of AdoHcy is saturable with incomplete conversion of MetH to the base-off form. Such behavior would be expected if AdoHcy binds preferentially, but not exclusively, to enzyme in the reactivation conformation, and the difference in free energy of binding to the base-off and base-on forms is insufficient to pull the equilibrium completely over to the base-off form. Comparison of the base-off/base-on ratios of cobalamin in the fragment (Fig. 4A) and the full-length protein (Fig. 4B) shows that the inclusion of two more conformations in the ensemble shifts the distribution toward the base-on form, indicating that these additional conformations are base-on when the cobalamin is methylated.
Fig. 4.

AdoHcy titrations of methylated Asp757Glu MetH (649–1227) (A) and Asp757Glu MetH (2–1227) (B) at 37°C. MetH (≈3 μM) was added to a solution containing varying concentrations of AdoHcy in 0.1 M KPi buffer (pH 7.2) at 37°C. Spectra were obtained 2 min after addition of the protein. In each case, only the initial (black) and final (blue) spectra that were obtained in the absence of AdoHcy and in the presence of saturating concentrations of AdoHcy are shown. (Insets) Shown is the decrease in the absorbance of the samples at 532 nm caused by conversion of base-on to base-off methylcobalamin as a function of the concentration of AdoHcy. The percent contributions of base-on and base-off cobalamin to the spectra were obtained by deconvolution, as described in the text, and are shown below the graphs.
Spectral changes associated with addition of AdoHcy to wild-type MetH (649–1227) and MetH (2–1227) are shown in Figs. 5 A and B, respectively. Addition of AdoHcy to the wild-type fragment shifts the equilibrium to the base-off form (Fig. 5A), whereas the spectrum of full-length MetH (2–1227) is almost unaffected by AdoHcy (Fig. 5B). The decreased population of the reactivation conformation in MetH (2–1227) relative to the C-terminal fragment is further support for the presence of the base-on states 2 and/or 3 in MetH (2–1227), which compete with the AdoMet-binding domain for interaction with the cobalamin-binding region. Comparison of the data in Fig. 5B with the behavior of the Asp757Glu mutant (Fig. 4B) establishes that the Asp757Glu mutation shifts the equilibrium between base-on and base-off forms to favor the latter.
Fig. 5.

AdoHcy titrations of methylated wild-type MetH (649–1227) (A) and MetH (2–1227) (B). MetH (≈4–5 μM) was added to a solution containing varying concentrations of AdoHcy in 0.1 M KPi buffer (pH 7.2) at 37°C. Spectra were obtained 2 min after addition of the protein. In each case, only the initial (black) and final (blue) spectra that were obtained in the absence of AdoHcy and in the presence of saturating concentrations of AdoHcy are shown. (A Inset) The decrease in the absorbance of the samples at 525 nm caused by conversion of base-on to base-off methylcobalamin as a function of the concentration of AdoHcy. The contributions of base-on and base-off cobalamin to the spectra are indicated below the graphs.
S-Adenosylmethionine Shifts the Equilibrium Toward Base-On Methylcobalamin. The presence of the methyl group of AdoMet in close proximity to the methyl group of methylcobalamin is predicted to lead to steric clashes that would disfavor the reactivation conformation. As expected, addition of AdoMet (0.2 mM) to Asp757Glu MetH (649–1227) increases the concentration of the base-on form of the cofactor (Keq ≈ 0.33, Fig. 6) even at 37°C where the fragment is in the base-off form in its absence (Keq ≈ 1). These observations with AdoMet are complementary to the results with the Asp757Glu variant in the presence of AdoHcy, and reinforce the assignment of the base-off UV-visible spectrum to the reactivation conformation of the protein (state 4).
Fig. 6.

Effect of AdoMet on the absorbance spectrum of methylated Asp757Glu MetH (649–1227). The spectra were obtained in 0.1 M KPi buffer (pH 7.2) with 4.2 μM Asp757Glu MetH (649–1227) in the presence (blue) or absence (black) of 0.2 mM AdoMet at 37°C. The contributions of base-on and base-off cobalamin to the spectra is indicated below the graph.
Modulation of Conformational Equilibria in MetH by CH3-H4folate. The experiments with AdoHcy and AdoMet suggest that MetH in solution behaves as an ensemble of conformations in equilibrium with one another. In the case of MetH (649–1227), the ensemble can be biased in favor of the reactivation conformation (state 4) by inclusion of AdoHcy, and in favor of the base-on conformation (state 1) in the presence of AdoMet. Full-length MetH (2–1227) was examined to determine whether inclusion of CH3–H4folate also alters the distribution among the conformational states. The rationale for these studies was that in the full-length protein, the AdoMet-binding domain competes with at least three other regions of the protein for access to the cobalamin (state 4 versus states 1–3). In wild-type MetH, this competition strongly favors the base-on forms, because AdoHcy by itself is not able to shift the population toward the reactivation complex (state 4). However, disfavoring one of these competing conformations could divert some of the protein into the conformation that juxtaposes the AdoMet and B12 binding domains. Our hypothesis was that steric clashes between the methyl groups of CH3–H4folate and methylcobalamin might destabilize the conformation where the folate and cobalamin binding domains are juxtaposed (state 2) in much the same way that AdoMet disfavors the formation of the reactivation conformation in methylated Asp757Glu MetH (649–1227) (state 4).
The interplay between AdoHcy and CH3–H4folate was first examined with MetH (2–1227). Although neither AdoHcy nor CH3–H4folate alone can alter the absorption spectrum of the wild-type full-length MetH (2–1227), when they are present together a base-off population can be observed in the UV-visible spectrum of MetH (2–1227) (Fig. 7A). Deconvolution of the spectrum of the wild-type MetH (2–1227) yields a measurable equilibrium (Keq ≈ 0.25) between base-on and base-off forms of the cobalamin. The effects of AdoHcy and CH3–H4folate are much more dramatic with the MetH Asp757Glu (2–1227) variant (Fig. 7B). In contrast to results obtained with wild-type MetH, either CH3–H4folate or AdoHcy, singly, increases the base-off component in the spectrum. In the presence of both ligands, significantly more of the base-off form is observed (62%), than with either ligand in isolation. The spectral changes reported here are reminiscent of a reversible equilibrium between base on/off forms of freshly reductively methylated MetH as a function of temperature, reported by Fujii and Huennekens (21), which we now attribute to the effects of bound AdoHcy and CH3–H4folate.
Fig. 7.

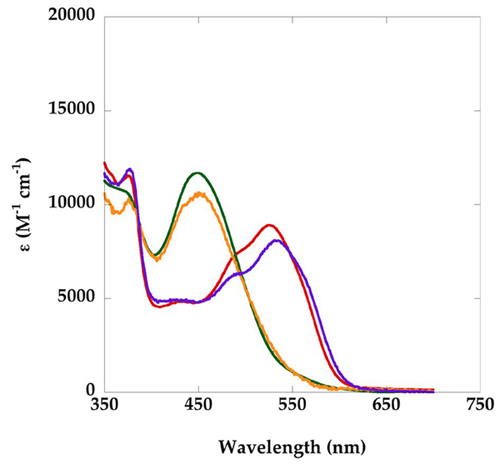
Effects of CH3–H4folate and/or AdoHcy on the absorbance spectra of methylated wild-type (A) and Asp757Glu (B) variants of MetH (2–1227). The spectra with wild-type MetH were obtained in 0.1 M KPi buffer (pH 7.2) with 4 μM MetH in the presence of 1 mM CH3–H4folate (red) or 1 mM CH3–H4folate and 1 mM AdoHcy (orange). The spectra with the Asp757Glu MetH (2–1227) were obtained in 0.1 M KPi buffer (pH 7.2) with 5 μM MetH in the absence of ligands (red), with 1 mM CH3–H4folate (black), 1 mM AdoHcy (purple), or 1 mM CH3–H4folate and 1 mM AdoHcy (orange). All spectra were obtained after 2 min incubation of the samples at 37°C. The contributions of base-on and base-off cobalamin to the spectra are indicated below the graphs.
The free energies derived from the equilibrium constants measured from the distributions of base-off and base-on wild-type and Asp757Glu variants of MetH (2–1227) and MetH (649–1227) are summarized in Fig. 8A. In MetH (649–1227) the base-on conformation is favored by ≈0.9 kcal·mol-1; mutation of Asp-757 to glutamate equalizes the base-on and base-off populations of the C-terminal fragment (ΔG ≈ 0 kcal·mol-1). Wild-type MetH (2–1227) is predominantly base-on at 37°C; however, for the Asp757Glu variant of MetH (2–1227) the ΔG is ≈1.2 kcal·mol-1. The ligands, CH3–H4folate and AdoHcy, further destabilize the base-on states of Asp757Glu MetH (2–1227) by 0.6 and 0.9 kcal·mol-1, respectively; in the presence of both ligands the base-on state is destabilized by 1.5 kcal·mol-1 (ΔG = -0.3 kcal·mol-1). Thus the ΔΔG values for AdoHcy and CH3–H4folate are additive, consistent with independent binding of the two molecules. In the wild-type protein, these ligands singly do not measurably alter the equilibrium between base-on and base-off states; however, in the presence of both, a ΔG of ≈0.85 kcal·mol-1 is measured. Assuming that the ligands shift the equilibria in the wild-type protein to the same extent as in Asp757Glu MetH (2–1227), the ΔG for the wild-type protein in the absence of any ligands would be ≈2.4 kcal·mol-1. Thus, one can explain the lack of measurable change in the spectrum of the wild-type protein in the presence of either ligand, in isolation, by noting that the 0.6 or 0.9 kcal·mol-1 that would result from the binding of either of these ligands singly would not alter the equilibrium for the base-on to base-off states sufficiently to result in measurable (>10%) build-up of base-off cobalamin.
Fig. 8.

(A) Summary of ΔG values for methylated wild-type (red) and Asp757Glu (orange) MetH (2–1227) or MetH (649–1227) at 37°C. The energy differences were obtained from the equilibrium constants for the base-on and base-off forms (Keq = [base-off]/[base-on]) at 37°C by using ΔG = -RT ln Keq.(B) Distribution of conformational states of methylated wild-type and Asp757Glu variants of MetH (2–1227) in the presence and absence of CH3–H4folate. The distributions for wild-type and Asp757Glu MetH (2–1227) in the absence (black) or presence (blue) of CH3–H4folate were calculated as described in the text.
In addition to revealing the free energies that are associated with the conformational changes, the effect of ligands on the conformational equilibria in MetH provide a quantitative measure of the distribution of the various conformational states shown in Fig. 1B. In the following analysis, we have made two assumptions. First, the ratio of the cap and reactivation states (Fig. 1B, states 1 and 4) is presumed to remain the same in the full-length protein as in the MetH (649–1227) fragment. Second, the effect of CH3–H4folate is to disfavor the formation of state 2, where the folate-bearing region caps the cobalamin-binding domain, and it is assumed that this fraction of the ensemble is redistributed among the remaining states in proportion to their relative abundance in the absence of CH3–H4folate. The boldest of these assumptions is the first one, that contacts with the catalytic domains in the intact enzyme do not alter the equilibrium between the reactivation and cap conformations that is seen in the C-terminal fragment. Using these assumptions, one can draw the distribution diagrams that are shown in Fig. 8B for wild-type and Asp757Glu variants of MetH (2–1227) in the presence and absence of CH3–H4folate. The first assumption and the experimentally determined equilibrium constants allow one to calculate the distribution of the protein between states 1 and 4, and estimate the sum of the distribution in states 2 and 3. The second assumption allows one to tease-apart the contributions of states 2 and 3 to the composite because we presume that CH3–H4folate depopulates state 2, without altering relative stability of the other states. The most striking feature of the resulting distributions is that states 2 and 3 dominate the ensembles that constitute both wild type and Asp757Glu MetH (28% and 60% in wild-type and 20% and 56% in Asp757Glu MetH). Indeed, these are the states that one would expect the protein to adopt during turnover.
Discussion
Conformational changes are integral to catalysis by a number of multimodular proteins, which require rapid association and dissociation of extensive interdomain interfaces to effect reactions or to transduce signals. Modular arrangement can afford flexibility, allowing active sites to be assembled as needed. A spectacular example of modular construction is found in the machinery for the production of polyketide antibiotics, where modules that are required for the biosynthesis of the metabolite are vectorially arranged within a single polypeptide. This ensemble of active sites catalyzes activation, condensation, and modification reactions required for the formation of the elaborate polyketide metabolites from simple building blocks (22–24).
In the case of MetH, deciphering the rules that dictate the interdomain interactions has been facilitated by the presence of the colored cobalamin cofactor. The spectral properties of the cobalamin are sensitive to the coordination environment of the cofactor, and differ between the catalytic (states 1–3) and reactivation (state 4) conformations of the protein. Hence, the coordination state of the cobalamin reports on distribution of protein amongst the states shown in Fig. 1B, and provides insights into the control of the rearrangements that allow each of the substrate-bearing regions of MetH to interact with the cobalamin-binding domain.
The picture that emerges is that MetH, in the methylcobalamin form, is an ensemble of interconverting conformational states that are distinct from one another (Fig. 1B). At any instant, only one of the substrate binding domains of the protein can interact productively with the cobalamin-binding region. Ligands may bias the distribution within the ensemble by selectively stabilizing or destabilizing a particular state. The oxidation and alkylation states of the cobalamin, which vary in the course of the catalytic cycle, will further bias the distributions within the ensemble by altering interactions between the cobalamin and each substrate-bearing domain. Catalysis entails dynamic redistribution among states of similar energy, and the rates of redistribution are likely to limit the rate of turnover. Thus, the behavior of MetH is intermediate between the two extremes posed in the Introduction: purely random rearrangements of the domains to access conformational states vs. a lock-step formation and breakdown of discrete states.
The perturbations in the conformational ensemble that are induced by ligands appear to depend critically on the strength of the coordination between cobalt and the nitrogen donated by His-759. Earlier studies of the cob(II)alamin form of MetH suggested that weakening of the coordination of His-759 to cobalt induced by the Asp757Glu mutation leads to increased concentration of the reactivation conformation (16, 17). This effect can be rationalized from the structure of the reactivation conformation, which indicates that the cobalt must be dissociated from His-759 (11). Thus weaker coordination should favor conversion to the reactivation conformation, and indeed the His759Gly mutant enzyme appears to be thermodynamically trapped in the reactivation conformation. The studies presented here confirm that the presence of the Asp757Glu mutation also shifts the distribution of methylcobalamin conformers to favor the reactivation conformation.
The methylation state of the cofactor is also an important determinant of the conformational distribution. When the protein is in the methylcobalamin state, CH3–H4folate disfavors state 2 (folate on) because of steric repulsion between the two methyl groups. However, when the cofactor is demethylated, methyltetrahydrofolate is expected to stabilize state 2. Similarly, binding of AdoMet disfavors the reactivation conformation of methylated enzyme, whereas preliminary experiments indicate that it favors the reactivation conformation of enzyme in the cob(II)alamin form (V.B., A. Fleischhacker, and R.G.M., unpublished observations).
When MetH is in the cob(II)alamin form the equilibrium distribution of conformers shifts to favor the reactivation conformation (17). This shift is important physiologically because it decreases the energy required for conversion to the reactivation conformation. Binding of flavodoxin can then result in state 4 becoming the dominant conformation. In fact, previous UV/visible spectrophotometric and electron paramagnetic resonance spectroscopic studies have shown that the addition of flavodoxin to MetH converts base-on to base-off cob(II)alamin, whereas it does not change the absorbance properties of methylcobalamin (25).
Surprisingly, during catalysis, the cob(I)alamin form of the enzyme is not predominately in the reactivation conformation even though this form of the cofactor is preferentially four coordinate (base-off). Previous experiments have shown that cob(I)alamin generated by demethylation with Hcy only reacts with CH3–H4folate, indicating that it is not in the reactivation conformation, whereas cob(I)alamin generated by chemical reduction of cob(II)alamin enzyme only reacts with AdoMet (19). These observations imply that the conformational ensemble is not at equilibrium in the cob(I)alamin state, although the reason for the blockade between state 4 and states 1–3 remains unknown.
We now have tools to measure the redistribution of conformational states induced by formation of cob(II)alamin and cob(I)alamin, and to measure the kinetics of redistribution. For instance, stopped-flow UV-visible spectrophotometric experiments involving demethylation of methylcobalamin protein with Hcy exhibit multiple phases of reaction (26), which we now interpret as arising from rate-limiting redistribution of conformations among the various forms of the protein. In addition, ligands such as AdoHcy, AdoMet, or CH3–H4folate may prove useful in obtaining x-ray crystal structures of MetH in various conformations. We believe that many of the principles deduced from the behavior of methionine synthase may be applicable to conformational interchange in other multimodular proteins. A more detailed description of our analysis of the conformational equilibria is available in ref. 27.
Supplementary Material
Acknowledgments
These studies were supported in part by National Institutes of Health Grants GM24908 (to R.G.M.) and GM16429 (to M.L.L.) and by National Research Service Award Postdoctoral Fellowship F32 GM 20524 (to V.B.).
This contribution is part of the special series of Inaugural Articles by members of the National Academy of Sciences elected on April 30, 2002.
Abbreviations: MetH, cobalamin-dependent methionine synthase; Hcy, homocysteine; AdoMet, S-adenosyl-l-methionine.
References
- 1.Fujii, K., Galivan, J. H. & Huennekens, F. M. (1977) Arch. Biochem. Biophys. 178 662-670. [DOI] [PubMed] [Google Scholar]
- 2.Taylor, R. T. & Weissbach, H. (1967) J. Biol. Chem. 242 1517-1521. [PubMed] [Google Scholar]
- 3.Banerjee, R. V., Johnston, N. L., Sobeski, J. K., Datta, P. & Matthews, R. G. (1989) J. Biol. Chem. 264 13888-13895. [PubMed] [Google Scholar]
- 4.Drummond, J. T., Huang, S., Blumenthal, R. M. & Matthews, R. G. (1993) Biochemistry 32 9290-9295. [DOI] [PubMed] [Google Scholar]
- 5.Goulding, C. W., Postigo, D. & Matthews, R. G. (1997) Biochemistry 36 8082-8091. [DOI] [PubMed] [Google Scholar]
- 6.Bandarian, V. & Matthews, R. G. (2001) Biochemistry 40 5056-5064. [DOI] [PubMed] [Google Scholar]
- 7.Drennan, C. L., Huang, S., Drummond, J. T., Matthews, R. G. & Ludwig, M. L. (1994) Science 266 1669-1674. [DOI] [PubMed] [Google Scholar]
- 8.Dixon, M. M., Huang, S., Matthews, R. G. & Ludwig, M. (1996) Structure (London) 4 1263-1275. [DOI] [PubMed] [Google Scholar]
- 9.Doukov, T., Seravalli, J., Stezowski, J. J. & Ragsdale, S. W. (2000) Structure (London) 8 817-830. [DOI] [PubMed] [Google Scholar]
- 10.Evans, J. C., Huddler, D. P., Jiracek, J., Castro, C., Millian, N. S., Garrow, T. A. & Ludwig, M. L. (2002) Structure (London) 10 1159-1171. [DOI] [PubMed] [Google Scholar]
- 11.Bandarian, V., Pattridge, K. A., Lennon, B. W., Huddler, D. P., Matthews, R. G. & Ludwig, M. L. (2002) Nat. Struct. Biol. 9 53-56. [DOI] [PubMed] [Google Scholar]
- 12.Garrow, T. A. (1996) J. Biol. Chem. 271 22831-22838. [DOI] [PubMed] [Google Scholar]
- 13.Roberts, D. L., Zhao, S., Doukov, T. & Ragsdale, S. W. (1994) J. Bacteriol. 176 6127-6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hall, D. A., Vander Kooi, C. W., Stasik, C. N., Stevens, S. Y., Zuiderweg, E. R. & Matthews, R. G. (2001) Proc. Natl. Acad. Sci. USA 98 9521-9526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hall, D. A., Jordan-Starck, T. C., Loo, R. O., Ludwig, M. L. & Matthews, R. G. (2000) Biochemistry 39 10711-10719. [DOI] [PubMed] [Google Scholar]
- 16.Jarrett, J. T., Hoover, D. M., Ludwig, M. L. & Matthews, R. G. (1998) Biochemistry 37 12649-12658. [DOI] [PubMed] [Google Scholar]
- 17.Jarrett, J. T., Amaratunga, M., Drennan, C. L., Scholten, J. D., Sands, R. H., Ludwig, M. L. & Matthews, R. G. (1996) Biochemistry 35 2464-2475. [DOI] [PubMed] [Google Scholar]
- 18.Amaratunga, M., Fluhr, K., Jarrett, J. T., Drennan, C. L., Ludwig, M. L., Matthews, R. G. & Scholten, J. D. (1996) Biochemistry 35 2453-2463. [DOI] [PubMed] [Google Scholar]
- 19.Jarrett, J. T., Huang, S. & Matthews, R. G. (1998) Biochemistry 37 5372-5382. [DOI] [PubMed] [Google Scholar]
- 20.Jarrett, J. T., Goulding, C. W., Fluhr, K., Huang, S. & Matthews, R. G. (1997) Methods Enzymol. 281 196-213. [DOI] [PubMed] [Google Scholar]
- 21.Fujii, K. & Huennekens, F. M. (1979) in Biochemical Aspects of Nutrition, ed. Yagi, K. (Japan Scientific Societies Press, Tokyo), pp. 173-183.
- 22.Khosla, C., Gokhale, R. S., Jacobsen, J. R. & Cane, D. E. (1999) Annu. Rev. Biochem. 68 219-325. [DOI] [PubMed] [Google Scholar]
- 23.Cane, D. E., Walsh, C. T. & Khosla, C. (1998) Science 282 63-68. [DOI] [PubMed] [Google Scholar]
- 24.Cane, D. E. & Walsh, C. T. (1999) Chem. Biol. 6 319-325. [DOI] [PubMed] [Google Scholar]
- 25.Hoover, D. M., Jarrett, J. T., Sands, R. H., Dunham, W. R., Ludwig, M. L. & Matthews, R. G. (1997) Biochemistry 36 127-138. [DOI] [PubMed] [Google Scholar]
- 26.Banerjee, R. V., Frasca, V., Ballou, D. P. & Matthews, R. G. (1990) Biochemistry 29 11101-11109. [DOI] [PubMed] [Google Scholar]
- 27.Bandarian, V. & Matthews, R. G. (2003) Methods Enzymol., in press. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}