

Abstract
The origin of Alzheimer's disease (AD) has been subjected to an intense amount of examination; however, a clear conclusion as to the nature of this crippling disease has yet to be identified. What is readily accepted is that a definitive marker of this disease is the aggregation of the amyloid β-peptide (Aβ) into neuritic plaques. The recent observation that nicotine exposure leads to delayed onset of AD has stimulated a flurry of research into the nature of this neuroprotective effect. This phenomenon has been debated, but no consensus has been reached, and although these studies have targeted nicotine, the primary alkaloid in tobacco, few studies have considered the physiological role of nicotine metabolites in disease states. Nornicotine is a major nicotine metabolite in the CNS and has been shown to participate in the aberrant glycation of proteins in vivo in a process termed nornicotine-based glycation. Herein is detailed a potentially fortuitous role of nornicotine-based glycation in relation to the pathology of AD. Specifically, nornicotine was found to covalently alter Aβ, leading to reduced peptide aggregation. Potential consequences of this reaction cascade include reduced plaque formation and/or altered clearance of the peptide, as well as attenuated toxicity of soluble Aβ aggregates. The findings described provide an alternative mechanism for nicotine neuroprotection in AD and a means for the alteration of amyloid folding based on a covalent chemical event.
Amyloidoses are a group of >20 human diseases that share the common feature of the assembly of naturally occurring soluble proteins into insoluble deposits with distinct tinctorial properties and fibrillar morphology (1). The protein inherent to each disorder varies widely by both molecular weight and sequence, yet all share a propensity for undergoing assembly into well-ordered β-pleated sheets termed fibrils. Among these disease states, Alzheimer's disease (AD) (2) is the most common amyloidosis of the brain and the leading cause of dementia, affecting >4 million elderly individuals in the United States alone (3). AD is characterized by the deposition of neurotoxic amyloid fibrils in areas of the brain important for memory and cognition (4). With the size of the aging baby boomer population, AD could rapidly reach epidemic proportions in the industrialized world; studies have estimated that by the year 2025, there will be ≈22 million cases of AD worldwide, with at least 10 million in the United States (5). Undoubtedly, there is an immediate need for effective treatments for AD as an ailing population of this magnitude would cause extreme stress on the health-care system, possibly leading to its eventual collapse.
Although extensive study into AD has been performed, the precise mechanism leading to the onset of the disease remains unclear. A popular hypothesis, the amyloid β-peptide (Aβ) hypothesis, states that CNS build-up of the Aβ is neurotoxic and has received increasing support (6). Repeated demonstrations have shown that the self-assembly of this peptide is critical to the pathogenesis of AD (7), and in particular, an oligomeric form of Aβ is necessary for in vitro and in vivo neurotoxicity (8). Aβ, a 39- to 43-aa peptide and the primary constituent of amyloid plaques, is produced from the cleavage of a much larger protein termed the amyloid precursor protein (APP) (9). In brain tissue, the most abundant form of Aβ is the 42-aa fragment of APP (10), and the production of Aβ1–42 in cell transfection experiments is increased by APP and presenilin mutations that cause AD (11, 12).
The use of small molecules and peptides as inhibitors of Aβ oligomerization has become increasingly popular as a promising pharmacotherapy for AD. Extensive biophysical measurements have demonstrated that the Aβ peptide can fold into α-helical or random, extended chain structures and the detrimental β-sheet structures that eventually lead to insoluble amyloids (13). The first example of chemical disruption of β-sheet containing polymers dates to the early 1990s when it was postulated that conformational modification of the pentamer “core” of Aβ, specifically the Aβ17–21 region, might lead to the development of anti-Aβ agents (14). This notion became known as the “binding surface hypothesis” and was later substantiated through the use of hexadecyl-N-methylpiperidinium bromide as a selective inhibitor of Aβ fibrillogenesis (15). Soon after those reports, biologically inactive peptides derived from the Val-Phe-Phe tripeptide residues of Aβ were shown to attenuate Aβ12–28-induced amnesia in a mouse model of Aβ toxicity (16). Additionally, studies using the pentapeptide ligand KLVFF have demonstrated that this region is essential in the polymerization of the full-length Aβ (17).
Recently, two reports describing the relevance of the Maillard reaction in AD have appeared (18, 19). This reaction, simply characterized as the reaction between reducing sugars and amines, was first described ≈90 years ago (20). Also known as nonenzymatic browning, the Maillard reaction has been extensively reviewed by food chemists for its role in the development and deterioration of flavor, taste, and the nutritional value of foods during processing and storage (21). In the past 20 years, the significant role of the Maillard reaction and the formation of so-called advanced glycation end products in biological systems has become linked to disease states ranging from diabetes (22) and normal aging (23) to cancer (24). The reaction takes place through a series of transformations, the first of which is the reversible formation of a Schiff base between the amine component and ring-opened form of the reducing sugar, followed by Amadori rearrangement (25) to the corresponding deoxyglucosone. These intermediates have known susceptibility to oxidation via aerobic and anaerobic pathways to give 1,2-dicarbonyl compounds, which then rapidly react with available nucleophiles (26).
Within our laboratories, we have demonstrated that nornicotine 1, a minor psychoactive (27) tobacco alkaloid (28) and nicotine metabolite (29, 30), can both catalyze aqueous aldol reactions (31) and participate in the in vitro and in vivo aberrant glycation of proteins in a process termed nornicotine-based glycation (32) (Fig. 1). Further study into the glycation of proteins by nornicotine revealed that one mechanism of action of this process is the covalent modification of lysine side-chain residues by nornicotine. This chemical potential of a secondary metabolite to participate in potentially detrimental covalent chemistry in vivo had gone unrecognized, and our discovery led us to explore the pathological consequences of nornicotine-based protein glycation. In the formation of neurotoxic fibrils from Aβ, it is well characterized that the KLVFF motif at residues 16–20 is critical in the formation of the β-pleated sheet structure of Aβ (17). Hence, we felt it was plausible that if nornicotine could covalently glycate Lys-16, the polymerization domain would become sterically occluded, thereby reducing the peptide's propensity to aggregate into fibrils (Fig. 2).
Fig. 1.

Aberrant nornicotine-based glycation. The process begins by reaction of nornicotine with the ring-opened form of glucose to give the corresponding Amadori product. Conversion of this product to 1,2-dicarbonyl-containing compounds gives a reactive electrophile capable of covalently modifying protein residues.
Fig. 2.

Participation of nornicotine and glucose in Aβ1–40 aggregation. Glycation with nornicotine leads to covalent modification of the KLVFF sequence, physically blocking a critical site in the formation of amyloid fibrils. The sphere represents the nornicotine-derived advanced glycation end product.
Materials and Methods
General Methods. All NMR spectra were recorded at 37.0°C ± 0.1°C on a Varian INOVA 400-MHz actively shielded spectrometer equipped with a Performa II gradient pulse amplifier and a 5-mm ASW probe capable of producing up to 30 G·cm-1 z-field gradient pulses. Matrix-assisted laser desorption/ionization Fourier transform MS (FTMS) experiments were performed on an IonSpec FTMS mass spectrometer. Electrospray ionization MS experiments were performed on an API 100 Perkin–Elmer SCIEX single quadrupole mass spectrometer.
Nornicotine Glycation Experiments. Glucose (200 mM) was added to 200 mM PBS (pH 7.4) to obtain a glucose-enriched buffer. Amyloid β-protein1–40 (0.43 mg, 0.1 μmol) was then dissolved in this buffer in a microcentrifuge tube to attain a final concentration of 0.1 mM (final reaction volume = 1 ml). Immediately after the addition of peptide, nornicotine was added to the reaction (0.4 mM) and the solution was filtered through a 0.2-μm syringe filter. The reactions were incubated at 37°C in the dark, and aliquots were removed at given time intervals and analyzed by thioflavin T staining of fibrils and MS. Samples for MS were first desalted with ZipTipC18 (Millipore) used according to the manufacturer's instructions. Control peptide/glucose and peptide-alone experiments were also performed and analyzed by using identical methods.
Thioflavin T Fluorescence Monitoring of Aβ1–40 Aggregation. An aliquot of the Aβ incubation reactions as described (1.16 μl) was added to 150 μl of thioflavin T (10 μM in 50 mM K3PO4, pH 6.0) in a black-walled 96-well plate. Fluorescence measurements were performed in duplicate at 25°C by using a Molecular Devices SPECTRAmax GEMINI plate reader. Control wells contained thioflavin T only, peptide only, or nornicotine only. The fluorescence intensity was measured by using a 450-nm excitation filter and a 482-nm emission filter. Each data point is the average of the intensity over six scans.
Diffusion-Ordered NMR Spectroscopy (DOSY) Experiments. The pulse and spectra width was adjusted to 6.8 ms and 6,395.9 Hz, respectively. The free induction decay contained 32,000 data points. For all of the experiments, the DgcsteSL pulse sequence was used, and 15 spectra of 640 transients each were collected with gradient pulse amplitudes ranging from 5 to 23 G·cm-1, where an ≈90–95% decrease in the intensity of nornicotine resonances was achieved at the largest gradient amplitudes. The baselines of all arrayed spectra were corrected before processing the data. After data acquisition, each free induction decay was apodized with line broadening of 1.0 Hz and Fourier-transformed. The processing program (the DOSY macro on Varian instruments) determines the peak heights of all of the signals above a pre-established threshold and fits the decay curve for each peak to a Gaussian function. The DOSY macro was run with data that was zero-filled to 131,072 data points. This macro calculates the diffusion coefficient by fitting the experimental data to the Stejskal–Tanner formula (33):
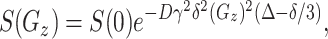 |
[1] |
where S(Gz) and S(0) are the signal intensities obtained with respective gradients strengths of Gz and 0, D is the diffusion coefficient, γ is the gyromagnetic constant, δ is the gradient pulse duration, and Δ is the diffusion delay. The results of the DOSY method of analysis are pseudo 2D spectra with NMR chemical shifts along one axis and calculated diffusion coefficients on the other axis.
All samples for high-resolution DOSY 1H NMR analysis were prepared in standard 5-mm tubes by dissolving glucose-d7 (18 mg, 96 μmol) in phosphate buffer (200 mM) prepared in D2O (pD = 7.0) and then adding Aβ12–28 (1 mg, 0.51 μmol) and 1.7 μl of a 300 mM stock solution of either nornicotine or nicotine prepared in deuterated phosphate buffer. The samples were vortexed and then incubated in the dark at 37°C. Controls samples of nornicotine and nicotine in buffer, with and without glucose, were also prepared and analyzed by using DOSY and direct exponential curve resolution algorithm (DECRA) processing. All reactions used for NMR analysis were also desalted with ZipTipC18 (Millipore), used according to the manufacturer's instructions, and analyzed by MS (see above).
DECRA Analysis. Sample spectra used for DOSY processing (see above) were also used for DECRA processing. The baselines of all arrayed spectra were corrected before processing the data. After data acquisition, each free induction decay was apodized with line broadening of 0.5 Hz and Fourier-transformed. The DECRA macro was used as acquired from the Varian VNMR user library. Data analyzed with DECRA were first zero-filled to 131,072 data points.
NOESY Spectrum of Aβ, Nornicotine, and Glucose Incubations. Two-dimensional phase-sensitive nuclear Overhauser effect spectra were acquired on Aβ12–28 glycation samples identical to those used in the DOSY experiments. The spectra were acquired by using 256 scans per increment and 185 increments. The mixing time was set at 200 ms. After acquisition, the spectra were zero-filled to 8,192 × 8,192 points, apodized with 1.5-Hz line broadening in the first dimension and a Gaussian apodization of 0.100 s in both dimensions, and Fourier-transformed.
Results and Discussion
Recently, it was reported that nicotine metabolism to nornicotine is higher centrally (≈20%) compared with peripheral metabolism (34). The significance of this finding has not been realized; however, based on our previous observations with norncotine and its role in aberrant protein glycation (32), we believe this increased concentration could lead to observable effects from nornicotine exposure. Earlier research has shown that cigarette smoking is negatively correlated with Parkinson's disease (35) and positively correlated with the delayed onset of AD (36). These findings have encouraged studies into the proposed neuroprotective effect of nicotine; however, data obtained in vivo have been inconclusive and heavily debated (ref. 37 and references therein). The beneficial effects of nicotine have been attributed to a number of factors including an up-regulation of nicotine receptors that are deficient in the brain, or possibly a protection from the Aβ-induced neurotoxicity (38). Others have postulated that nicotine also serves another role by inhibiting β-amyloidosis through noncovalent binding to either the α-helical structure of Aβ (39) or a small, soluble β-sheet aggregate (40).
We propose that nornicotine can inhibit amyloidosis through similar mechanisms to nicotine and is also capable of covalently inhibiting oligomerization through protein glycation (Fig. 2). Amazingly, this observed increase in reactivity of nornicotine can be attributed simply to the loss of one methyl group from nicotine. Although nornicotine is present in minor amounts, we believe its long half-life and increased concentration in the CNS should have significant biological consequences.
Covalent Modification of Aβ by Nornicotine and Inhibition of Peptide Aggregation. To prepare covalently glycated amyloid peptide, incubations were prepared in which nornicotine, glucose, and Aβ1–40 were incubated in phosphate buffer. After given periods of time, the extent of fibril formation was assessed by using thioflavin T staining of fibrils and subsequent fluorescence detection (41). After incubation for 14 days, a 18% reduction in fluorescence was observed in samples in which nornicotine, glucose, and the peptide were present. This decrease could not be observed in corresponding control incubations containing nornicotine and peptide or nicotine and peptide, leading us to suspect that covalent modification of the amyloid peptide was leading to a detectable decrease in the ability of the peptide to aggregate. In an effort to ascertain the structure of the nornicotine-advanced glycation end product in these reactions, we attempted to use MS to analyze glycation reactions with both the full-length amyloid peptide and the Aβ12–28 fragment used in our subsequent diffusion NMR experiments. Although a distinctive peak is present in the nornicotine glycation reactions by electrospray MS, the change in mass is not readily attributable to common advanced glycation end product structures. The observed mass change roughly corresponds with the addition of one glucose molecule and one nornicotine molecule to the peptide, yet this evidence is not conclusive proof of covalent modification of Aβ by nornicotine. Hence, we began to search for other techniques that allow direct observation of covalently bound nornicotine moieties.
It has been demonstrated that Aβ can give rise to small toxic derivatives other than the highly aggregated species found in amyloid deposits (42, 43). Among these toxic forms are curvi-linear structures known as protofibrils (44) and soluble low-order species secreted from cultured cells (45). Small aggregates, such as dimer and trimers of Aβ, have been shown to impair synaptic function when injected into rat brains; however, Aβ monomers, protofibrils, and fibrils do not share in this property. The critical glycated lysine residue of Aβ is expected to be buried in a hydrophobic core upon fibril formation and not accessible to either soluble glucose or nornicotine. Therefore, we anticipated that nornicotine-based glycation can occur only with soluble low-molecular-weight Aβ species. Incubation of nornicotine and glucose with preformed Aβ fibrils resulted in no detectable decrease in the formation of fibrils by thioflavin T staining. This finding suggests that nornicotine is capable of reacting only with potentially cytotoxic soluble Aβ aggregates, thereby altering the toxicological profile of these compounds.
DOSY and DECRA Analysis of Aβ, Glucose, and Nornicotine Incubations. Pulsed-field gradient (PFG) NMR spectroscopy has become a well-established technique for the accurate measurement of diffusion coefficients of a wide variety of molecules (46, 47). In a basic PFG NMR experiment, two gradient pulses along the z axis (the length of the NMR tube) are used to encode and decode spatial positions of the individual molecules of the sample. As a technique for both biological and organic chemists, PFG NMR is extremely powerful as information regarding the diffusion of sample components can be acquired without chromatographic purification using standard NMR equipment. The use of this technique to study protein aggregation phenomenon has been demonstrated with a wide variety of biological systems (48–50). We chose to use this spectroscopic technique to monitor the interaction of nornicotine with Aβ as opposed to other techniques such as ultracentrifugation and dynamic light scattering as PFG-NMR allows direct measurements to be made rapidly on experimental samples, even with low-molecular-weight samples not readily amenable to other techniques.
In our diffusion NMR studies, we chose to use the Aβ fragment VHHQKLVFFAEDVGSNK (Aβ12–28) as it has been shown to be a suitable model in the study of peptide aggregation. Previous studies using this fragment have shown that, like full-length Aβ, this peptide forms amyloid fibrils above pH 4 (51, 52). Additionally, the reduced size and increased solubility of this peptide relative to the parent peptide increases signal-to-noise ratios in the NMR spectra while also simplifying spectral inter- pretation. Spectra were acquired by using parameters optimized such that maximum decay of the nornicotine resonances were observed.
Assuming a two-state equilibrium in which nornicotine is bound either covalently or noncovalently to the peptide or free in solution, an estimate can be made of the percentage of total nornicotine that is bound to the peptide. In the DOSY analysis, overlapped peaks are not resolved in the diffusion dimension, but instead the observed diffusion coefficient is the weighted average of the two or more species present at a given chemical shift. Based on our diffusion data (Fig. 3A), ≈10% of the nornicotine becomes bound to the peptide after 24 h, presumably because of a noncovalent mechanism. However, after extended incubation, the amount increases to 16% of the total nornicotine as can clearly be observed by the shift in the observed diffusion coefficient for the aromatic resonances of nornicotine (Fig. 3B). Interestingly, this percentage of bound nornicotine roughly correlates with the decrease in thioflavin T fluorescence previously observed in nornicotine glycation reactions (see above).
Fig. 3.

Nornicotine glycation of Aβ12–28 diffusion data. (A) Stacked plot of the obtained 1H PFG-NMR spectra. (B) Pseudo 2D DOSY plot of diffusion data showing the observed differences in diffusion of nornicotine and amyloid peptide. It is important to note that the observed diffusion coefficient of nornicotine in this spectrum is the weighted average of both bound and unbound nornicotine. (C) Results of DECRA analysis of diffusion data showing two distinct populations of nornicotine, one bound to the peptide and the other free in solution.
In cases where spectral overlap occurs or low signal-to-noise ratios are observed, the DOSY analysis is extremely limited. In these cases, spurious results can arise because of time-averaged diffusion coefficients of overlapped components. However, other methods, including DECRA, have been implemented to overcome these obstacles (34). A key advantage of this algorithm resides in its ability to resolve overlapped components without the use of a resonance that is attributable to a single pure component. Using the DECRA method, the output is not a pseudo 2D plot, but instead a stacked plot is generated with each constituent comprising a single 1D NMR spectrum.
We felt nornicotine glycation of Aβ should be readily applicable to DECRA analysis as one would expect the nornicotine molecules that are free in solution and covalently bound to the peptide to have very similar chemical shifts. This fact is corroborated by closely examining a 1D 1H NMR spectrum of the glycation reactions; no distinct resonances can be observed for bound and unbound nornicotine moieties. However, upon applying DECRA to the data, it is very apparent that two different populations of nornicotine moieties are present, a faster moving component comprised of unbound nornicotine, and a component that moves with the diffusion coefficient of the amyloid peptide (Fig. 3C), implying these nornicotine molecules are covalently bound to the peptide. Gratifyingly, the diffusion coefficients resulting from the DECRA analysis are in agreement with those obtained from DOSY analysis of the data. Further corroborative evidence of covalent modification can been seen by analyzing nicotine-Aβ incubations as nicotine can only inhibit aggregation via a noncovalent mechanism (39, 40). In this data set, two populations of nicotine molecules cannot be observed (data not shown).
Detection of Specific Interactions Between Aβ and Nornicotine. To examine the specific nature of the interactions between the amyloid peptide and nornicotine, we conducted NOESY experiments to observe peptide side chains that were in spatial proximity to the nornicotine alkaloid nucleus. Upon analysis of the data, a clear interaction between the pyrrolidine ring of nornicotine and the peptide is visible (Fig. 4A). These data, in concert with the described diffusion NMR experiments (see above), are highly suggestive of a covalent interaction between nornicotine and the amyloid peptide. Examination of the NOESY spectrum from an incubation of Aβ12–28 with glucose and nicotine revealed no visible cross-peaks (Fig. 4B). This finding strongly implicates a covalent mechanism for the interaction of nornicotine with the amyloid peptide. Cross-peaks were visible between residues of the peptide, as well as peaks attributable to nicotine–nicotine interactions, indicating that sufficient signal is present to observe any potential interactions between nicotine and the peptide.
Fig. 4.

Phase-sensitive NOESY spectra of nornicotine protein glycation reactions. (A) Reaction of nornicotine, glucose-d7, and Aβ12–28. The cross-peaks observed between 2.59 and 2.68 ppm (marked with an arrow) are between the peptide and pyrrolidine resonances of nornicotine. (B) Reaction of nicotine, glucose-d7, and Aβ12–28. Critically, the cross-peaks observed with nornicotine are no longer present, whereas other internal nuclear Overhauser effects remain visible.
The purported ability of nicotine as a neuroprotective agent in AD is a hotly contested area of research, with hypotheses as to the nature of this effect ranging from up-regulation of deficient nicotine receptors to noncovalent binding of nicotine preferentially to helical Aβ structures. Our studies may provide an alternative mechanism for this phenomenon that depends on a nicotine metabolite instead of the parent compound itself. Although the significance of nicotine metabolism to nornicotine in the CNS is not understood, perhaps a fortuitous consequence of this increased production of nornicotine is a beneficial effect resulting from protein glycation, a process generally considered to be detrimental. Additionally, our studies have demonstrated a mechanism for the alteration of amyloid protein aggregation through a previously unidentified covalent chemical event. Interestingly, this mechanism of action could also preferentially alter the neurotoxicity of potentially toxic soluble aggregates. Nicotine and nornicotine treatments are intriguing and potentially valuable treatments for AD; however, both compounds have significant toxicity and known psychoactivity. These results argue for increased study into new therapeutic compounds and their libraries capable of covalently modifying Aβ while displaying reduced toxicity and psychoactivity. Finally, nornicotine-based glycation may also play a role in the formation of amyloid plaques resulting from other disease states. For example, the onset of Parkinson's disease has been negatively correlated with smoking (35), and the aggregation of α-synuclein in this disease into amyloid plaques is well characterized.
Acknowledgments
We gratefully acknowledge Dr. Laura Pasternack for helpful discussions and the National Institute on Drug Abuse (Grant F31 DA-15973 to T.J.D.) and The Skaggs Institute for Chemical Biology for financial support.
Abbreviations: AD, Alzheimer's disease; Aβ, amyloid β-peptide; DOSY, diffusion-ordered NMR spectroscopy; DECRA, direct exponential curve resolution algorithm; PFG, pulsed-field gradient.
References
- 1.Maggio, J. E. & Mantyh, P. W. (1996) Brain Pathol. 6 147-162. [DOI] [PubMed] [Google Scholar]
- 2.Alzheimer, A. (1906) Neurol. Centr. 23 1129-1136. [Google Scholar]
- 3.Iqbal, K. (1991) Prevalence and Neurobiology of Alzheimer's Disease (Wiley, New York).
- 4.Heininger, K. (2000) Rev. Neurosci. 11 213-328. [DOI] [PubMed] [Google Scholar]
- 5.Usen, K. E. & Morgan, D. (2001) DNA Cell Biol. 20 677-678. [DOI] [PubMed] [Google Scholar]
- 6.Sisodia, S. S. & Price, D. J. (1995) FASEB J. 9 366-370. [DOI] [PubMed] [Google Scholar]
- 7.Citron, M., Oltersdorf, T., Haass, C., McConlogue, L., Hung, A. Y., Seubert, P., Vigo-Pelfrey, C., Lieberburg, I. & Selkoe, D. J. (1992) Nature 360 672-674. [DOI] [PubMed] [Google Scholar]
- 8.Frautschy, S. A., Baird, A. & Cole, G. M. (1991) Proc. Natl. Acad. Sci. USA 91 8362-8366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldgaber, D., Lerman, M. I., McBride, O. W., Saffiotti, U. & Gajdusek, D. C. (1987) Science 235 877-880. [DOI] [PubMed] [Google Scholar]
- 10.Selkoe, D. J. (1994) Annu. Rev. Cell. Biol. 10 373-403. [DOI] [PubMed] [Google Scholar]
- 11.Scheunner, D., Eckman, C., Jensen, M., Song, X. & Citron, M. (1996) Nat. Med. 2 864-870. [DOI] [PubMed] [Google Scholar]
- 12.Suzuki, C., Golde, T. E. & Younkin, S. G. (1994) Science 264 1336-1340. [DOI] [PubMed] [Google Scholar]
- 13.Zagorski, M. G. (1999) in The Biology-Chemistry Interface, eds. Snyder, J. K. & Cooper, R. (Dekker, New York), Vol. 14, pp. 397-430. [Google Scholar]
- 14.Hilbich, C., Kisters-Woike, B., Reed, J., Masters, C. L. & Beyreuther, K. (1991) J. Mol. Biol. 2118 149-163. [DOI] [PubMed] [Google Scholar]
- 15.Woods, S. J., MacKenzie, L., Maleeff, B., Hurle, M. R. & Wetzel, R. (1996) J. Biol. Chem. 271 4086-4092. [DOI] [PubMed] [Google Scholar]
- 16.Flood, J. F., Roberts, E., Sherman, M. A., Kaplan, B. E. & Morley, J. E. (1994) Proc. Natl. Acad. Sci. USA 91 380-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tjernberg, L. O., Naslund, J., Lindqvist, F., Johansson, J., Karlstrom, A. R., Thyberg, J., Terenius, L. & Nordstedt, C. (1996) J. Biol. Chem. 271 8545-8548. [DOI] [PubMed] [Google Scholar]
- 18.Vitek, M. P., Bhattacharya, K., Glendening, J. M., Stopa, E., Vlassara, H., Bucala, R., Manogue, K. & Cerami, A. (1994) Proc. Natl. Acad. Sci. USA 91 4766-4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith, M. A., Tandea, S., Richey, P. L., Miyata, S., Yan, S.-D., Stern, D., Sayre, L. M., Monnier, V. M. & Perry, G. (1994) Proc. Natl. Acad. Sci. USA 91 5710-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maillard, L. C. (1912) C. R. Acad. Sci. Ser. II 154 66-68. [Google Scholar]
- 21.Ledl, F. & Schleicher, E. (1990) Angew. Chem. Int. Ed. Engl. 29 565-594. [Google Scholar]
- 22.Monnier, V. M., Kohn, R. R. & Cerami, A. (1984) Proc. Natl. Acad. Sci. USA 81 583-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Monnier, V. M. (1990) J. Gerontol. 45 B105-B111. [DOI] [PubMed] [Google Scholar]
- 24.Bucala, R. & Cerami, A. (1996) in The Maillard Reaction: Consequences for the Chemical and Life Sciences, ed. Ikan, R. (Wiley, New York), pp. 161-181.
- 25.Hodge, J. E. (1955) Adv. Carbohydr. Chem. 10 169-205. [DOI] [PubMed] [Google Scholar]
- 26.Ho, C.-T. (1996) in The Maillard Reaction: Consequences for the Chemical and Life Sciences, ed. Ikan, R. (Wiley, New York), pp. 27-53.
- 27.Bardo, M. T., Green, T. A., Crooks, P. A. & Dwoskin, L. P. (1999) Psychopharmacology 146 290-296. [DOI] [PubMed] [Google Scholar]
- 28.Kisaki, T. & Tamaki, E (1961) Arch. Biochem. Biophys. 92 351-355. [DOI] [PubMed] [Google Scholar]
- 29.Curvall, M. & Kazeni, V. E (1993) in Nicotine and Related Alkaloids: Absorption, Distribution, Metabolism, and Excretion, eds. Gorrod, J. W. & Wahren, J. (Chapman & Hall, Stockholm), pp. 147-179.
- 30.Crooks, P. A., Li, M. & Dwoskin, L. P (1997) Drug Metab. Dispos. 25 47-54. [PubMed] [Google Scholar]
- 31.Dickerson, T. J. & Janda, K. D (2002) J. Am. Chem. Soc. 124 3220-3221. [DOI] [PubMed] [Google Scholar]
- 32.Dickerson, T. J. & Janda, K. D (2002) Proc. Natl. Acad. Sci. USA 99 15084-15088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stejskal, E. O. & Tanner, J. E (1965) J. Chem. Phys. 42 288-292. [Google Scholar]
- 34.Antalek, B. & Windig, W. (1996) J. Am. Chem. Soc. 118 10331-10332. [Google Scholar]
- 35.Bharucha, N. E., Stokes, L., Schoenberg, B. S., Ward, C., Ince, S., Nutt, J. G., Eldridge, R., Calne, D. B., Mantel, N. & Duvoisin, R. (1986) Neurology 36 284-288. [DOI] [PubMed] [Google Scholar]
- 36.van Duijn, C. M. & Hofman, A. (1991) Br. Med. J. 302 1491-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zamani, M. R. & Allen, Y. S. (2001) Biol. Psychiatry 49 221-232. [DOI] [PubMed] [Google Scholar]
- 38.Kihara, T., Shimohama, S., Sawada, H., Kimura, J., Kume, T., Kochiyama, H., Maeda, T. & Akaike, A. (1997) Ann. Neurol. 42 159-163. [DOI] [PubMed] [Google Scholar]
- 39.Salomon, A. R., Marcinowski, K. J., Friedland, R. P. & Zagorski, M. G. (1996) Biochemistry 35 13568-13578. [DOI] [PubMed] [Google Scholar]
- 40.Zeng, H., Zhang, Y., Peng, L.-J., Shao, H., Menon, N. K., Yang, J., Salomon, A. R., Freidland, R. P. & Zagorski, M. G. (2001) Biol. Psychiatry 49 248-257. [DOI] [PubMed] [Google Scholar]
- 41.LeVine, H., III (1993) Protein Sci. 2 404-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oda, T., Wals, P., Osterburg, H. H., Johnson, S. A. & Pasinehi, G. M. (1995) Exp. Neurol. 136 22-31. [DOI] [PubMed] [Google Scholar]
- 43.Oda, T., Pasinehi, G. M., Osterberg, H. H., Anderson, D., Johnson, S. A. & Finch, C. E. (1994) Biochem. Biophys. Res. Commun. 204 1131-1136. [DOI] [PubMed] [Google Scholar]
- 44.Walsh, D. M., Hartley, D. M., Kusumoto, Y., Fezoui, Y., Condron, M. M., Lomakin, A., Benedek, G. B., Selkoe, D. J. & Teplow, D. B. (1999) J. Biol. Chem. 274 25945-25952. [DOI] [PubMed] [Google Scholar]
- 45.Walsh, D. M., Klyubin, I., Fadeeva, J. V., Cullen, W. K., Anwyl, R., Wolfe, M. S., Rowan, M. J. & Selkoe, D. J. (2002) Nature 416 535-539. [DOI] [PubMed] [Google Scholar]
- 46.Johnson, C. S., Jr. (1999) Progr. Nuclear Magn. Reson. Spectrosc. 34 203-256. [Google Scholar]
- 47.Antalek, B. (2002) Concepts Magn. Reson. 14 225-258. [Google Scholar]
- 48.Mansfield, S. L., Jayawickrama, D. A., Timmons, J. S. & Larive, C. K. (1998) Biochim. Biophys. Acta 1382 257-265. [DOI] [PubMed] [Google Scholar]
- 49.Altieri, A. S., Hinton, D. P. & Byrd, R. A. (1995) J. Am. Chem. Soc. 117 7566-7567. [Google Scholar]
- 50.Krishnan, V. V. (1997) J. Magn. Reson. 124 468-473. [DOI] [PubMed] [Google Scholar]
- 51.Kirschner, D. A., Indouye, H., Duffy, L. K., Sinclair, A., Lind, M. & Selkoe, D. J. (1987) Proc. Natl. Acad. Sci. USA 84 6953-6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Castano, E. M., Ghiso, J., Prelli, F., Gorevic, P. D., Migheli, A. & Frangione, B. (1986) Biochem. Biophys. Res. Commun. 141 782-789. [DOI] [PubMed] [Google Scholar]