

Abstract
Asymmetric cell division in Caulobacter crescentus yields daughter cells that have different cell fates. Compartmentalization of the predivisional cell is a critical event in the establishment of the differential distribution of regulatory factors that specify cell fate. To determine when during the cell cycle the cytoplasm is compartmentalized so that cytoplasmic proteins can no longer diffuse between the two nascent progeny cell compartments, we designed a fluorescence loss in photobleaching assay. Individual cells containing enhanced GFP were exposed to a bleaching laser pulse tightly focused at one cell pole. In compartmentalized cells, fluorescence disappears only in the compartment receiving the bleaching beam; in noncompartmentalized cells, fluorescence disappears from the entire cell. In a 135-min cell cycle, the cells were compartmentalized 18 ± 5 min before the progeny cells separated. Clearance of the 22000 CtrA master transcriptional regulator molecules from the stalked portion of the predivisional cell is a controlling element of Caulobacter asymmetry. Monitoring of a fluorescent marker for CtrA showed that the differential degradation of CtrA in the nascent stalk cell compartment occurs only after the cytoplasm is compartmentalized.
The bacterium Caulobacter crescentus divides asymmetrically, producing two different daughter cells: the swarmer cell and the stalked cell (Fig. 1A). These cells differ in their transcription programs, protein composition, and behavior (1, 2). A critical event in Caulobacter cell division is the compartmentalization of the nascent daughter cells. Logically, compartmentalization must occur before separation of the progeny cells, but there has been no direct measurement of the timing of this event. In electron micrographs of dividing Caulobacter cells, a septum is not detected before the completion of cell division (3). The Caulobacter cell is thought to divide by progressive constriction of the FtsZ ring until the two daughter cells eventually separate (4, 5). FtsZ is a highly conserved tubulin-like GTPase that forms a ring at the division plane ≈90 min into a 180-min Caulobacter cell cycle; ≈30 min later, constriction of the cell is visible. Constriction takes ≈60 min, from the first visible constriction to cell separation (5). The detailed mechanisms of the last stages of bacterial cell division, including closing off the inner and outer membranes, are unknown.
Fig. 1.

(A) C. crescentus cell cycle. The motile swarmer cell (SW) has a polar flagellum (wavy line) and pili (straight lines), contains the CtrA protein (gray shading), and does not replicate its DNA (nonreplicating DNA represented as a ring). During differentiation into a stalked cell (ST), the flagellum is shed and a stalk is built at the same pole, CtrA is proteolyzed, and DNA replication initiates (theta structure). CtrA accumulates in the predivisional cell (PD), then is cleared from the stalked compartment of the late PD cell. The PD cell divides to yield a SW cell and a ST cell. (B and C) Two representative FLIP experiment results demonstrating the absence (B) and the presence (C) of compartmentalization. (B) A fluorescence image of a C. crescentus cell expressing EGFP, before (Left) and 2 sec after (Right) photobleaching. The cellwide loss of fluorescence indicates that this cell is not yet compartmentalized. (C) The same experiment performed on a different cell. In this case, retention of fluorescence in the portion of the cell distal to the focused laser beam shows that this cell is compartmentalized. White circles, focused laser used for bleaching. The diameter of the circles corresponds to the full width half maximum size of the laser beam. (Scale bar = 1 μm.)
The two-component signal transduction response regulator, CtrA, controls multiple events in the Caulobacter cell cycle (6). CtrA directly regulates at least 95 genes organized in 55 operons (7). Phosphorylated CtrA binds to the origin of replication, repressing initiation of chromosome replication (8, 9). CtrA is present and phosphorylated in the swarmer cell, preventing chromosome replication. Proteolysis of CtrA at the swarmer-to-stalked cell transition allows initiation of replication in the stalked cell. CtrA accumulates in the predivisional cell, and then is cleared by proteolysis from the stalked compartment of the late predivisional cell (Fig. 1 A). Activated (i.e., phosphorylated) CtrA in the swarmer compartment maintains the repression of replication, whereas removal of CtrA from the stalked compartment allows a new round of replication to begin. Differential degradation and phosphorylation of CtrA in the nascent daughter cells has been shown to be essential for establishment of their distinct fates (10).
Solution of the diffusion equation (11) describing the time evolution of protein concentration in bacteria-sized cells shows that any transient heterogeneity in the concentration of free cytoplasmic proteins will disappear by diffusion in ≈100 msec (assuming a diffusion coefficient on the order of 10 μm2/sec). Therefore, before compartmentalization, untethered cytoplasmic proteins should be uniformly distributed throughout the predivisional cell by diffusion. In the late predivisional stage, when Caulobacter contains a nonuniform distribution of CtrA protein, we expect that either the CtrA protein is not freely diffusing, or that a barrier blocks diffusion between the two compartments of the predivisional cell. To distinguish between these possibilities, we investigated the timing of predivisional cell compartmentalization, relative to asymmetric CtrA degradation and progeny cell separation.
Materials and Methods
For more details, see Supporting Materials and Methods, which is published as supporting information on the PNAS web site, www.pnas.org.
Bacterial Strains and Plasmids. Strain LS3008 (M. Laub and J. Skerker, personal communication) has enhanced GFP (EGFP) (BD Biosciences) under the control of the xylose promoter (Pxyl) (12) integrated into the chromosome of CB15N (a synchronizable derivative of the wild-type strain CB15) at the Pxyl locus. Plasmid pEJ165 contains enhanced yellow fluorescent protein (EYFP) (BD Biosciences) fused to the first 117 aa of the CtrA gene, followed by the last 15 aa of CtrA (YFP-CtrA RD + 15). pEJ165 is derived from pEJ146 (13) and is pMR10 based. Plasmid pEJ176 contains a red fluorescent protein (mRFP1.1, a derivative of mRFP1; ref. 14) (R. Tsien and R. Campbell, personal communication) under Pxyl in the pJS14 plasmid. pEJ165 and pEJ176 were transferred from Escherichia coli S17–1 into C. crescentus CB15N by conjugation (15) to make strain EJ188.
Microscopy and Photobleaching. Fluorescence microscopy was performed on a Nikon TE300 inverted microscope with a Nikon S Fluor ×100 objective with a numerical aperture of 1.3. Images were recorded with a Princeton Instruments Intensified PentaMAX charge-coupled device camera. Either the 488-nm or the 514-nm line of a Coherent Innova 200 Argon Ion laser was used for illumination. A lens of focal length 2.5 cm was placed 2.5 cm from the back of the microscope, on a mount that allowed the lens to be moved in and out of the beam path. With the lens in the path, the laser formed a Gaussian spot with a full-width-half-maximum (FWHM) size of 9 μm. Without the lens, the FWHM size was 1 μm. The compartmentalization assay was performed as follows: a cell, immobilized on an agarose pad, was positioned with one end at the focal point of the laser. The cell was imaged in widefield mode with the lens in the beam path. The lens was removed from the path and the cell was bleached with the focused beam for 5 sec. During the bleaching pulse, the camera intensifier was switched off to avoid damage. The lens was put back into the beam, the intensifier was switched on, and the cell was imaged again. The exposure time for each widefield image was 1 sec. The time from the start of the bleaching pulse to the second image was 7 sec.
Results
Measurement of the Timing of Compartmentalization of Predivisional Cells. The time in the cell cycle when cytoplasmic diffusion no longer occurs between the two compartments of a predivisional cell was measured by a fluorescence loss in photobleaching (FLIP) assay (16) using a fluorescence microscope with a 488-nm laser as the illumination source. Illumination was switched from wide field for fluorescent imaging to a focused spot for bleaching by removing a lens in the light path. Caulobacter cells expressing EGFP (strain LS3008) were imaged and then bleached with the laser focused at one end of the cell for 5 sec. A second fluorescence image was then taken of the bleached cells two sec later (Fig. 1 B and C). We refer to the end of the cell that received the laser pulse as the proximal end, and the other end of the cell as the distal end. In a cell containing no cytoplasmic diffusion barrier, rapid diffusion of EGFP results in bleaching of all EGFP in the cell during the duration of the polar-focused laser pulse. After bleaching, some cells displayed no EGFP signal (Fig. 1B); in others, however, the distal compartment of the cell remained fluorescent (Fig. 1C), indicating that a physical barrier was preventing diffusion between the two compartments.
For each cell assayed, we calculated the decrease in fluorescence intensity of the distal half of the cell after the bleaching pulse (ΔId). As a control, LS3008 cells were treated identically to those in the FLIP assay, except that the laser was focused ≈1.5 μm away from the cell. Aside from nonspecific bleaching by the tails of the laser spot and by the widefield illumination used for imaging, the cells in this control experiment should remain fluorescent after the laser pulse. The percent decrease in fluorescence intensity (ΔI) of these control cells after the “bleaching” pulse was calculated and found to range from 3% to 31% (Fig. 2A). The variation in ΔI for the control cells is attributable to variation in the focal plane and variation in the distance from the focused laser to the cell. Of the 115 cells treated with the FLIP assay, 112 had ΔId < 35% (indicating compartmentalization) or ΔId > 87% (indicating no compartmentalization) (Fig. 2 A). Only three cells showed intermediate values of ΔId, indicating reduced but nonzero diffusion between the two halves of the cell. Those cells with ΔId > 35% were said not to be compartmentalized, because they were distinguishable from the control population. The remaining cells were said to be compartmentalized.
Fig. 2.

(A) Histogram of decrease in average fluorescence intensity of the distal portion of the cell after the bleaching pulse (ΔId), for strain LS3008. Experimental cells were bleached with a laser focused at one pole, as in Fig. 1. Control cells were treated identically, except that the laser was focused ≈1.5 μm to the side of the cell. Cells with ΔId < 35% were said to be compartmentalized. (B) Fraction of swarmer cells, stalked cells, and early and late predivisional cells present in the synchronized population as a function of time. Cell type was determined by morphology (see Supporting Materials and Methods). Seventy-five minutes into the cell cycle, the population consists primarily of stalked cells. As the cell cycle progresses, stalked cells develop into predivisional cells, which then divide to form new swarmer and stalked cells (hatching). (C) Time lag between compartmentalization and cell division. Triangles show the fraction of cells that are compartmentalized. Circles show the fraction of the originally isolated swarmer cells that have completed cell division and separated into two progeny cells (Fd). Fd = Nd/2(Nt - Nd/2), where Nd is the number of divided cells counted and Nt is the total number of cells counted. This equation corrects for the fact that, after a cell has divided, it will be counted as two cells. Each point on the “compartmentalized” curve is calculated from measurements on 22–34 cells, except the 75-min time point (14 cells). Error bars show the standard deviation of the binomial distribution (33). Each point on the “divided” curve is based on at least 300 cells. Error bars on these points show an estimate of the error due to cells whose stage in the cell cycle cannot be unambiguously determined based on their morphology.
To determine when in the cell cycle the barrier appears, the FLIP assay was performed on samples taken every 15 min from a synchronized population of Caulobacter cells grown in liquid media (Fig. 2C). A separate aliquot of cells was fixed at each time point where the FLIP assay was performed, and the percentage of swarmer cells, stalked cells, and early and late predivisional cells (categorized by cell morphology) was determined (Fig. 2B). At 120 min we began to observe newly divided progeny cells in the synchronized culture. Because we want to determine the fraction of the originally isolated swarmer cells that have become compartmentalized, we assayed only predivisional cells (based on morphology) after the 105-min time point. However, the 75- to 105-min time points still represent the most accurate measurements. Based on the data from this time period, we conclude that the cytoplasmic barrier forms 18 ± 5 min before the progeny cells separate (Fig. 2C). Barriers were detected only in highly pinched predivisional cells, such as the cell in Fig. 1C, and never in unpinched or slightly pinched cells, such as the left cell in Fig. 1B. However, it was not possible to determine whether a cell had a barrier based on morphology alone, because some highly pinched predivisional cells were fully bleached, indicating diffusion between the two halves of the cell.
To determine whether the observed diffusion barrier also prevented diffusion on longer time scales, we imaged some compartmentalized cells again 5 or 10 min after the FLIP assay. If slow diffusion was still occurring between the two halves of the cell, then the unbleached GFP in the distal half would be expected to diffuse into the proximal half, and the cell should appear uniformly fluorescent at the end of 10 min. If no intercompartment diffusion occurs, then the image taken after 10 min should be identical to the picture taken immediately after bleaching; the distal half should be fluorescent and the proximal half should be dark. Twenty of twenty cells that showed a barrier in the initial FLIP assay showed no diffusion after 5 min, and 10 of 10 cells that showed a barrier in the initial FLIP assay showed no diffusion after 10 min. We conclude that the vast majority of the cells classified as compartmentalized with the FLIP assay had no measurable diffusion of EGFP between the two halves of the cell on a 10-min time scale.
Simulation Analysis of Diffusion in Pinched Cells. A diffusion simulation model was used to address two questions: (i) what is the largest hole size at the point of constriction consistent with the FLIP assay results? (ii) Could the barrier block the diffusion of protein-sized molecules but permit diffusion of smaller molecules? The cell was modeled as a cylinder of length 3 μm and diameter 0.5 μm. The barrier was modeled as a disk across the middle of the cell, with a circular hole in the center. Cases with various combinations of disk width, w, and hole diameter, d, were simulated. The laser was modeled as having a one-dimensional Gaussian profile, centered at one end of the cell, with a full-width-half-maximum size of 1 μm and a maximum intensity of 13 kW/cm2. The GFP diffusion coefficient was taken to be 8 μm2/sec (17). The photobleaching quantum yield of EGFP was taken to be 8.3 × 10-6 (18). Published values of the photobleaching quantum yield for EGFP vary (18, 19), but we found that the results of the simulation were dominated by diffusion effects and not strongly dependent on bleaching parameters. The diffusion equation (DE) in cylindrical coordinates, reduced to two dimensions owing to radial symmetry, and with an added term to account for photobleaching is
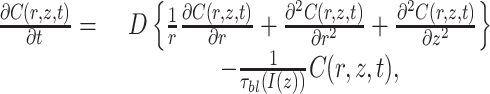 |
[1] |
where C is the concentration of unbleached EGFP, D is the diffusion coefficient, and τbl is the 1/e bleaching time of EGFP for laser intensity I. The DE was solved numerically using the Matlab “PDE Tools” package (see Data Sets 1 and 2, which are published as supporting information on the PNAS web site, for Matlab files). Results show that a cell could appear compartmentalized in the FLIP assay while still allowing diffusion of EGFP between the two compartments that would be observable on longer time scales. For example, a cell with w = 100 nm and d = 12 nm would appear compartmentalized in the FLIP assay. Specifically, 2 sec after photobleaching, the fluorescence signal would be gone from the proximal cell compartment but remain in the distal compartment, as in Fig. 1C Right. However, if the same cell were imaged 10 min after bleaching, the cell would appear uniformly fluorescent due to diffusion between the two compartments. To maintain an asymmetric distribution of unbleached EGFP for 10 min, as observed in our experiments, the intercompartment hole would have to be smaller than 2.4 nm. A GFP molecule has an approximately cylindrical shape of length 4.2 nm and diameter 2.4 nm (20). Therefore, we predict that, for the cells classified as compartmentalized in our experiments, the residual hole, if any, between the two compartments of the predivisional cell is smaller than a GFP molecule. The three cells with intermediate values of ΔId in Fig. 2 A may represent cells with intercompartment holes slightly larger than the diameter of EGFP, allowing slow diffusion between the two compartments. CtrA has a molecular weight of 26 kDa, compared with 27 kDa for EGFP. Therefore, a barrier that blocks EGFP diffusion should also block CtrA diffusion. It is possible that a small hole could block the passage of EGFP but still allow small molecules to pass through; however, diffusion through such a small hole would be slow. For example, for w = 100 nm and d = 2.4 nm, a molecule with a diffusion coefficient five times that of EGFP (as might be expected for a molecule of molecular mass = 200 Da) (21) could equilibrate its concentration in the two compartments in ≈3 min. The equilibration time was calculated by simulating diffusion in a cell with initial GFP concentration of 1 in one half and 0 in the other half and determining the time required for the concentration difference between the two compartments to become <0.1.
Correlation of Compartmentalization with CtrA Asymmetry. We determined the number of CtrA molecules in swarmer and predivisional cells by probing Western blots of Caulobacter cell extracts and known quantities of purified CtrA protein with a CtrA antibody (6). There are 9,500 ± 2,000 molecules of CtrA in a swarmer cell and 22,000 ± 4,000 molecules in a predivisional cell. The 22,000 molecules of CtrA are preferentially degraded in the stalked portion of the predivisional cell, by a proteolytic mechanism either localized or specifically activated in this compartment. To determine the relative timing of the formation of the diffusion barrier and the appearance of an asymmetric CtrA distribution in predivisional cells, we analyzed cells containing both a red fluorescent protein (mRFP1.1) and a yellow fluorescent fusion protein as a marker for CtrA degradation (YFP-CtrA RD + 15) (strain EJ188). The amount of the YFP-CtrA RD + 15 fusion protein has been shown to follow the amount of native CtrA protein in cells (13). The spatial distribution of YFP-CtrA RD + 15 protein also corresponds to the spatial distribution of native CtrA as assayed by immunofluorescence (10, 13). Therefore we used this fusion protein to assay the distribution of CtrA in the same cells where the FLIP assay was performed on the mRFP1.1 molecules. We performed this two-color assay on late predivisional cells (selected from a mixed population by morphology), by using a fluorescence microscope configured to acquire simultaneous images of the red and yellow fluorescence. These images were assessed for compartmentalization and for symmetry or asymmetry of CtrA and placed into one of four possible categories (Fig. 3). The mRFP1.1 images were analyzed as for the EGFP images described above. The same control experiment of bleaching a region outside the cell was performed. In this case the decrease in intensity in the control cells (ΔI) was 15–53% (Fig. 3E). Experimental cells with ΔId < 60% were said to be compartmentalized. The YFP-CtrA RD + 15 images were analyzed by computing the average fluorescence intensity of each compartment of the cell, then calculating the percent difference in intensity between the two compartments. As a control, the difference in intensity between the two compartments of cells containing EGFP was computed and found to range from 0% to 25% (Fig. 3F). Asymmetry in the control cells is attributable to imperfect correction for uneven illumination, and focal plane variation. Experimental cells with a >25% difference in intensity between the two compartments were classified as having an asymmetric CtrA distribution, because they were distinguishable from the control cells. Of 164 cells, 51 had symmetric CtrA and were not compartmentalized; 42 had symmetric CtrA and were compartmentalized; 71 had asymmetric CtrA and were compartmentalized; and 0 had asymmetric CtrA and were not compartmentalized. Because we never observed a noncompartmentalized cell with asymmetric CtrA, we conclude that the cytoplasmic diffusion barrier forms before the appearance of an asymmetric distribution of CtrA. In the case of the 42 compartmentalized cells with a symmetric distribution of CtrA, we surmise that we caught the cells at the start of or just before initiation of CtrA degradation.
Fig. 3.

Simultaneous observation of compartmentalization and asymmetric CtrA localization. (A–C) Representative images. (Top) Images are false colored; yellow images represent signal from YFP-CtrA RD + 15. (Middle and Bottom) Gray images represent signal from cytoplasmic mRFP1.1. (Top and Middle) Images were taken before bleaching. White circles show the placement of the focused laser used for bleaching. The diameter of the circles is 1 μm, corresponding to the full width half maximum size of the laser beam. (Bottom) Images show the same cells after bleaching. (D) Schematic showing the expected results from a cell that had asymmetric CtrA but no diffusion barrier. We did not observe any cells of this type. (E) Histogram of decrease in red fluorescence intensity of the distal portion of the cell after the bleaching pulse (ΔId) for strain EJ188. Experimental cells were treated with the FLIP assay in the two-color compartmentalization/CtrA degradation experiment as shown in A–D. Control cells were treated identically, except that the laser was focused ≈1.5 μm to the side of the cell. Cells with ΔId < 60% were said to be compartmentalized. (F) Histogram of fluorescence asymmetry. Percent fluorescence asymmetry is defined as Af = (Ibright - Idim)/Ibright, where Ibright and Idim are the average fluorescence intensities of the bright and dim compartments of the cell, respectively. Experimental cells are strain EJ188. Af for the experimental cells was calculated by using the yellow fluorescence signal from the YFP-CtrA RD + 15 fusion protein. Control cells are strain LS3008, containing cytoplasmic EGFP. Af for the control cells was calculated by using the green fluorescence signal from EGFP. EJ188 cells with Af > 25% were said to have asymmetric CtrA.
Discussion
The two compartments of the Caulobacter predivisional cell differ in protein composition and transcriptional activity. Specifically, CtrA is present only in the swarmer compartment of the late predivisional cell where it functions to silence the origin of replication. Several genes are selectively transcribed in the swarmer compartment, including the flagellar fliK (formerly flbG) operon and the fljK gene (22, 23), and in the stalked compartment, including hemE (9), gyrB (24), lon, and dnaK (25). A cytoplasmic reporter protein transcribed from the hemE promoter in the ≈20 min preceding cell division is found preferentially in the stalked progeny cells (9), suggesting that a diffusion barrier exists between the two compartments of the predivisional cell before progeny cell separation. Transcription of the hemE gene is repressed by CtrA∼P, accounting for the restriction of hemE transcription to the stalked half of the late predivisional cell. However, gyrB, lon, dnaK, fliK, and fljK are not directly regulated by CtrA (7), implying that other transcriptional regulators are present or active in only one compartment of the late predivisional cell. Barrier formation may determine the time at which differential transcription, degradation, and activation of cytoplasmic proteins can be established in the two compartments of the predivisional cell.
Here, we demonstrate the presence of a barrier to diffusion between the two compartments of the late predivisional cell, and show that it occurs 18 ± 5 min before cell separation. The physical composition of this barrier is unknown, but may be the final fusion of the membrane layers. The Bacillus subtilis gene SpoIIIE and its E. coli homologue FtsK have been postulated to have a role in the translocation of DNA through the closing septum (26). Although Caulobacter does not have a septum that is visible in electron micrographs, the final step in the creation of the diffusion barrier 18 min before cell separation may be the interaction of FtsK multimers with the constriction ring, ensuring the creation of two compartments.
A mechanism by which compartmentalization of a predivisional cell could lead to differential gene transcription has been postulated for activation of fliK transcription by the FlbD response regulator in the swarmer compartment of the late predivisional cell. FlbD is required for activation of the class III and IV flagellar genes, including fliK. Activation of fliK by FlbD requires the MS-ring, a membrane-embedded part of the flagellar structure. Muir and Gober (27) hypothesize that in early predivisional cells, FlbD is activated by the presence of the MS ring, and activated FlbD is free to diffuse throughout the cell; after barrier formation, only the FlbD in the swarmer compartment remains activated, because only the swarmer cell contains a flagellum. In this example, the formation of the diffusion barrier, combined with the asymmetrically located flagellar structure, directly causes different transcription patterns in the swarmer and stalked compartments. It does so by blocking cell-wide distribution, via diffusion, of the signal originating at the flagellated pole.
The C. crescentus predivisional cell, containing phosphorylated CtrA, divides into a swarmer cell, containing phosphorylated CtrA, and a stalked cell, lacking CtrA (Fig. 1A). Experiments on a nonproteolyzable CtrA mutant show that even when CtrA is present in stalked cells, it is not phosphorylated (10). Therefore, the cell has to selectively degrade CtrA in the nascent stalked cell and selectively phosphorylate CtrA in the nascent swarmer cell. Degradation and phosphorylation of CtrA are thought to be separately regulated. Does the cell sense that is has become compartmentalized, and only then initiate CtrA degradation and dephosphorylation in the stalked compartment? The cell could sense the onset of compartmentalization using a mechanism similar to the one proposed above for selective activation of fliK transcription by FlbD. If the kinase responsible for phosphorylating CtrA were present only in the swarmer compartment, then barrier formation would isolate the CtrA in the stalked compartment from the kinase, leading to its dephosphorylation. Similarly, if an inhibitor of CtrA degradation were located in the swarmer compartment, then on compartmentalization of the predivisional cell, repression of CtrA degradation would be removed in the stalked compartment, initiating CtrA degradation. It is also possible that the completion of compartmentalization could be signaled by release of a regulatory factor from the site of cell division. This regulatory factor could turn on an activator of CtrA degradation located in the stalked compartment. Recent work suggests that CtrA degradation requires an activating factor (13).
Finally, it is possible that regulation of CtrA degradation and dephosphorylation is unrelated to compartmentalization. In this case, CtrA degradation and dephosphorylation could begin before compartmentalization under some conditions. We investigated the consequences to the cell of a lack of synchronization of CtrA degradation, CtrA dephosphorylation, and compartmentalization. If CtrA degradation were to begin before the barrier formed, the amount of CtrA in the whole cell would be reduced, assuming that most of the CtrA in the cell is freely diffusing. The early predivisional cell contains 22,000 molecules of CtrA, or 62 μM. The dissociation constants for the five CtrA binding sites at the origin of replication range from 0.2 μMto0.6 μM for unphosphorylated CtrA and from 0.003 μM to 0.015 μM for CtrA∼P (28). We calculated the probability of having all five origin CtrA sites fully occupied, for various concentrations of phosphorylated and unphosphorylated CtrA. We assume that two copies of CtrA bind cooperatively to each site with a Hill coefficient of 2, and that there are 110 additional CtrA sites on the chromosome (7), with a dissociation constant of 0.35 μM for unphosphorylated CtrA and 0.008 μM for CtrA∼P. We find that the five origin CtrA sites will be fully occupied with 99% probability if the cell contains 0.52 μM of phosphorylated CtrA or 8.7 μM of unphosphorylated CtrA. CtrA is stable in swarmer cells but at the swarmer-to-stalk transition the half-life of CtrA abruptly switches to 4 min. CtrA∼P reverts to the unphosphorylated form with a half-life of 5 min in asynchronous cultures (10). If we assume that these half-lifes apply in the stalked compartment of the predivisonal cell, then the cell could tolerate 15 min of premature degradation and dephosphorylation and still have all five CtrA sites at the origin occupied with 99% probability. The concentration of CtrA∼P required to repress DNA replication initiation is unknown. However, because binding of CtrA∼P at the origin represses initiation, it seems unlikely that DNA replication could initiate when all of the origin sites are bound by CtrA. We used the same rationale to predict a lag of at least 15 min between the start of CtrA degradation and the initiation of DNA replication in the stalked compartment, suggesting that replication in the nascent stalked cell compartment would begin near the time of cell separation. This prediction is consistent with data showing that the replisome forms in the stalked compartment of late predivisional cells near the time of progeny cell separation (29).
Diffusion barriers are crucial for generating and maintaining asymmetric distributions of molecules in many organisms. For example, a membrane diffusion barrier exists between the mother cell and the bud in Saccharomyces cerevisiae, and this diffusion barrier is required for the localization of the IST2 protein to the bud (30). In sporulating Bacillus subtilis cells, the septum prevents diffusion between the mother cell and the forespore, allowing the protein composition of the two compartments to diverge (31). Photobleaching has been used to demonstrate the presence of compartments in the periplasm of Escherichia coli, though the function of these compartments is unknown (32). The photobleaching technique described in this work could be used to determine the presence and timing of compartmentalization in a wide range of systems. In Caulobacter, the detection of a barrier to cytoplasmic diffusion and the determination of the time in the cell cycle at which it forms has provided insights into the process of asymmetric cell division.
Supplementary Material
Acknowledgments
We are grateful to R. Tsien and R. Campbell for providing a plasmid containing mRFP1.1 in advance of publication. We thank J. Deich and members of the Moerner laboratory for technical assistance. We thank members of the McAdams and Shapiro laboratories for critical reading of the manuscript. E.M.J. and H.H.M. are supported by Office of Naval Research Grant N00014-02-1-0538. K.R.R. and L.S. are supported by National Institutes of Health Grant GM32506/5120M2. W.E.M. is supported by National Science Foundation Grant MCB-0212503. This work was supported by Defense Advanced Research Projects Agency Grant MDA972-00-1-0032.
Abbreviations: EGFP, enhanced GFP; YFP, yellow fluorescent protein; RFP, red fluorescent protein; FLIP, fluorescence loss in photobleaching.
References
- 1.Ryan, K. R. & Shapiro, L. (2003) Annu. Rev. Biochem. 72 367-394. [DOI] [PubMed] [Google Scholar]
- 2.Shapiro, L., McAdams, H. H. & Losick, R. (2002) Science 298 1942-1946. [DOI] [PubMed] [Google Scholar]
- 3.Poindexter, J. S. & Hagenzieker, J. G. (1981) Can. J. Microbiol. 27 704-719. [DOI] [PubMed] [Google Scholar]
- 4.Quardokus, E., Din, N. & Brun, Y. V. (1996) Proc. Natl. Acad. Sci. USA 93 6314-6319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quardokus, E. M., Din, N. & Brun, Y. V. (2001) Mol. Microbiol. 39 949-959. [DOI] [PubMed] [Google Scholar]
- 6.Quon, K. C., Marczynski, G. T. & Shapiro, L. (1996) Cell 84 83-93. [DOI] [PubMed] [Google Scholar]
- 7.Laub, M. T., Chen, S. L., Shapiro, L. & McAdams, H. H. (2002) Proc. Natl. Acad. Sci. USA 99 4632-4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quon, K. C., Yang, B., Domian, I. J., Shapiro, L. & Marczynski, G. T. (1998) Proc. Natl. Acad. Sci. USA 95 120-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marczynski, G. T., Lentine, K. & Shapiro, L. (1995) Genes Dev. 9 1543-1557. [DOI] [PubMed] [Google Scholar]
- 10.Domian, I. J., Quon, K. C. & Shapiro, L. (1997) Cell 90 415-424. [DOI] [PubMed] [Google Scholar]
- 11.Crank, J. (1998) The Mathematics of Diffusion (Oxford Univ. Press, Oxford).
- 12.Meisenzahl, A. C., Shapiro, L. & Jenal, U. (1997) J. Bacteriol. 179 592-600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ryan, K. R., Judd, E. M. & Shapiro, L. (2002) J. Mol. Biol. 324 443-455. [DOI] [PubMed] [Google Scholar]
- 14.Campbell, R. E., Tour, O., Palmer, A. E., Steinbach, P. A., Baird, G. S., Zacharias, D. A. & Tsien, R. Y. (2002) Proc. Natl. Acad. Sci. USA 99 7877-7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ely, B. (1991) Methods Enzymol. 204 372-384. [DOI] [PubMed] [Google Scholar]
- 16.White, J. & Stelzer, E. (1999) Trends Cell Biol. 9 61-65. [DOI] [PubMed] [Google Scholar]
- 17.Elowitz, M. B., Surette, M. G., Wolf, P. E., Stock, J. B. & Leibler, S. (1999) J. Bacteriol. 181 197-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peterman, E. J. G., Brasselet, S. & Moerner, W. E. (1999) J. Phys. Chem. A 103 10553-10560. [Google Scholar]
- 19.Harms, G. S., Cognet, L., Lommerse, P. H., Blab, G. A. & Schmidt, T. (2001) Biophys. J. 80 2396-2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ormo, M., Cubitt, A. B., Kallio, K., Gross, L. A., Tsien, R. Y. & Remington, S. J. (1996) Science 273 1392-1395. [DOI] [PubMed] [Google Scholar]
- 21.Freifelder, D. (1982) Physical Biochemistry (Freeman, New York).
- 22.Gober, J. W., Champer, R., Reuter, S. & Shapiro, L. (1991) Cell 64 381-391. [DOI] [PubMed] [Google Scholar]
- 23.Milhausen, M. & Agabian, N. (1983) Nature 302 630-632. [DOI] [PubMed] [Google Scholar]
- 24.Rizzo, M. F., Shapiro, L. & Gober, J. (1993) J. Bacteriol. 175 6970-6981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reuter, S. H. & Shapiro, L. (1987) J. Mol. Biol. 194 653-662. [DOI] [PubMed] [Google Scholar]
- 26.Errington, J., Daniel, R. A. & Scheffers, D. J. (2003) Microbiol. Mol. Biol. Rev. 67 52-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muir, R. E. & Gober, J. W. (2002) Mol. Microbiol. 43 597-615. [DOI] [PubMed] [Google Scholar]
- 28.Siam, R. & Marczynski, G. T. (2000) EMBO J. 19 1138-1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jensen, R. B., Wang, S. C. & Shapiro, L. (2001) EMBO J. 20 4952-4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takizawa, P. A., DeRisi, J. L., Wilhelm, J. E. & Vale, R. D. (2000) Science 290 341-344. [DOI] [PubMed] [Google Scholar]
- 31.Levin, P. A. & Losick, R. (2000) in Prokaryotic Development, eds. Brun, Y. V. & Shimkets, L. J. (Am. Soc. Microbiol., Washington, DC), p. 477.
- 32.Foley, M., Brass, J. M., Birmingham, J., Cook, W. R., Garland, P. B., Higgins, C. F. & Rothfield, L. I. (1989) Mol. Microbiol. 3 1329-1336. [DOI] [PubMed] [Google Scholar]
- 33.Taylor, J. R. (1982) An Introduction to Error Analysis (University Science Books, Mill Valley, CA).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.