

Abstract
Cerebrospinal fluid (CSF) from healthy individuals contains between 1,000 and 3,000 leukocytes per ml. Little is known about trafficking patterns of leukocytes between the systemic circulation and the noninflamed CNS. In the current study, we characterized the surface phenotype of CSF cells and defined the expression of selected adhesion molecules on vasculature in the choroid plexus, the subarachnoid space surrounding the cerebral cortex, and the cerebral parenchyma. Using multicolor flow cytometry, we found that CSF cells predominantly consisted of CD4+/CD45RA-/CD27+/CD69+-activated central memory T cells expressing high levels of CCR7 and L-selectin. CD3+ T cells were present in the choroid plexus stroma in autopsy CNS tissue sections from individuals who died without known neurological disorders. P- and E-selectin immunoreactivity was detected in large venules in the choroid plexus and subarachnoid space, but not in parenchymal microvessels. CD4+ T cells in the CSF expressed high levels of P-selectin glycoprotein ligand 1, and a subpopulation of circulating CD4+ T cells displayed P-selectin binding activity. Intercellular adhesion molecule 1, but not vascular cell adhesion molecule 1 or mucosal addressin cell adhesion molecule 1, was expressed in choroid plexus and subarachnoid space vessels. Based on these findings, we propose that T cells are recruited to the CSF through interactions between P-selectin/P-selectin ligands and intercellular adhesion molecule 1/lymphocyte function-associated antigen 1 in choroid plexus and subarachnoid space venules. These results support the overall hypothesis that activated memory T cells enter CSF directly from the systemic circulation and monitor the subarachnoid space, retaining the capacity to either initiate local immune reactions or return to secondary lymphoid organs.
Between 175,000 and 500,000 cells are present in the cerebrospinal fluid (CSF) of healthy individuals. Their functions and trafficking patterns are obscure, although it is believed that they participate in the immune defense of the CNS. Leukocytes traffic rapidly between blood and subarachnoid space (SAS), as indicated by studies in patients treated with anti-CD2 Abs, which demonstrated Ab-labeled cells in the CSF 18 h after infusion (1). The cellular composition of CSF, characterized by a predominance of lymphocytes but few erythrocytes, mononuclear phagocytes, or polymorphonuclear neutrophils, is not a simple reflection of peripheral blood (PB), suggesting a stringently regulated control over cell migration into the SAS.
Despite an extensive literature delineating the formation of the fluid component of CSF, the sites of entry and exit of leukocytes to the CSF are not well characterized. Based on results of studies conducted in rodents, it has been proposed that lymphocytes migrate into the brain and spinal cord through the blood–brain barrier surrounding deep parenchymal vessels and subsequently drain into the CSF (2). This concept was challenged by a recent study using intravital microscopy to study cerebral microcirculation, which failed to detect any interactions between highly activated neuroantigen-specific encephalitogenic T cells and resting vascular endothelium in the brain, indicating that migration through the healthy blood–brain barrier is a low-efficiency event (3). Interestingly, an intravital microscopy study of rodent spinal cord documented vascular “capture” of activated lymphocytes, raising the possibility of regional variability in trafficking mechanisms (4). For trafficking into CSF, an alternative possibility is that cells may extravasate directly from the systemic circulation into the SAS surrounding meningeal vessels or may enter ventricular CSF through the choroid plexus. Migration through these two pathways was suggested by the finding of fluorescence-labeled splenocytes in the SAS and choroid plexus stroma 2 h after i.v. injection in mice (5).
To elucidate pathways for leukocyte migration to the CSF, we characterized the surface phenotype of CSF cells and defined the expression of selected adhesion molecules on vasculature in the choroid plexus, the SAS surrounding the cerebral cortex, and the cerebral parenchyma.
Materials and Methods
CSF Samples. Blood and CSF were obtained from 69 consecutive patients (50 women) referred for diagnostic lumbar puncture. The collection of blood and CSF samples was approved by the Institutional Review Board of the Cleveland Clinic Foundation, and written consent was obtained from all subjects. Patients with inflammatory disorders of the CNS were excluded from the study. Study patients had normal results during evaluation (n = 20) or were diagnosed with noninflammatory neurological diseases (NIND; n = 49) and were analyzed together as an NIND cohort. Patient ages ranged from 16 to 81 years (mean 49). Because of limited cell numbers (mean 0.7, range from 0 to 7 cells per μl), the analysis of cell phenotypes in each sample was restricted to one or two trafficking determinants, along with lineage and activation markers. Diagnoses and specification of cell surface determinants analyzed in each individual are provided in Table 1, which is published as supporting information on the PNAS web site, www.pnas.org.
Brain Tissue. Choroid plexus was isolated from the lateral ventricles, and the arachnoid membrane with adjacent SAS stroma was dissected from the surface of the brain from four patients who died with non-neurological disorders. An additional six choroid plexus samples were obtained from consecutive autopsies. The ages of the patients were 1–95 years (median 57 years). The samples were fixed in formalin and embedded in paraffin. In addition, autopsy sections of brain parenchyma were obtained from archival material from four patients without neurological or inflammatory disorders. Two sections with active CNS inflammation were selected from a series of well characterized cases of multiple sclerosis at the University of Vienna. Both sections exhibited pattern II pathology (6). Under the guidelines established by the National Institutes of Health, the collection of brain tissue for immunohistochemistry was exempt for review, as determined by the Institutional Review Board of the Cleveland Clinic Foundation.
mAbs. CD4 PerCP (SK3), CD26 phycoerythrin (PE) (L272), and L-selectin PE (SK11) were from BD Biosciences; CCR7 PE (150503) was from R & D Systems; CCR7 (2H4), CD4 FITC/APC (RPA-T4), CD8 biotin (RPA-T8), CD25 PE (MA251), CD27 FITC (M-T271), CD45RA PE (HI100), CD45RO FITC/APC (UCHL1), CD69 PE (FN50), and peripheral node addressin (MECA-79) were from BD PharMingen; CD3 PE-Texas red (UCHT1) was from Beckman Coulter; CD19 PE-Cy7 (SJ25-C1) was from Caltag Laboratories (Burlingame, CA); CCR7 (7H12) was from Millennium Pharmaceuticals (Cambridge, MA); P-selectin glycoprotein ligand 1 (PSGL-1) PE (KPL-1) was from Research Diagnostics (Flanders, NJ); P-selectin (1E3) and platelet-endothelial cell adhesion molecule 1 (JC70A) were from DAKO; intercellular adhesion molecule 1 (ICAM-1) (G5), vascular cell adhesion molecule 1 (VCAM-1) (H276), and mucosal addressin cell adhesion molecule 1 (K19) were from Santa Cruz Biotechnology; and CD3 (PS1) and E-selectin (16G4) were from NovoCastra (Newcastle, U.K.).
Flow Cytometry. Immunostainings for flow cytometry were performed as described (7). Ten-milliliter aliquots of CSF were collected and stained within 20 min of sampling, in parallel with 100 μl of PB from the same patient. Erythrocytes were lysed after staining by using FACS lysing solution (BD Biosciences). Staining was performed at room temperature for 15 min. Titrations were performed for each mAb in blood samples to define the concentration that resulted in saturating conditions and an optimal signal-to-noise ratio. Identical concentrations of mAbs were used for blood and CSF samples, as numbers of CSF cells per staining never exceeded the number of PB cells.
P-selectin binding activity was detected on PB mononuclear cells obtained through density centrifugation on Ficoll (Lymphocyte Separation Media, Mediatech, Herndon, VA). Cells were incubated with a P-selectin–IgG fusion protein (8) in the presence of cations (Mg and Ca) or EDTA (negative control) for 45 min on ice, followed by incubation with a polyclonal anti-IgG Ab and directly conjugated mAbs against cell lineage markers.
Cells were acquired on an LSR (BD Immunocytometry Systems; four-color stainings) or a MoFlo flow cytometer (Cytomation, Fort Collins, CO; six-color stainings) and analyzed by using winlist software (Verity Software House, Topsham, ME). Cells were gated according to forward and side light-scattering properties and were positively selected for CD3/CD19 or CD4 expression. Isotype-matched control mAbs were used to define background fluorescence.
Statistical Methods. As the results were not normally distributed, the nonparametric Mann–Whitney U test was used for statistical analysis. Reported P values are two-tailed and considered statistically significant at P < 0.05.
Immunohistochemistry. Immunohistochemistry was performed as described (9). In brief, paraffin-embedded tissue sections were deparaffinized and steamed in citrate buffer (pH 6.0) or Tris buffer (pH 9.0) with 1 mM EDTA (for E-selectin). Slides intended for P-selectin staining were immersed in methanol. All slides were incubated overnight with primary Ab at 4°C and incubated with biotinylated secondary Ab at room temperature for 40 min. Incubation with antimucosal addressin cell adhesion molecule 1 was performed in the presence of 0.1% Triton X (Sigma). After staining, slides were incubated with avidin–biotin–perioxidase complex (ABC; Vectastain Elite, Vector Laboratories) and were developed by using diaminobenzidine (DAB) substrate (Sigma). Two-color stainings were performed by combining alkaline phosphatase (AP)-labeled and biotinylated secondary Abs. After development with ABC and DAB, slides were incubated with the AP substrate Fast Red (DAKO). Staining patterns for all Abs were verified on positive control tissue.
Results
Phenotype of CSF Lymphocytes. We used six-color flow cytometry to characterize lymphocyte subpopulations in paired blood and CSF samples from eight NIND patients by using the following definitions: CD4 Tnaïve (CD3+, CD4+, CD8-, CD45RA+, CD27+), CD4 Tcentral memory (TCM) (CD3+, CD4+, CD8-, CD45RA-, CD27+), CD4 Teffector memory (TEM) (CD3+, CD4+, CD8-, CD45RA-, CD27-), CD8 Tnaïve (CD3+, CD4-, CD8+, CD45RA+, CD27+), CD8 TCM (CD3+, CD4-, CD8+, CD45RAlo/neg, CD27+), CD8 TEM (CD3+, CD4-, CD8+, CD45RA+, CD27-), Bnaïve (CD19+, CD3-, CD27-), and Bmemory (CD19+, CD3-, CD27+).
Whereas PB leukocytes consisted of a mixture of all lymphocyte subpopulations, we found a clear predominance of memory CD4+ T cells in the CSF [83.4 ± 16.0% (mean ± SD) of all lymphocytes (CD3+ plus CD19+ cells)] (Fig. 1). CSF contained a small population of CD8+ memory T cells (11.2 ± 13.8%), whereas <1% of all CD4+ or CD8+ T cells displayed a naïve phenotype. In the CSF CD4+ T cell population, approximately one-third of all cells were CD27- TEM, whereas only a small percent of CD8+ T cells were CD27- TEM. B cells were sparsely represented in the CSF, with an equal distribution between Bnaïve (0.8 ± 1.6%) and Bmemory (0.4 ± 1.2%).
Fig. 1.

Six-color flow cytometry was used to characterize the main subpopulations of lymphocytes in paired blood (Upper) and CSF (Lower) samples. The graph shows mean and SD of eight patients with noninflammatory neurological diseases.
These results confirmed an earlier report showing that the majority of CSF CD4+ T cells are CD45RA-/CD27+ (10), a phenotype associated with reduced immediate effector functions and low expression of organ-specific homing receptors (11–13). We extended these results by using three-color flow cytometry in a group of 23 NIND patients and determined that 91.3 ± 4.7% of all CD4+/CD45RO+ memory T cells in the CSF expressed CD27, a frequency that was slightly higher than in PB (89.2 ± 6.5%; P < 0.05; Fig. 2A). Taken together, our analysis indicates that <10% of CSF T cells are terminally committed tissue-invasive effector cells.
Fig. 2.

(A) The majority of CD4+ T cells in both PB (Left) and CSF (Right) were CD45RO+/CD27+ TCM. Small, but distinct, populations of CD45RO+/CD27- TEM were observed in both compartments. Although ≈50% of PB CD4+ T cells were CD45RO-, this phenotype was rare in the CSF. (B) Expression of CCR7, L-selectin, and CD69 on CD4+/CD45RO+ T cells in PB (hatched plot) and CSF (open plot). Isotype-matched irrelevant Abs were used as negative controls in each experiment (filled plot). A representative histogram (n = 5–14) is shown.
Expression of CCR7 and L-Selectin on CSF T Cells. We stained CSF T cells for CCR7, a chemokine receptor required for homing to secondary lymphoid tissues. CCR7 is highly expressed on TCM, whereas absence of CCR7 identifies TEM (14). The vast majority of CD4+/CD45RO+ T cells in the CSF from NIND patients were CCR7-positive (83.3 ± 9.8%; n = 9; Fig. 2B). The percentages of CCR7+ cells in the population of CD45RO+/ CD4+ T cells in CSF were similar to those found in PB (88.4 ± 4.2%). The expression of CCR7 on CSF T cells was confirmed with three different CCR7 mAbs (clones 7H12, 2H4, and 150503). By using an RT-PCR assay, ample CCR7 transcripts were demonstrated in mRNA prepared from CSF cells from a patient with postinfectious myelitis (data not shown).
The expression of CCR7 on CSF T cells implied a capacity to traffic to secondary lymphoid organs, a possibility that would be supported by coexpression of L-selectin (15, 16). We observed that a majority of CD4+/CD45RO+ T cells in CSF from NIND patients expressed L-selectin (72.4 ± 12.0%; n = 7). Although these percentages were similar to levels in PB (71.9 ± 4.9%), significantly higher L-selectin mean fluorescence intensities were present in the CSF (blood, 82.1 ± 12.4; CSF, 621.8 ± 198.4; P < 0.05; Fig. 2B).
Activated lymphocytes exhibit increased capacity for migration, both in vitro and in vivo. CSF CD4+/CD45RO+ T cells were 10-fold enriched for expression of CD69, a marker for recent activation (<72 h), compared with PB (CSF, 46.5 ± 16.7%; blood, 4.1 ± 1.6%; P < 0.0001; n = 10; Fig. 2B). In contrast, CD4+/CD45RO+ T cells in the CSF did not express higher levels of CD25 or CD26, as compared with PB (data not shown).
These results demonstrate that the majority of human CSF leukocytes are CD4+ TCM, which maintain capacity for homing to secondary lymphoid organs.
CD3+ T Cells in Choroid Plexus Stroma. Pioneer lymphocytes, proposed to perform immune surveillance, were detected in murine choroid plexus and leptomeninges (5, 17). To assess whether human T cells might traffic similarly, we analyzed sections of choroid plexus from seven individuals. By using two-color immunohistochemistry, vascular endothelium was defined with an anti-platelet-endothelial cell adhesion molecule 1 mAb, whereas T cells were detected with anti-CD3. CD3+ T cells were observed in the choroid plexus parenchyma from all donors, both adjacent to small vessels in choroid plexus villi and near large venules in the choroidal stroma (Fig. 3).
Fig. 3.
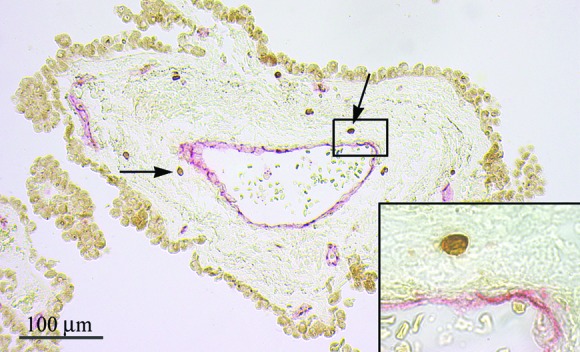
CD3+ T cells (dark brown) were detected in the choroid plexus stroma, both adjacent to small vessels in choroid plexus villi (not shown) and surrounding large venules in the choroidal stroma (arrows). CD3+ T cells were localized outside of choroid plexus vessels as demonstrated by staining platelet-endothelial cell adhesion molecule 1 positive endothelial cells (red; Inset).
Expression of P- and E-Selectin on CNS Endothelial Cells. We next investigated the expression of selected adhesion molecules in the vasculature of noninflamed CNS. These experiments focused on counterreceptors for adhesion ligands detected on CSF leukocytes. Three potential sites of T cell entry into the CSF were evaluated: brain parenchymal microvessels, postcapillary venules in the SAS, and choroid plexus vasculature.
Surprisingly, constitutive P-selectin immunoreactivity was detected in the stromal vasculature in choroid plexus samples of 10/10 individuals (Fig. 4A). The expression of P-selectin was localized to the endothelial cells of large venules in the choroidal stroma, whereas small vessels in choroid plexus villi were negative (Fig. 4C). P-selectin expression was also detected in large venules in the SAS (Fig. 4B). The continuous and linear distribution of P-selectin immunoreactivity along the vascular endothelium was consistent with surface expression of the protein rather than storage in cytoplasmic Weibel–Palade bodies (Fig. 4G). P-selectin expression was not observed on endothelial cells in histologically normal brain parenchyma or inflammatory parenchymal multiple sclerosis lesions, despite the presence of perivascular leukocyte aggregates (Fig. 4D). Providing an internal positive control, punctate P-selectin immunoreactivity was localized on platelets within the lumina of parenchymal vasculature.
Fig. 4.

P-selectin (A and B) and E-selectin (E and F) immunoreactivity was detected in large venules in choroid plexus (A and E) and SAS (B and F) from individuals who died without neurological or inflammatory disorders. The staining was predominantly detected in large vessels in the choroidal stroma, whereas smaller vessels in choroid plexus villi were negative (C; staining shows P-selectin). In inflammatory parenchymal lesions, punctate P-selectin immunoreactivity was localized to intravascular platelets, whereas endothelial cells were negative (D). The continuous and linear distribution of P-selectin immunoreactivity along the vascular endothelium indicated surface expression of the protein (G; staining shows vessel in choroid plexus). ICAM-1 expression was detected in endothelial cells at multiple localizations in the noninflamed CNS. Shown is characteristic staining of small vessels in the choroid plexus villi (J), large venules in choroidal stroma (H), and SAS (I).
Immunoreactivity for E-selectin was detected in endothelial cells of large venules in the choroidal stroma (Fig. 4E) and the SAS (Fig. 4F), whereas smaller vessels in choroid plexus villi were E-selectin negative. Expression of E-selectin was not detected on endothelial cells in histologically normal brain parenchyma or multiple sclerosis lesions (data not shown).
Expression of P- and E-selectin was highly selective, because neither peripheral node addressin nor mucosal addressin cell adhesion molecule 1, counterreceptors for L-selectin, was detected in endothelial cells in any of the CNS localizations under study (data not shown).
P-Selectin Binding. P-selectin binding through ligands presented on PSGL-1 is required for circulating murine T cells to enter the intrathecal compartment, in a migration pattern that is proposed to support immune surveillance of the CNS (17). We found that all CD4+/CD45RO+ T cells in CSF from patients with NIND, as in PB, expressed PSGL-1 (Fig. 5A). In each case (n = 5), higher PSGL-1 mean fluorescence intensity was observed in the CSF (2,202.6 ± 85.8) as compared with PB samples (1,263.6 ± 248.0; P < 0.05). P-selectin only binds to PSGL-1 when it presents appropriate carbohydrate residues. Therefore, we analyzed P-selectin binding activity on circulating cells by using a P-selectin–IgG fusion protein and found that a substantial population of CD4+/CD45RA- T cells in PB (28.0 ± 9.3%; n = 4) bound P-selectin in vitro (Fig. 5B).
Fig. 5.

(A) All CD4+/CD45RO+ T cells from PB (hatched plot) and CSF (open plot) express PSGL-1. (B) Approximately one-third of CD4+/CD45RA- T cells in PB bound P-selectin in a solution-phase adhesion assay (hatched plot). Filled histogram shows assay performed in the presence of EDTA as negative control.
ICAM-1 Expression on CNS Endothelial Cells. Leukocyte extravasation requires integrin-mediated arrest on cell adhesion molecules. ICAM-1 was expressed abundantly in multiple localizations of the CNS: in small vessels in the choroid plexus villi (Fig. 4J), large venules in the choroidal stroma (Fig. 4H), and postcapillary venules in the meninges (Fig. 4I). In addition, a fraction of parenchymal microvessels in histologically normal brains was ICAM-1 positive as reported (18).
VCAM-1 was expressed by a few branched cells with a morphology suggestive of microglia in normal brain parenchyma, whereas endothelial cells of brain parenchymal microvessels were consistently VCAM-1 negative as reported (19). No VCAM-1 immunoreactivity was detected in endothelial cells in the SAS or choroid plexus (data not shown).
Discussion
During the last several years, successful characterization of lymphocyte homing to the small intestine, skin, and lymphoid organs provided models for cell migration through the body in health and disease. Despite these advances, little is known about recruitment of lymphocytes to the healthy CNS. The present study addressed the phenotype of CSF cells from individuals without CNS inflammation, paying particular attention to the expression of trafficking determinants. The routine clinical availability of CSF provides an opportunity to obtain leukocytes present in a sterile tissue fluid in the absence of inflammation. In other locations such as joint, pleura, and peritoneal cavity, small fluid volumes and scarcity of cells limit detailed phenotypic characterization. It has often been proposed that cells in such fluids are derived from the tissue and reflect processes ongoing in the tissue compartment. Our findings suggest that this assumption may be incomplete and that CSF lymphocytes can be directly recruited from blood to perform immune surveillance of the CNS. Whether this model of immune surveillance is pertinent for other sterile tissue compartments remains an open question.
Our current results demonstrated that cells in the noninflamed CSF are predominantly CD4+/CD45RO+/CD27+/ CD69+ recently activated TCM expressing high levels of CCR7, L-selectin, and PSGL-1. This phenotype is consistent with a primary role for CSF cells in surveillance, rather than for effector functions. The expression of CD45RO, CD27, CCR7, and L-selectin is characteristic of previously activated memory T cells that have engaged antigen in secondary lymphoid organs and acquired a capacity to migrate through extralymphoid tissues, but still retain receptors allowing the cells to return to the lymphoid compartment (20). CD27+ memory CD4+ T cells lack immediate effector functions and express low levels of organ-specific homing receptors (11). Similarly, memory CD4+ T cells expressing L-selectin proliferate well in response to tetanus toxoid, have longer telomeres than L-selectin negative cells, and express genes and proteins consistent with immune surveillance functions (16). Absence of CCR7 on CD4+ memory T cells identifies tissue-infiltrating effector cells (14), but the presence of CCR7 does not exclude retention of effector functions. CD4+ T cells in a variety of tissues, including skin and rheumatoid synovium, express CCR7, and equal proportions of polarized cytokine-producing cells were detected in populations of cells sorted for the presence or absence of CCR7 (21, 22). Further, immediate cytokine production was demonstrated in CCR7+ cells (22, 23). Based on these data, it has been suggested that CD4+/CCR7- T cells are recently activated and have not yet up-regulated receptors necessary for homing to peripheral lymphoid organs (21). We found concurrent presence of CD69 and CCR7 on CSF T cells, suggesting an intermediate state of activation, consistent with requirements to execute immune surveillance and react rapidly to challenge.
We also analyzed autopsy tissue sections from individuals that died without inflammatory or neurological disorders, to define the expression of relevant endothelial counterreceptors for leukocyte adhesion molecules. The results demonstrated a selective and spatially restricted pattern of adhesion molecule expression in the noninflamed CNS. In particular, constitutive expression of P- and E-selectin was detected in large venules associated with arachnoid membranes and choroid plexus stroma, whereas cerebrovascular endothelial cells of parenchymal vessels were consistently negative. This finding was unexpected because P- and E-selectin are not routinely detected in human noninflamed tissue, nor in parenchymal vessels of the healthy rodent brain or by resting brain microvascular endothelial cells in vitro (24–27). However, recent reports provided functional and descriptive data consistent with the importance of P-selectin to CNS lymphocyte trafficking. Through the use of intravital microscopy, it has uniformly been shown that blockade of PSGL-1 or P-selectin abrogated lymphocyte rolling and tethering during various experimental models of CNS inflammation (3, 28, 29). Physiological relevance of P-selectin-mediated rolling on CNS endothelium was further suggested by the fact that P-selectin-deficient animals displayed significantly reduced influx of leukocytes to the CSF in a model of cytokine-induced meningitis (30). Additionally, a recent report described P-selectin expression in the choroid plexus of healthy mice (5).
The expression of selectins as observed in this study could potentially result from changes associated with circulatory arrest or other terminal events. However, the consistent pattern of P- and E-selectin expression in large stromal venules of all analyzed tissues provides evidence against such mechanisms. The selective and spatially restricted pattern of adhesion molecule expression in endothelial cells of arachnoid vessels and choroidal stroma suggests that these structures are involved in the recruitment of lymphocytes to the CSF. Although most studies of CNS cell trafficking have focused on lymphocyte migration into the brain parenchyma through the blood–brain barrier surrounding deep parenchymal vessels, such migration seems to be of low efficiency in the noninflamed brain (3). Further evidence supporting leukocyte migration through vessels in the meninges and choroid plexus was provided by the presence of fluorescently labeled lymphocytes in these structures 2 h after adoptive transfer of activated splenocytes (5). In the present study, T cells were detected in the choroidal stroma, providing indirect proof that lymphocytes traffic through this location in humans. Cell recruitment through the SAS and choroid plexus has been proposed to be of particular importance during immune surveillance of the healthy CNS because blockade of P-selectin inhibited early migration of labeled T cells into the noninflamed brain at a time point when anti-very late antigen 4 antibodies had no suppressive effect (17). Our studies demonstrated that the majority of CD4+/CD45RO+ T cells in both blood and CSF expressed PSGL-1, one of the proteins capable of presenting the carbohydrate ligands for P- and E-selectin. In all cases, higher PSGL-1 mean fluorescence intensity was observed in the CSF as compared with paired blood samples. Using a solution-phase adhesion assay, we demonstrated that a subpopulation of CD4+/ CD45RO+ T cells in blood expressed functional P-selectin ligand, favoring the hypothesis that interactions between endothelial P-selectin and its lymphocyte ligand presented on PSGL-1 are physiologically relevant.
Involvement of ICAM-1 in T cell recruitment to the CSF is less surprising. ICAM-1 is expressed on resting endothelium in various tissues including parenchymal vessels in noninflamed human brain (18, 31). ICAM-1 mediates firm arrest of circulating T cells through binding of lymphocyte function-associated antigen 1 (integrin αLβ2), which is expressed by a majority of CSF T cells from both healthy volunteers and patients with CNS inflammation, and on virtually all CNS-infiltrating leukocytes in inflammatory multiple sclerosis lesions (18, 32, 33). ICAM-1 was constitutively expressed in the choroid plexus of healthy mice and mediated binding of lymphocytes in vitro with a Stamper–Woodroof assay (34, 35). In contrast to previous studies in rodents (34, 36), we did not detect VCAM-1 in choroid plexus endothelium or epithelium. Although the reason for this difference remains speculative, it may be related to the sensitivity of the techniques in perfused animal tissue versus human autopsy tissue, or to species-related variation.
Soluble antigens from brain white matter follow the drainage of brain interstitial fluid through the ependyma into ventricular CSF (37). The presence of a plentiful assortment of potential antigen-presenting cells in the CSF compartment, such as ventricular epiplexus cells and meningeal, choroid plexus, and perivascular macrophages, would facilitate local restimulation of activated memory CSF T cells by presentation of cognate antigens, providing a mechanism for immune surveillance of the CSF compartment (38). Consistent with this hypothesis, inflammatory reactions occur earlier in the SAS than in the brain parenchyma in animal models. For instance, lymphocytes are first detectable in the leptomeninges 5–6 days postimmunization during experimental autoimmune encephalomyelitis, well before leukocytes are present in the brain parenchyma (39). A brisk response to pathogenic microbes in the SAS is of critical importance in the defense against spread of meningeal infections to the CNS parenchyma.
In summary, our results support the overall hypothesis that activated memory T cells enter CSF directly from the systemic circulation and monitor the SAS, retaining the capacity either to initiate local immune reactions or to return to secondary lymphoid organs. Further analysis of gene expression and functional capacity of these cells should provide insight into the pathogenesis of neuroinflammatory disorders.
Note Added in Proof. While this manuscript was under review, Giunti et al. (40) reported that CSF T cells from patients with inflammatory neurological diseases are of a CD45RO+/CD27+/CCR7+ memory phenotype.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grant PO1NS38667 (to R.M.R.) and European Union Grant QLG3-CT-2002-00612 (to H.L.).
Abbreviations: CSF, cerebrospinal fluid; NIND, noninflammatory neurological diseases; PB, peripheral blood; SAS, subarachnoid space; TCM, central memory T cells; TEM, effector memory T cells; ICAM-1, intercellular adhesion molecule 1; VCAM-1, vascular cell adhesion molecule 1; PE, phycoerythrin; PSGL-1, P-selectin glycoprotein ligand 1.
References
- 1.Hafler, D. A. & Weiner, H. L. (1987) Ann. Neurol. 22 89-93. [DOI] [PubMed] [Google Scholar]
- 2.Prat, A., Biernacki, K., Wosik, K. & Antel, J. P. (2001) Glia 36 145-155. [DOI] [PubMed] [Google Scholar]
- 3.Piccio, L., Rossi, B., Scarpini, E., Laudanna, C., Giagulli, C., Issekutz, A. C., Vestweber, D., Butcher, E. C. & Constantin, G. (2002) J. Immunol. 168 1940-1949. [DOI] [PubMed] [Google Scholar]
- 4.Vajkoczy, P., Laschinger, M. & Engelhardt, B. (2001) J. Clin. Invest. 108 557-565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carrithers, M. D., Visintin, I., Viret, C. & Janeway, C. S., Jr. (2002) J. Neuroimmunol. 129 51-57. [DOI] [PubMed] [Google Scholar]
- 6.Lucchinetti, C., Brück, W., Parisi, J., Scheithauer, B., Rodriguez, M. & Lassmann, H. (2000) Ann. Neurol. 47 707-717. [DOI] [PubMed] [Google Scholar]
- 7.Kivisäkk, P., Liu, Z., Trebst, C., Tucky, B. H., Wu, L., Stine, J., Mack, M., Rudick, R. A., Campbell, J. J. & Ransohoff, R. M. (2003) Methods 29 319-325. [DOI] [PubMed] [Google Scholar]
- 8.Maly, P., Thall, A., Petryniak, B., Rogers, C. E., Smith, P. L., Marks, R. M., Kelly, R. J., Gersten, K. M., Cheng, G., Saunders, T. L., et al. (1996) Cell 86 643-653. [DOI] [PubMed] [Google Scholar]
- 9.Sørensen, T. L., Tani, M., Jensen, J., Pierce, V., Lucchinetti, C., Folcik, V. A., Qin, S., Rottman, J., Sellebjerg, F., Strieter, R. M., et al. (1999) J. Clin. Invest. 103 807-815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hintzen, R. Q., Fiszer, U., Fredrikson, S., Rep, M., Polman, C. H., Van Lier, R. A. & Link, H. (1995) J. Neuroimmunol. 56 99-105. [DOI] [PubMed] [Google Scholar]
- 11.De Jong, R., Brouwer, M., Hooibrink, B., Pouw-Kraan, T., Miedema, F. & Van Lier, R. A. (1992) Eur. J. Immunol. 22 993-999. [DOI] [PubMed] [Google Scholar]
- 12.Hintzen, R. Q., De Jong, R., Lens, S. M., Brouwer, M., Baars, P. & Van Lier, R. A. (1993) J. Immunol. 151 2426-2435. [PubMed] [Google Scholar]
- 13.Hamann, D., Baars, P. A., Rep, M. H., Hooibrink, B., Kerkhof-Garde, S. R., Klein, M. R. & Van Lier, R. A. (1997) J. Exp. Med. 186 1407-1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sallusto, F., Lenig, D., Forster, R., Lipp, M. & Lanzavecchia, A. (1999) Nature 401 708-712. [DOI] [PubMed] [Google Scholar]
- 15.Warnock, R. A., Askari, S., Butcher, E. C. & von Andrian, U. H. (1998) J. Exp. Med. 187 205-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hengel, R. L., Thaker, V., Pavlick, M. V., Metcalf, J. A., Dennis, G., Jr., Yang, J., Lempicki, R. A., Sereti, I. & Lane, H. C. (2003) J. Immunol. 170 28-32. [DOI] [PubMed] [Google Scholar]
- 17.Carrithers, M. D., Visintin, I., Kang, S. J. & Janeway, C. A., Jr. (2000) Brain 123, 1092-1101. [DOI] [PubMed] [Google Scholar]
- 18.Bö, L., Peterson, J. W., Mørk, S., Hoffman, P. A., Gallatin, W. M., Ransohoff, R. M. & Trapp, B. D. (1996) J. Neuropathol. Exp. Neurol. 55 1060-1072. [PubMed] [Google Scholar]
- 19.Peterson, J. W., Bö, L., Mørk, S., Chang, A., Ransohoff, R. M. & Trapp, B. D. (2002) J. Neuropathol. Exp. Neurol. 61 539-546. [DOI] [PubMed] [Google Scholar]
- 20.Butcher, E. C. & Picker, L. J. (1996) Science 272 60-66. [DOI] [PubMed] [Google Scholar]
- 21.Campbell, J. J., Murphy, K. E., Kunkel, E. J., Brightling, C. E., Soler, D., Shen, Z., Boisvert, J., Greenberg, H. B., Vierra, M. A., Goodman, S. B., et al. (2001) J. Immunol. 166 877-884. [DOI] [PubMed] [Google Scholar]
- 22.Kim, C. H., Rott, L., Kunkel, E. J., Genovese, M. C., Andrew, D. P., Wu, L. & Butcher, E. C. (2001) J. Clin. Invest. 108 1331-1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Debes, G. F., Hopken, U. E. & Hamann, A. (2002) J. Immunol. 168 5441-5447. [DOI] [PubMed] [Google Scholar]
- 24.Barkalow, F. J., Goodman, M. J., Gerritsen, M. E. & Mayadas, T. N. (1996) Blood 88 4585-4593. [PubMed] [Google Scholar]
- 25.Engelhardt, B., Vestweber, D., Hallmann, R. & Schulz, M. (1997) Blood 90 4459-4472. [PubMed] [Google Scholar]
- 26.Navratil, E., Couvelard, A., Rey, A., Henin, D. & Scoazec, J. Y. (1997) Neuropathol. Appl. Neurobiol. 23 68-80. [PubMed] [Google Scholar]
- 27.Love, S. & Barber, R. (2001) Neuropathol. Appl. Neurobiol. 27 465-473. [DOI] [PubMed] [Google Scholar]
- 28.Carvalho-Tavares, J., Hickey, M. J., Hutchison, J., Michaud, J., Sutcliffe, I. T. & Kubes, P. (2000) Circ. Res. 87 1141-1148. [DOI] [PubMed] [Google Scholar]
- 29.Kerfoot, S. M. & Kubes, P. (2002) J. Immunol. 169 1000-1006. [DOI] [PubMed] [Google Scholar]
- 30.Tang, T., Frenette, P. S., Hynes, R. O., Wagner, D. D. & Mayadas, T. N. (1996) J. Clin. Invest. 97 2485-2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sobel, R. A., Mitchell, M. E. & Fondren, G. (1990) Am. J. Pathol. 136 1309-1316. [PMC free article] [PubMed] [Google Scholar]
- 32.Svenningsson, A., Hansson, G. K., Andersen, O., Andersson, R., Patarroyo, M. & Stemme, S. (1993) Ann. Neurol. 34 155-161. [DOI] [PubMed] [Google Scholar]
- 33.Oreja-Guevara, C., Sindern, E., Raulf-Heimsoth, M. & Malin, J. P. (1998) Acta Neurol. Scand. 98 310-313. [DOI] [PubMed] [Google Scholar]
- 34.Deckert-Schlüter, M., Schlüter, D., Hof, H., Wiestler, O. D. & Lassmann, H. (1994) J. Neuropathol. Exp. Neurol. 53 457-468. [DOI] [PubMed] [Google Scholar]
- 35.Steffen, B. J., Breier, G., Butcher, E. C., Schulz, M. & Engelhardt, B. (1996) Am. J. Pathol. 148 1819-1838. [PMC free article] [PubMed] [Google Scholar]
- 36.Wolburg, K., Gerhardt, H., Schulz, M., Wolburg, H. & Engelhardt, B. (1999) Cell Tissue Res. 296 259-269. [DOI] [PubMed] [Google Scholar]
- 37.Weller, R. O. (1998) J. Neuropathol. Exp. Neurol. 57 885-894. [DOI] [PubMed] [Google Scholar]
- 38.McMenamin, P. G. (1999) J. Comp. Neurol. 405 553-562. [PubMed] [Google Scholar]
- 39.Waksman, B. H. & Adams, R. D. (1962) Am. J. Pathol. 41 135-162. [PMC free article] [PubMed] [Google Scholar]
- 40.Giunti, D., Borsellino, G., Benelli, R., Marchese, M., Capello, E., Valle, M. T., Pedemonte, E., Noonan, D., Albini, A., Bernardi, G., et al. (2003) J. Leukocyte Biol. 73 584-590. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.