

Abstract
Previously we described a reliable method based on immunodepletion for isolating mesenchymal stem cells (MSCs) from murine bone marrow and showed that, after intracranial transplantation, the cells migrated throughout forebrain and cerebellum and adopted neural cell fates. Here we systemically administered MSCs purified by immunodepletion from male bleomycin (BLM)-resistant BALB/c mice into female BLM-sensitive C57BL/6 recipients and quantified engraftment levels in lung by real-time PCR. Male DNA accounted for 2.21 × 10-5% of the total lung DNA in control-treated mice but was increased 23-fold (P = 0.05) in animals exposed to BLM before MSC transplantation. Fluorescence in situ hybridization revealed that engrafted male cells were localized to areas of BLM-induced injury and exhibited an epithelium-like morphology. Moreover, purification of type II epithelial cells from the lungs of transplant recipients resulted in a 3-fold enrichment of male, donor-derived cells as compared with whole lung tissue. MSC administration immediately after exposure to BLM also significantly reduced the degree of BLM-induced inflammation and collagen deposition within lung tissue. Collectively, these studies demonstrate that murine MSCs home to lung in response to injury, adopt an epithelium-like phenotype, and reduce inflammation and collagen deposition in lung tissue of mice challenged with BLM.
Idiopathic pulmonary fibrosis is a crippling disease characterized by progressive dyspnea and is associated with a high mortality rate (1). Presently, no effective therapies to reverse or retard the course of the disease are available (1). Several recent studies have demonstrated that stem cells derived from adult tissues can home to and/or participate in the development of lung tissue (2–4), raising the possibility that stem cell-based therapies may be developed for effective intervention of lung diseases. However, the nature of the signals involved in the recruitment of stem cells into the lung, the extent of stem cell engraftment, and the effect of injury or disease on these processes remains undetermined.
To begin to address these issues we compared the engraftment of systemically administered mesenchymal stem cells (MSCs) in lung tissue of normal mice to that of mice exposed to bleomycin (BLM), which represents a well established model of lung injury resulting in pulmonary fibrosis (5, 6). We also evaluated whether MSC administration altered the clinical course of BLM-induced lung injury. Importantly, these studies used MSCs isolated from murine bone marrow by immunodepletion, a method that removes contaminating hematopoietic cells, which possess an appreciable engraftment potential in vivo, from plastic adherent marrow cultures (7). Moreover, we used a real-time PCR assay that specifically targets sequences on the mouse Y chromosome to quantify levels of male MSCs in the lungs of female transplant recipients (8). Our results reveal that MSCs engraft in the lungs of normal mice at low levels, but engraftment is increased significantly in response to BLM-induced injury. In the latter case engrafted donor cells adopted an epithelium-like morphology and also copurify with type II epithelial cells. Our data also demonstrate that early but not late administration of MSCs ameliorated the fibrotic injuries observed in the lungs of BLM-treated mice.
Materials and Methods
Isolation of Murine MSCs and Alveolar Type II Cells. MSCs were isolated from mouse bone marrow as described (7) except that whole bone marrow was plated at a density of 1.46 × 106 cells per cm2 and cultured for 8–10 days before harvest. MSCs (up to 40 × 106 cells) were added to M-280 Dynabeads (five beads per cell; Dynal, Oslo) conjugated to an anti-CD11b antibody (10 μg per mg of beads; PharMingen) in a volume of 1 ml and incubated on a rotator at 4°C for 45 min. Successive rounds of immunodepletion by using antibodies against CD34 and CD45 (PharMingen) were conducted similarly. Immunodepleted cells were suspended in Hanks' balanced salt solution and used for further experiments as described. Alveolar epithelial type II cells were isolated from the lung tissue as described (9). Approximately 2 × 106 cells were used to prepare genomic DNA for real-time PCR. Alternatively, 5 × 104 cells were cultured and fixed on positively charged slides for analysis by fluorescence in situ hybridization (FISH).
Fluorescence-Activated Cell Sorting. Aliquots (2.5 × 105) of immunodepleted MSCs were suspended in 50 μl of wash buffer (0.1% sodium azide/1.0% BSA in PBS) containing a rat anti-mouse CD16/CD32 antibody (Fc Block, PharMingen) at a concentration of 1 μg per 1 × 106 cells and incubated for 5 min at 4°C in the dark. Wash buffer (50 μl) containing 5 μg of the appropriate fluorochrome-conjugated primary antibody (PharMingen) was added, and the cells were incubated for an additional 20 min. Cells were washed twice with 200 μl of wash buffer, and the extent of cell labeling was evaluated by using a Beckman Coulter Model Epics XL. Isotype controls were run in parallel by using the same concentration of each antibody tested.
BLM-Induced Lung Injury and MSC Administration. Female 6- to 10-week-old C57BL/6 mice (Charles River Breeding Laboratories) were anesthetized via an i.p. injection of tribromoethanol (Aldrich) and then exposed to BLM as described (10, 11). Immunodepleted male MSCs (5 × 105 in 200 μl of PBS) were injected into the jugular vein immediately after or 7 days after challenge with BLM. Animals were killed 14 days after BLM exposure by an injection of sodium pentobarbital (120 mg/kg), perfused with cold 0.9% NaCl, and the left lungs were removed and used for evaluation of collagen content or RNA isolation. The right lungs were fixed in situ for 2 h by the intratracheal instillation of 10% neutral formalin (Sigma) and then postfixed for 24 h. Sagittal sections (4-μm) of paraffin-embedded lung tissue were used for FISH. Collagen deposition was estimated by measuring the total hydroxyproline content of the lung as described (10, 11). Results were expressed as micrograms of hydroxyproline per lung. All experiments involving live animals were approved by the animal use and care committee of Tulane University Health Sciences Center.
Real-Time PCR. Real-time PCR was carried out as described (8) on a 7700 sequence detection system (Applied Biosystems) by using the PCR primers 5′-TTTTGCCTCCCATAGTAGTATTTCCT-3′ and 5′-TGTACCGCTCTGCCAACCA-3′ and the TaqMan probe 5′-FAM-AGGGATGCCCACCTCGCCAGATAMRA-3′.
Standard curves were generated by serially diluting male mouse genomic DNA into female mouse genomic DNA prepared from liver.
FISH. Localization of male Y chromosome sequences in 4-μm paraffin-embedded sections of mouse lung tissue was done by using the Universal ISH kit and a mouse Y chromosome paint probe (Innogenex, San Ramon, CA) as described (8). Sections were counterstained with 4′,6-diamidino-2-phenylindole or ethidium bromide (1 mg/ml), photographed by using a Leica (Deerfield, IL) RX-DMV upright fluorescent microscope attached to a digital camera (Cooke Sensicam), and rendered by using slidebook software (Intelligent Imaging Innovations, Denver).
Osteopontin and Metalloproteinase RNA Expression in Mouse Lung. Microarray analysis was used to quantify the effect of BLM exposure on osteopontin mRNA levels in lung as described (12). Labeled cRNA was hybridized to the GeneChip Mu6500 array (Affymetrix, Santa Clara, CA). Data points for C57BL/6 mice represent total RNA pooled from six animals, whereas data points for 129/J mice represent total RNA isolated from individual mice (n = 3–5). Fold changes were determined by dividing the mean of the average differences in each experimental condition by that for the control-treated group. Metalloproteinase RNA expression was evaluated by using a RiboQuant kit (PharMingen) following manufacturer recommendations.
Results
BLM-Induced Lung Injury Enhances MSC Engraftment. We exploited a well characterized murine model of BLM-induced pulmonary fibrosis to evaluate how injury effects engraftment of stem cells in lung. Accordingly, we isolated by immunodepletion murine MSCs from the bone marrow of male BLM-resistant BALB/c mice and transplanted them via the i.v. route into female BLM-sensitive C57BL/6 mice that were challenged with BLM. We then quantified engraftment levels of male, donor-derived cells in the lungs of female transplant recipients 14 days after BLM challenge using real-time PCR. No male DNA was detected in lung tissue isolated from control female mice, and male DNA accounted for ≈2.21 × 10-5% of total lung DNA in female mice (n = 7) that were administered MSCs. In contrast, male DNA accounted for 5.18 × 10-4% of total lung DNA in female transplant recipients (n = 10) exposed to BLM before injection of MSCs. This result represents a 23-fold increase in engraftment levels of donor-derived cells as compared with mice not exposed to BLM (P < 0.05) (Fig. 1).
Fig. 1.

Quantification of MSC engraftment in mouse lung by real-time PCR. (A) Relationship between threshold cycle number and the percentage of male genomic DNA within the indicated samples. The standard curve (blue dots) was generated by using samples containing from 100% to 0% male genomic DNA. (B) Histogram of the data in A showing the percentage of male genomic DNA within lung tissue or type II epithelial cells isolated from female mice 14 days after exposure to BLM, MSC administration (MSCs), or MSC administration after BLM exposure (BLM+MSC). Plotted values represent the arithmetic mean, and error bars represent the standard deviation (*, P = 0.05, and **, P = 0.001 by Student's t test).
To confirm these findings we localized engrafted male cells in the lungs of BLM-treated female transplant recipients by FISH using a mouse Y chromosome paint probe. Relatively few cells were detected in lung tissue that demonstrated a positive hybridization signal against the Y chromosome, which was anticipated based on our quantitative analyses. The detected donor-derived cells were localized in areas of BLM-induced lung injury and appeared to conform to the morphology of epithelial cells (Fig. 2). To evaluate this engraftment further, we isolated epithelial type II cells from the lungs of female transplant recipients and measured their content of male DNA by real-time PCR. Epithelial type II cells isolated from the lungs of female mice exposed to BLM contained no detectable male DNA. In contrast, male DNA accounted for 1.37 × 10-3% of the total DNA content of epithelial type II cells isolated from the lungs of BLM-treated female mice administered male MSCs (Fig. 1 A). Therefore, purification of epithelial type II cells resulted in a 3-fold enrichment of male donor MSCs as compared with that contained in whole lung tissue. This engraftment level also represents a 62-fold increase as compared with mice not exposed to BLM (P = 0.001) (Fig. 1B).
Fig. 2.

(A)A4-μm section of lung tissue from a male mouse hybridized with an FITC-conjugated Y chromosome paint probe and counterstained with ethidium bromide. The arrows indicate representative nuclei that contain the Y chromosome. (B) Localization of Y chromosome-positive nuclei (arrow) within lung tissue of a female mouse 14 days after BLM exposure and administration of male MSCs. (C and D) A series (8–20) of images through the z axis were rendered from lung sections (as in B) to demonstrate that the probe hybridization signals (arrows) are associated specifically with cell nuclei.
Freshly isolated epithelial type II cells were identified by Papanicolaou staining of lamellar bodies (Fig. 3A). Culture of the cells resulted in adhesion and loss of lamellar bodies. The cultured cells also expressed cytokeratins (Fig. 3B), reacted with osmium tetroxide, but did not express vimentin and desmin (data not shown), indicating that the isolation procedure yielded a homogeneous population. FISH with a Y chromosome paint probe resulted in a positive hybridization signal for most of the type II epithelial cells isolated from the lungs of male mice (Fig. 3C). We also identified a few cells that demonstrated a positive hybridization signal in preparations of type II epithelial cells from female mice exposed to BLM and administered MSCs (Fig. 3D). These results are consistent with our real-time PCR data and confirm that male donor MSCs copurified with type II epithelial cells from lung.
Fig. 3.
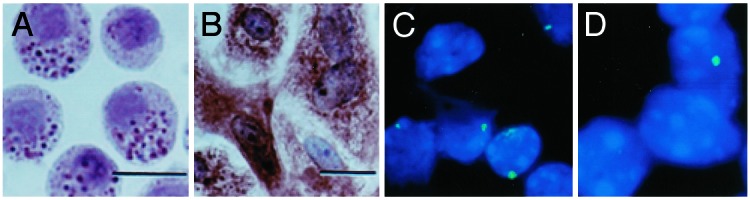
Copurification of engrafted MSCs with type II epithelial cells. (A) Freshly isolated alveolar epithelial type II cells show darkly stained cytoplasmic granules after pap staining. (B) Cultured epithelial type II cells stain positively with an anticytokeratin antibody. (C) FISH analysis of cytospin preparations of alveolar type II cells (50,000 per slide) isolated from the lungs of a male mouse. (D) FISH analysis of alveolar epithelial type II cells isolated from female mice 14 days after exposure to BLM and administration of male MSCs. (Scale bars, 100 μM; magnification, ×40.)
BLM Enhances Osteopontin Expression in Mouse Lung. Immunodepleted murine MSCs uniformly express CD44 but do not express the v4 and v6 isoforms of this polymorphic glycoprotein that functions as a cell-surface receptor for hyaluronan (HA) (13) and osteopontin (14). Interaction between CD44 and HA is fundamental for the normal development of the lung and the repair of lung injury (15). Although BLM exposure is known to induce expression of HA in lung (16, 17), its effect on osteopontin expression is not well characterized. Accordingly, we evaluated the effects of BLM exposure on expression levels of osteopontin mRNA in the lungs of BLM-sensitive strains (C57BL/6 and 129/J). Endotracheal challenge with BLM induce a time-dependent increase in osteopontin mRNA levels in lung, resulting in a 3- and 5-fold increase as compared with untreated mice at 7 and 14 days after BLM exposure, respectively (Fig. 4A). Therefore, two different ligands that bind to the CD44 receptor are induced in lung tissue by BLM.
Fig. 4.
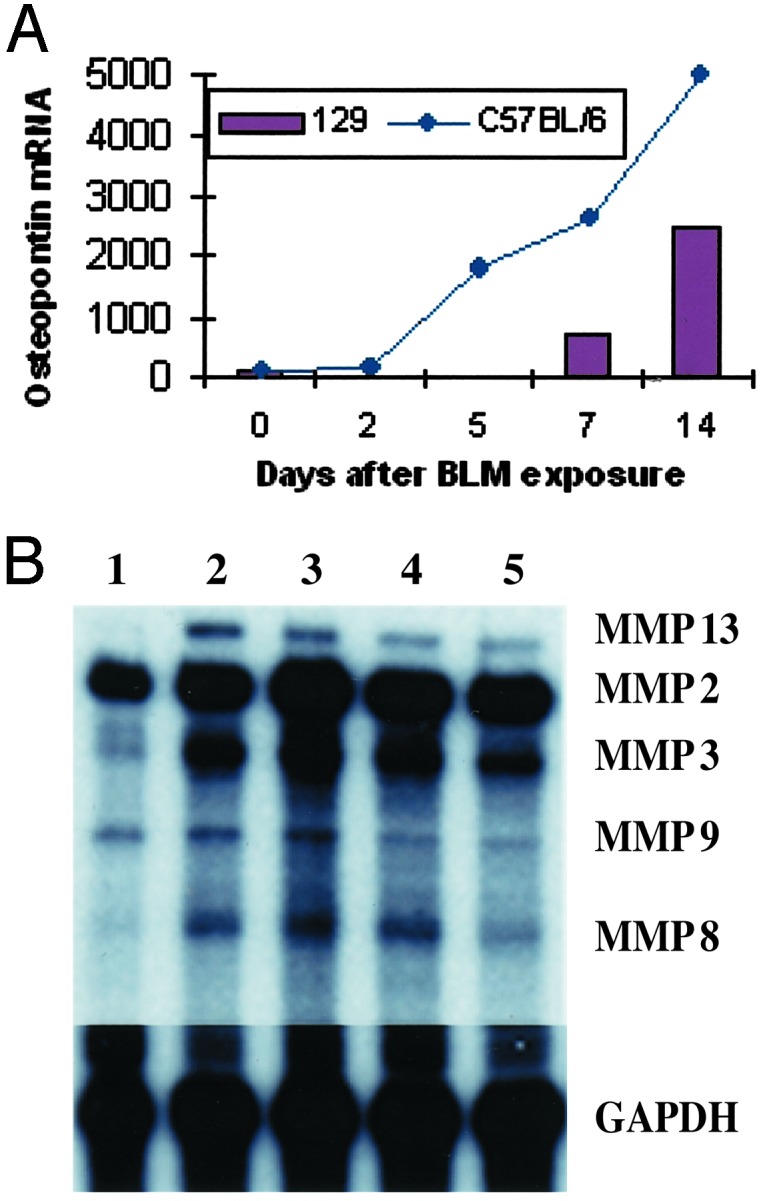
Effect of BLM on osteopontin and MMP mRNA levels in lung. (A) Changes in osteopontin expression levels in lung tissue at various times after BLM exposure. The data points for C57BL/6 mice represent total RNA pooled from lung tissue of six animals. The data points for 129/J mice represent the mean of three to five separate expression values analyzing total RNA prepared from individual mice. (B) Total RNA was isolated from the lungs of untreated C57BL/6 mice (lane 1) or mice 14 days after exposure to BLM alone (lanes 2 and 3) or BLM exposure followed by MSC administration (lanes 4 and 5) and subjected to ribonuclease protection analysis as described in Materials and Methods. Relative levels of each MMP transcript were determined by densitometry and normalized to the levels of GAPDH mRNA.
Engrafted MSCs Alter BLM-Induced Lung Injury in C57BL/6 Mice. The intratracheal exposure of mice to BLM but not to saline resulted in the development of subpleural areas of inflammation that encompassed ≈55–60% of the lung parenchyma, which was consolidated with loss of normal alveolar architecture, and also involved the bronchi and vasculature (Fig. 5). Administration of MSCs immediately after challenge with BLM reduced the extent of inflammation within the lung as evidenced by large areas of undamaged tissue with normal alveolar architecture. In contrast, no significant difference in the degree of inflammation was observed in animals administered MSCs 7 days after challenge with BLM as compared with those exposed to BLM alone (Fig. 5).
Fig. 5.

Effect of MSC engraftment on BLM-induced lung injury in mice. Low-magnification photomicrographs of tissue sections prepared from the lungs of a C57BL/6 mouse 14 days after saline exposure (A), 14 days after BLM exposure (B), and 14 days after BLM exposure and MSC administration (C). Note that the extent of BLM-induced inflammation, as evidenced by the wedge-shaped area of pneumonitis in B, is greatly reduced in C. TB, terminal bronchiole; PL, pleural surface. (Scale bar, 20 μm.)
BLM exposure in mice also results in an increase in collagen deposition in the lung. Therefore, we compared the amount of hydroxyproline, a modified amino acid specifically found in collagen, in lung tissue of normal mice to those challenged with BLM with or without MSC administration. The hydroxyproline content of lung tissue from control-treated mice was 48.1 ± 7 μg per lung. At 14 days after exposure to BLM this valued increased significantly to 93 ± 20 μg per lung (P < 0.05). In contrast, the hydroxyproline content of lung tissue from animals that were administered MSCs immediately after BLM challenge was 67 ± 1 μg per lung, which was also significantly different from that of control-treated mice but represented a statistically significant decrease (n = 7; P < 0.05) compared with animals exposed to BLM alone. Administration of MSCs 7 days after BLM exposure also resulted in a reduction in hydroxyproline content of lung 14 days later (81 ± 18 μg per lung), but this difference was not statistically significant as compared with animals exposed to BLM alone (P > 0.05).
In addition to modulating the extent of collagen deposition, we found that MSC administration also altered the expression level of various matrix metalloproteinases (MMPs) in BLM-treated mice. Compared with control animals, BLM exposure increased the expression level of transcripts encoding MMP2 [27.6 ± 6.9 vs. 96.5 ± 24.6 arbitrary units (AU), respectively] and MMP9 (5.48 ± 1 vs. 7.51 ± 1 AU, respectively) within lung tissue (Fig. 4B). In contrast, administration of MSCs to animals immediately after BLM exposure reduced the extent of BLM-induced expression of MMP2 (67.3 ± 16 AU) and completely attenuated BLM-induced expression of MMP9 (5 ± 0.5 AU) in lung tissue. BLM exposure also increased transcript levels of interstitial collagenase (MMP13) in lung as compared with untreated mice (9.83 ± 2 AU vs. 1.63 ± 0.2 AU, respectively), which was reduced modestly in animals that were administered MSCs after BLM exposure (6.43 ± 0.6 AU). Collectively, these results indicate that early but not late administration of MSCs reduces the degree of inflammation and fibrosis in the lungs of mice challenged with BLM.
Discussion
In this report we demonstrate that engraftment in lung tissue of systemically administered MSCs occurs at low levels in normal mice but is increased significantly in response to BLM-induced lung injury. We also show that administration of MSCs immediately after BLM challenge protects lung tissue from BLM-induced injury, as evidenced by a significant reduction in inflammation, collagen deposition, and MMP activation within lung tissue. Collectively, these findings indicate that systemic administration of MSCs may be beneficial in the treatment of lung disease even if engraftment levels of the administered cells are comparatively low.
An important aspect of this work is that the MSCs used in this study were enriched from plastic adherent cultures of murine bone marrow by immunodepletion, which is in contrast to several previous reports that evaluated the engraftment and therapeutic potential of MCs by transplantation in vivo of plastic adherent marrow cells (4, 18, 19). We and others (20–22) have shown that plastic adherent cultures elaborated from murine bone marrow contain a variety of hematopoietic cell types, which persist in the cultures even after serial passage and exhibit an appreciable engraftment potential in vivo. Therefore, the aforementioned studies are confounded in that transplantation of plastic adherent cells precludes a direct measure of the contribution made by MSCs to the experimental outcome. Our experimental approach, in contrast, provides a more direct measure of the engraftment and therapeutic potential of MSCs in lung.
At present, the mechanism by which BLM-induced lung injury augments MSC engraftment in lung tissue is unclear. Endotracheal challenge of mice with BLM leads to lung fibrosis and occurs in three stages. The first stage, acute pulmonary toxicity, results from BLM-induced DNA-strand scission and is characterized by apoptosis and necrosis of alveolar epithelial cells (23–25). This is followed by an inflammatory phase, wherein activated immune cells migrate into the lung and release a variety of cytokines including tumor necrosis factor α, which promotes the development of lung fibrosis (26, 27). The final stage is characterized by enhanced collagen deposition within lung, expansion of the lung interstitium due to the proliferation of fibroblasts and smooth muscle cells, and increased expression of MMPs that participate in the remodeling of the injured tissue (28). Each of these phases may affect engraftment of MSCs in lung uniquely. For example, although we isolated MSCs from a BLM-resistant strain of mice, it is possible that a significant percentage of administered cells were lost because of the acute toxicity of BLM or immune rejection by the host, because our population of MSCs constitutes an allotransplant. Conceivably, optimizing the dose of MSCs and timing of administration and transplanting autologous stem cells after BLM challenge may enhance engraftment levels further. The homing and engraftment of MSCs in lung may have been augmented by engagement of their CD44 receptor by HA and osteopontin. BLM exposure is known to increase expression of HA in lung (16, 17), and we have shown in this report that osteopontin mRNA levels are also increased under these conditions. Alternatively, release of cytokines and mitogens by immune-infiltrating cells may also affect the homing, engraftment, and proliferative status of MSCs in lung. Last, breakdown of the microvasculature due to BLM toxicity may provide a nonspecific mechanism by which MSCs gain increased access to lung tissue.
Our data also indicate that MSC administration after BLM exposure protects lung tissue from injury by significantly reducing the extent of inflammation and fibrosis. Several explanations may account for this effect of MSC administration. For example, MSCs may limit the injurious effects of BLM by replacing alveolar epithelial type II cells, which are thought to function as stem cells in lung and are known targets of apoptotic signals induced in lung by BLM (29). Differentiation of MSCs into epithelial type II cells may partially restore the stem cell pool, leading to increased genesis of alveolar cells for the resolution of disrupted alveolar surfaces, thereby augmenting the repair process. Generation of lung stem cells may also explain how low levels of MSC engraftment in lung can produce a positive effect on the health status of mice exposed to BLM. Notably, several recent reports have shown that fusion with somatic cells may enable stem cells to adopt unorthodox phenotypes (30, 31). At present we have not determined whether fusion of MSCs with somatic cells in lung is responsible for their epithelium-like morphology or whether the cells change fate in vivo in response to transplantation to a novel residence. If a significant fraction of engrafted MSCs do fuse with somatic cells, these events do not seem to limit their ability to ameliorate the injurious effects of BLM in lung.
Alternatively, MSCs may protect against BLM-induced injury by altering the microenvironment of lung at sites of engraftment. For example, MSCs may produce antagonists of tumor necrosis factor α or other cytokines that disrupt signal pathways leading to fibrosis (32, 33). Recent studies have shown that scavenging of HA degradation products in lung by CD44-expressing cells is necessary for the resolution of BLM-induced lung injury (14). Scavenging by MSCs of HA and osteopontin, which plays an important role in the pathogenesis of BLM-induced fibrosis by promoting the migration, adhesion, and proliferation of fibroblasts in lung, may be an important mechanism of MSC action. Clearly, the timing of MSC administration is an important determinant of their biological response. Delaying MSC administration by 7 days after BLM challenge did not inhibit engraftment but eliminated the ability of the cells to alter the course of disease progression. Therefore, MSCs may produce factors that impinge on molecules expressed early but not late during the course of BLM-induced injury.
In summary, our data indicate that MSCs, given their propensity to engraft in lung tissue and ability to ameliorate the injurious effects of BLM, constitute an effective cellular vehicle for the treatment of lung disease.
Acknowledgments
This work was supported in part by U.S. Public Health Service Grant ES10859-01A1 (to L.A.O.) from the National Institutes of Environmental Health, and by the Louisiana Gene Therapy Research Consortium (New Orleans) and HCA-The Health Care Company (Nashville, TN).
Abbreviations: BLM, bleomycin; MSC, mesenchymal stem cell; FISH, fluorescence in situ hybridization; HA, hyaluronan; MMP, matrix metalloproteinase; AU, arbitrary units.
References
- 1.American Thoracic Society/European Respiratory Society (2002) Am. J. Respir. Crit. Care Med. 165 277-304. [DOI] [PubMed] [Google Scholar]
- 2.Jiang, Y., Balkrishna, N., Reinhardt, R. L., Schwartz, R. E., Keene, C. D., Ortiz-Gonzalez, X. R., Reyes, M., Lenvik, T., Lund, T., Blackstad, M., et al. (2002) Nature 418 41-49. [DOI] [PubMed] [Google Scholar]
- 3.Krause, D. S., Theise, N. D., Collector, M. I., Henegariu, O., Hwang, S., Gardner, R., Neutzel, S. & Sharkis, S. J. (2001) Cell 105 369-377. [DOI] [PubMed] [Google Scholar]
- 4.Kotton, D. N., Yang-Ma, B., Cardoso, W. B., Sanderson, E. A., Summer, R. S., Williams, M. C. & Fine, A. (2001) Development (Cambridge, U.K.) 128 5181-5188. [DOI] [PubMed] [Google Scholar]
- 5.Bowden, D. H. (1984) Lab. Invest. 50 487-488. [PubMed] [Google Scholar]
- 6.Harrison, J. H. & Lazo, J. S. (1987) J. Pharmacol. Exp. Ther. 243 1185-1194. [PubMed] [Google Scholar]
- 7.Kopen, G. C., Prockop, D. J. & Phinney, D. G. (1999) Proc. Natl. Acad. Sci. USA 96 10711-10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McBride, C., Gaupp, D. & Phinney, D. G. (2003) Cytotherapy 5 7-18. [DOI] [PubMed] [Google Scholar]
- 9.Corti, M., Brody, A. R. & Harrison, J. H. (1996) Am. J. Respir. Cell Mol. Biol. 14 309-315. [DOI] [PubMed] [Google Scholar]
- 10.Ortiz, L. A., Champion, H., Lasky, J. A., Gozal, E., Hoyle, G., Friedman, M., Hyman, A. L. & Kadowitz, P. J. (2002) Am. J. Physiol. 282 L1209-L1221. [DOI] [PubMed] [Google Scholar]
- 11.Ortiz, L. A., Lasky, J. A., Lungarella, G., Cavarra, E., Martorana, P., Banks, W., Peschon, J., Brody, A. R. & Friedman, M. (1999) Am. J. Respir. Cell Mol. Biol. 20 825-833. [DOI] [PubMed] [Google Scholar]
- 12.Kaminski, N., Allard, J., Zuo, F., Griffiths, M. J. D., Morris, D., Huang, X., Sheppard, D. & Heller, R. A. (2002) Proc. Natl. Acad. Sci. USA 97 1778-1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aruffo, A., Stamenkovic, I., Melnick, M., Underhill, C. B. & Seed, B. (1990) Cell 61 1303-1313. [DOI] [PubMed] [Google Scholar]
- 14.Weber, G. F., Ashkhar, S., Glimcher, M. J. & Cantor, H. (1996) Science 271 509-512. [DOI] [PubMed] [Google Scholar]
- 15.Teder, P., Vandivier, R. W., Jiang, D., Liang, J. Cohn, L., Pure, E., Henson, P. M. & Nobel, P. W. (2002) Science 296 155-158. [DOI] [PubMed] [Google Scholar]
- 16.Teder, P. & Heldin, P. (1997) Am. J. Respir. Cell Mol. Biol. 17 367-385. [DOI] [PubMed] [Google Scholar]
- 17.Savani, R. C., Hou, G., Liu, P., Wang, C., Simmons, E., Grimm, P. C., Stern, R., Greenberg, A. H., DeLisser, H. M. & Khalil, N. (2000) Am. J. Respir. Cell Mol. Biol. 23 475-484. [DOI] [PubMed] [Google Scholar]
- 18.Pereira, R. F., O'Hara, M. D., Laptev, A. V., Halford, K. W., Pollard, M. D., Class, R., Simon, D., Livezey, K. & Prockop, D. J. (1998) Proc. Natl. Acad. Sci. USA 95 1142-1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin, H. K., Carter, J. E., Huntley, G. W. & Schuchman, E. H. (2002) J. Clin. Invest. 109 1183-1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kerk, D. K., Henry, E. A., Eaves, A. C. & Eaves, C. J. (1985) J. Cell. Physiol. 125 127-134. [DOI] [PubMed] [Google Scholar]
- 21.Witte, P. L., Robinson, M., Henley, A., Low, M. G., Stiers, D. L., Perkins, S., Fleischman, R. A. & Kincade, P. W. (1987) Eur. J. Immunol. 17 1473-1484. [DOI] [PubMed] [Google Scholar]
- 22.Phinney, D. G., Kopen, G., Isaacson, R. L. & Prockop, D. J. (1999) J. Cell. Biochem. 72 570-585. [PubMed] [Google Scholar]
- 23.Harrison, J. H., Hoyt, D. G. & Lazo, J. S. (1989) Mol. Pharmacol. 36 231-238. [PubMed] [Google Scholar]
- 24.Hagimoto, N., Kuqano, K., Nomoto, Y., Junitake, R. & Hara, N. (1997) Am. J. Respir. Cell Mol. Biol. 16 91-101. [DOI] [PubMed] [Google Scholar]
- 25.Ortiz, L. A., Moroz, K., Liu, J.-Y., Hoyle, G. W., Hammond, T., Banks, W., Hamilton, R. F., Holian, A., Brody, A. R. & Friedman, M. (1998) Am. J. Physiol. 275 L1208-L1218. [DOI] [PubMed] [Google Scholar]
- 26.Phan, S. H. & Kunkel, S. L. (1992) Exp. Lung Res. 18 29-43. [DOI] [PubMed] [Google Scholar]
- 27.Piguet, P. F., Collart, M. A., Grau, G. E., Kapanci, Y. & Vassalli, P. (1989) J. Exp. Med. 170 655-663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoyt, D. G. & Lazo, J. S. (1998) J. Pharmacol. Exp. Ther. 246 765-771. [PubMed] [Google Scholar]
- 29.Fehrenbach, H. (2001) Respir. Res. 2 33-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ying, Q.-L., Nichols, J., Evans, E. P. & Smith, A. G. (2002) Nature 416 545-548. [DOI] [PubMed] [Google Scholar]
- 31.Terada, N., Hamazaki, T., Oka, M., Hoki, M., Mastalerz, D. M., Nakano, Y., Meyer, E. M., Laurence, M., Petersen, B. E. & Scott, E. W. (2002) Nature 416 542-545. [DOI] [PubMed] [Google Scholar]
- 32.Piquet, P. F. & Vesin, C. (1994) Eur. Respir. J. 7 515-518. [DOI] [PubMed] [Google Scholar]
- 33.Ortiz, L. A., Lasky, J. A., Hamilton, R. F., Holian, A., Hoyle, G. W., Banks, W., Peschon, J., Brody, A. R. & Friedman, M. (1998) Exp. Lung Res. 24 721-743. [DOI] [PubMed] [Google Scholar]