

Abstract
Infection by Kaposi's sarcoma (KS)-associated herpesvirus (KSHV) is a key factor in the development of KS. Both latent and lytic KSHV infection is observed in KS tumor cells, and both genetic programs contribute importantly to KS pathogenesis. The viral replication and transcription activator (RTA) protein is a transcription factor that controls the switch from latency to lytic replication. We have previously shown that RTA can activate the expression of several lytic viral genes in transfected cells by interaction with recombination signal sequence-binding protein-Jκ (RBP-Jκ, also called CSL), which in uninfected cells is a transcriptional repressor that is the target of the Notch-signaling pathway. The recognition that many KSHV lytic genes, including RTA itself, contain RBP-Jκ-binding sites raised the possibility that RBP-Jκ-mediated repression may be central to the establishment of latency. Here, we have tested this hypothesis by examining KSHV infection of RBP-Jκ-null murine fibroblasts. Our results show that KSHV latency is efficiently induced in such cells; however, the reactivation of lytic gene expression, viral DNA replication, and the release of progeny viruses are dramatically inhibited in the absence of RBP-Jκ. RBP-Jκ-mediated repression is therefore not essential for establishment of latent infection, but the RTA-mediated redirection of RBP-Jκ activity from repression to activation is critical for lytic viral replication.
Kaposi's sarcoma (KS), the most common neoplasm of untreated AIDS patients, is a complex lesion characterized by endothelial proliferation, neoangiogenesis, and inflammatory cell infiltration (1, 2). In 1994, a novel herpesvirus, now termed KS-associated herpesvirus (KSHV) or human herpesvirus 8 (HHV8), was identified in KS lesions (3), and compelling epidemiological and molecular evidence strongly links KSHV infection to KS tumorigenesis (see refs. 4 and 5 for review). Like all herpesviruses, KSHV deploys two alternative genetic programs in infection: (i) latency, in which only a handful of viral genes is expressed and no infectious progeny are produced, and (ii) lytic infection, in which most viral genes are expressed in an ordered cascade, viral DNA is amplified, and infectious virions are released. Examination of KS biopsy specimens shows that KSHV infection is localized to the tumorous endothelial (spindle) cells (6), the majority of which are latently infected (7). A small subpopulation, however, are lytically infected (7), and recent evidence suggests that both latency and lytic infection contribute importantly to KS pathogenesis. The latency program encodes numerous proteins that can affect cell survival and proliferation in vitro (8–12), whereas the lytic cycle encodes a variety of signaling proteins that can either directly mediate angiogenesis and inflammation (13–16) or induce expression of host proteins that can do likewise (17). In addition, KSHV latency is somewhat unstable, in that proliferating cells frequently give rise to uninfected segregants (A. Grundhoff and D.G., unpublished results); ongoing lytic infection also serves as a source of virus that can sustain the population of latently infected cells via de novo infection/reinfection.
In most cultured cells, the default pathway of KSHV infection is latency; that is, newly infected cells do not express the full lytic cascade, but rather maintain the viral DNA in the nucleus as a low copy number, circular episome whose expression is tightly restricted (18, 19). Little is known about the mechanisms by which latency is established. Formally speaking, the absence of lytic gene expression might be due to the absence of one or more positive regulators, or to the presence of active repression (or both).
A single KSHV lytic-cycle viral gene (ORF50) controls the switch from latency to lytic replication (20, 21). Its product, the replication and transcription activator (RTA), is a transcription factor that is both necessary (22) and sufficient (20, 22, 23) to trigger lytic reactivation. The mechanisms by which KSHV RTA acts have been extensively studied in cells transiently transfected by reporter gene constructs. RTA harbors a potent C-terminal activation domain, deletion of which results in a loss of transactivation activity (22, 24). In addition, it has an N-terminal DNA-binding motif that can mediate sequence-specific DNA binding, and high-affinity sites for RTA recognition have been identified in several delayed-early promoters (25–27). However, some DE genes lacking high-affinity RTA sites are nonetheless still responsive to RTA transactivation, raising the possibility that RTA may also interact with host DNA-binding factors. Recently, we (28) discovered that RTA can directly interact with the host protein recombination signal sequence-binding protein-Jκ [RBP-Jκ; also called CBF1 (Cp-binding factor 1) or CSL], a sequence-specific transcription factor that is the key target of the Notch signaling pathway. In the ground state, RBP-Jκ is a repressor that recruits corepressor complexes [containing histone deacetylase (HDAC) 1 and -2, CBF1-interacting corepressor (CIR), SAP30, silencing mediator of retinoid and thyroid hormone receptors (SMRT), and SMRT/ HDAC1-associated repressor protein (SHARP)] to target promoters, inhibiting their expression (29–31). Although RBP-Jκ has been shown to interact with latent proteins of Epstein–Barr virus (EBV; refs. 32–35), KSHV RTA is the first lytic cycle herpesviral protein to be involved with the RBP-Jκ/Notch pathway. The discovery that many KSHV lytic-cycle genes that respond to RTA harbor RBP-Jκ sites that are required for RTA-mediated activation raised the possibility that RBP-Jκ may be a key repressor involved in the establishment and maintenance of latency. To test this hypothesis, and to explore the functional significance of the RTA/RBP-Jκ interaction in lytic viral replication as well, we have examined the effects of mutational ablation of RBP-Jκ on latent and lytic KSHV replication, using cultured fibroblasts from RBP-Jκ-/- mice (36).
Materials and Methods
Cells and Viruses. Mouse RBP-Jκ-/- (OT11) and WT (OT13) fibroblast cell lines were kindly provided by T. Honjo (Kyoto University, Kyoto) and were grown in high-glucose DMEM supplemented with 10% FBS and 100 units of mouse IFN-γ (Life Technologies, Grand Island, NY) per ml at 32°C. BCBL-1 cells were grown in RPMI medium 6140 supplemented with 10% FBS. Human foreskin fibroblast (HFF), SLK, and 293T cells were grown in DMEM supplemented with 10% FBS. Telomerase-immortalized microvascular endothelial (TIME) cells were maintained in an EBM-2 medium bullet kit (Clonetics, San Diego). KSHV was concentrated by centrifugation at 28,000 × g for 2 h from supernatant of BCBL-1 cells 5 days after phorbol 12-myristate 13-acetate (PMA)/ionomycin treatment. KSHV infection was carried out in the presence of 8 μg/ml polybrene for 2 h. Construction of adenovirus Ad-RTA, the recombinant adenovirus expressing RTA (ORF50), was described in details elsewhere (18). Briefly, the full-length RTA cDNA was subcloned to pShuttle (CLONTECH) at XbaI site with correct orientation. The RTA-expression cassette from pShuttle-ORF50 was then transferred to Adeno-X viral DNA (CLONTECH) by using unique PI-SceI/I-CeuI sites. The resulted recombinant Adeno-ORF50 DNA was packaged into infectious adenovirus by transfecting HEK293 cells. The recombinant adenoviruses were amplified on HEK293 cells and purified from crude lysate by CsCl gradient centrifugation twice. The virus titer was determined by A260 (A260 × 20 × 1012 = particles per ml). For infection, adenovirus was preincubated with 1 μg/ml polylysine (Sigma) in media for 100 min at room temperature and infected cells at a density of 1–5 × 103 particles per cell. Retroviral vector expressing RBP-Jκ was constructed by cloning RBP-Jκ cDNA (from pcDNA3.1-RBP-Jκ; described in ref. 28) between BamHI and XhoI sites of retroviral vector pBMN. Recovered retrovirus was used to infect OT11 cells, and stable cells OT11-RBP-Jκ were obtained after puromycin selection.
Immunofluorescence Assay. Rabbit anti-latency-associated nuclear antigen (LANA), anti-RTA, and anti-KbZIP antibodies have been described (21, 37, 38). Mouse anti-ORF59 antibody was purchased from Advanced Biotechnologies (Columbia, MD). Cells grown on chamberslides were washed once in PBS, and fixed with 4% paraformaldehyde for 10 min. After being blocked in 1% BSA/PBS for 15 min, cells were incubated with primary antibody (at a dilution of 1:400) in the presence of 0.25% saponin for 1 h and washed three times in PBS. Cells were then incubated with either FITC- or rhodamine-conjugated secondary antibody (Santa Cruz Biotechnology) (at a dilution of 1:300) in the presence of 0.25% saponin for 30 min and washed three times in PBS. Cells were mounted with media containing 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories) and observed under fluorescence microscopy.
Gardella Gel. The native agarose system used to separate herpesviral DNA was first described by Gardella (39). KSHV genomic DNA was characterized as described (40). Briefly, the horizontal agarose gel consisted of two parts, the 0.8% lysis gel (18.8 × 5 × 2.5 cm) containing 1 mg/ml protease K and 2% SDS at the left of loading wells and the 0.75% separation gel (18.8 × 24 × 2.5 cm) at the right. Cells (2 × 106) resuspended in a 50-μl loading buffer (15% Ficoll, 40 μg/ml RNase A, and 0.01% bromophenol blue in Tris-borate-EDTA buffer) after PBS washing were loaded directly to the wells. The electrophoresis was first carried out at 40 V for 2 h for the cells to be lysed by protease K and SDS in the lysis gel, and later at 160 V for 12 h for the released DNA to be separated. After transferring to a nylon membrane, both circular and linear forms of KSHV genomic DNA were detected by KSHV-specific probe.
Luciferase (LUC) Assay. The 4×RBPJrev-LUC vector has been described (28). The RTA-promoter-driven LUC reporter RTA-LUC was cloned by amplifying the 3-kb sequence upstream of RTA initiation codon and subcloning the fragment between XhoI and NcoI sites of pGL3-basic. Cells were plated onto 12-well dishes and grown to 70–80% confluency. DNA transfection was performed by using Fugene 6 (Roche, Indianapolis, IN) according to the manufacturer's protocol. For each transfection, 0.25 μg of reporter plasmid and 0 or 0.75 μg of transactivator were used, and total DNA was normalized to 1 μg with vector pcDNA3.1. pcDNA3.1-lacZ was also included in each transfection as an internal control and β-galactosidase assay was performed as instructed (Promega). Cell extracts were prepared and subjected to LUC assay by using a Luciferase Assay Kit (Promega). Each LUC assay was done in duplicate on at least two independent experiments.
Electrophoretic Mobility-Shift Assays (EMSA). EMSA was performed essentially as described (28). Nuclear extracts were prepared according to the manufacturer's manual (Active Motif, Carlsbad, CA). The 32P-labeled oligonucleotides containing the RBP-Jκ recognition site were incubated with nuclear extracts for 30 min at room temperature, in the presence or absence of unlabeled competitor oligonucleotides, in total volume of 20 μl of binding buffer (20 mM Hepes, pH7.4/50 mM KCl/1 mM EDTA/1 mM MgCl2/10 mM DTT/250 ng of dIdC/8.5% glycerol). Complexes were resolved on a 4% native polyacrylamide gel in 0.5× Tris-borate-EDTA. The gel was dried and exposed onto autoradiographic film.
Results
To explore the role of RBP-Jκ in KSHV replication, we examined the ability of KSHV to infect cells lacking this transcriptional repressor. Because this protein is ubiquitously expressed in human tissues, no human cell lines are available that lack RBP-Jκ activity. However, a line of mouse embryo fibroblasts (termed OT11) has been established from mice whose RBP-Jκ genes have been ablated by homologous recombination (36). We have recently shown that, despite the narrow host range of KSHV in vivo, murine cells can be infected in culture (18); as in most human lines, initial infection results in latency, from which lytic replication can be induced by ectopic expression of RTA (18). Accordingly, we used OT11 cells (and their WT counterparts, OT13; both kindly provided by T. Honjo) for these studies. As shown in Fig. 1A, nuclear extracts from OT13 cells specifically supershifted a 32P-labeled oligonucleotide containing RBP-Jκ site (Fig. 1 A, lane 2), which can be efficiently competed by cold oligonucleotide of WT (Fig. 1 A, lanes 3–5) but not mutant (Fig. 1 A, lanes 6–8) RBP-Jκ site. In contrast, nuclear extracts from OT11 cells failed to produce a specific supershifted band (Fig. 1 A, lane 9). Moreover, in transiently transfected OT11 cells, an RBP-Jκ-dependent reporter gene (four repeats of RBP-Jκ recognition sequence upstream of LUC gene) cannot be activated by RTA coexpression, a defect that can be restored by RBP-Jκ expression (Fig. 1B). Thus, OT11 cells specifically lack functional RBP-Jκ activity.
Fig. 1.

OT11 cells lack functional RBP-Jκ activity, and the RTA promoter contains functional RBP-Jκ sites. (A) Complexes able to specifically supershift RBP-Jκ oligos are present in OT13 but absent in OT11 nuclear extracts. Nuclear extracts were prepared as described in Materials and Methods. 32P-labeled RBP-Jκ oligos were incubated with nuclear extracts from OT13 (lanes 2–8), OT11 (lane 9), or OT11-RBPJκ (lane 10) in the absence (lane 2) or presence of increasing amounts of competitor oligos, WT (lanes 3–5) or mutant (mut) competitors (lanes 6–8). (B) RTA activation of RBP-Jκ-driven LUC reporter in OT11 cells is defective, and the defect can be partially corrected by cotransfecting an RBP-Jκ expression vector. Cells were cotransfected with template DNA 4×RBPJrev-Luc and effector DNA pcDNA3-RTA. In OT11 cells, RTA transactivation was tested in the absence (open bar) or presence (filled bar) of cotransfected pcDNA3.1-RBP-Jκ. LUC assay was performed as in Materials and Methods. The fold induction by RTA was plotted, with the error bars representing SDs of the results from at least two independent experiments. (C) Schematic depiction of putative RBP-Jκ-binding sites in the RTA promoter. The start of the RTA transcript is indicated by an arrow. The orientation and location of each RBP-Jκ-binding site upstream of the RTA transcript start site are shown. (D) RTA activation of RTA-promoter-driven LUC reporter was examined in either OT11 or OT13 cells. Cells were cotransfected with RTA-LUC and RTA (open bar) or empty vector (filled bar). pcDNA3.1-lacZ was included as an internal control. The LUC activities, normalized for transfection efficiency by β-galactosidase activities, are plotted, with the error bars representing SDs of the results from at least two independent experiments.
Fig. 1C shows that the promoter region of the KSHV RTA gene has seven candidate RBP-Jκ-binding sites, located at 430, 616, 1,430, 1,478, 2,056, 2,374, and 2,548 bp upstream of the RTA transcript start site. To examine the impact of these sites, we cloned 3 kb of potential RTA promoter sequences upstream of a LUC reporter gene and examined the activity of this construct in WT and RBP-Jκ-/- cells (Fig. 1D). The basal level of LUC expression in this construct was very low in WT and not appreciably elevated in RBP-Jκ-/- cells. Thus, we could not demonstrate active repression of this reporter gene in the context of transient transfection; however, this experimental situation is known to mimic authentic expression from the herpesviral chromosome very inexactly (41–44). Nonetheless, this result raised the possibility that the RBP-Jκ sites in the RTA promoter might not be functional. This possibility was eliminated when we examined the autoregulation of the RTA-LUC construct. RTA has previously been shown to up-regulate the activity of its own promoter in transient assays (45, 46). Consistent with those reports, we observed that, in WT cells, RTA induced expression from RTA-LUC ≈30-fold (Fig. 1D). Notably, however, this induction was diminished by 90% in RBP-Jκ-/- cells (Fig. 1D), indicating (i) that the RBP-Jκ sites in the RTA promoter are functional for RBP-Jκ binding and (ii) that RBP-Jκ may play a significant role in RTA-mediated autoregulation. The presence of multiple, functional RBP-Jκ sites in the regulatory region of the RTA gene therefore raises the possibility that RBP-Jκ-mediated repression of RTA (and possibly of other downstream lytic genes) might be important in the establishment of latency in de novo infection.
To explore this possibility, OT11 and OT13 cells were exposed to a concentrated stock of KSHV, and 4 days later the cells were assayed for expression of LANA, a specific marker of latent infection. As shown in Fig. 2, both WT and RBP-Jκ-/- cells expressed LANA in comparable numbers of cells. In both cases, LANA was correctly localized to the nucleus in its characteristic punctate pattern. By contrast, expression of markers specific for the lytic cycle (e.g., RTA itself), the polymerase processivity factor ORF59 (Fig. 2), or the envelope glycoprotein K8.1 (not shown) was rare in WT cells and absent in RBP-Jκ-/- cells.
Fig. 2.

Latent but not lytic genes are expressed in KSHV-infected OT11 or OT13 cells. Cells were infected by KSHV for 2 h and further incubated for 4 days. Latent marker LANA and lytic markers RTA and ORF59 were detected by immunofluorescence assay.
Another marker of authentic latency concerns the state of the viral genomic DNA. Encapsidated virion DNA is a linear duplex; however, on infection and establishment of latency, the incoming linear DNA is converted to a circular form that is maintained as an episome. This circular DNA form can be differentiated from linear viral DNA in an electrophoretic system originally described by Gardella (39) in which infected cells are lysed directly in the gel as electrophoresis is initiated. In this system, circular DNAs migrate more slowly than linears, from which they can be readily separated. Fig. 3A shows Gardella gel analysis of KSHV genomes liberated from latently infected WT (lane 4) and mutant (lane 6) cells; in both cases, circular DNA is formed with comparable efficiency.
Fig. 3.

Gardella gel analysis of KSHV genomic DNA. (A) OT13 cells (lanes 3 and 4) or OT11 cells (lanes 5 and 6) were mock-infected (lanes 3 and 5) or KSHV-infected (lanes 4 and 6). At 48 h postinfection, infected cells (2 × 106 cells) were subjected to Gardella gel electrophoresis. As a control for linear and circular KSHV DNA, BCBL-1 cells uninduced (-) or induced (+) with phorbol 12-myristate 13-acetate (PMA) were loaded (lanes 1 and 2). The linear DNA observed in latently infected cells (lanes 1, 4, and 6) likely derives primarily from fragmentation of circular episomes during handling. (B) KSHV genomic DNA failed to replicate after RTA induction in KSHV-infected OT11 cells. OT13 (lanes 1–4) or OT11 (lanes 5–8) were mock-infected (lanes 1 and 5) or KSHV-infected (lanes 2–4 and 6–8), followed 2 h later by either Ad-GFP (lanes 3 and 7) or Ad-RTA (lanes 1, 4, 5, and 8) infection. Cells were then subjected to Gardella gel analysis 2 days later.
Repression by RBP-Jκ in the ground state is only one of two possible roles for RBP-Jκ in the KSHV life cycle. As noted in the introduction, in transient transfection experiments and in cell-free electrophoretic mobility-shift assay reactions, RBP-Jκ can also target the key lytic cycle transactivator RTA to lytic promoters to allow their activation (28). However, herpesviral gene expression during authentic lytic infection often displays more complex regulation than that observed in transient assays, owing to the presence of the full repertoire of viral gene products. To explore the impact of the loss of RBP-Jκ on lytic infection, we infected WT and mutant cells bearing latent KSHV genomes with an adenovirus vector that constitutively expresses the KSHV RTA protein (or a control Ad vector expressing GFP). As expected, Ad-RTA infection results in a prompt and efficient induction of lytic cycle gene expression in WT cells, as judged by immunofluorescent staining for the early gene products of ORF59 and KbZIP (Fig. 4) and the late protein K8.1 (not shown). By contrast, despite efficient expression of RTA from the adenovirus vector in RBP-Jκ-/- cells, virtually no expression of these lytic markers was observed (Fig. 4). This result suggests that the absence of RBP-Jκ results in a dramatic early block to lytic induction: exactly the locus one would expect given the significant number of early viral genes whose promoters harbor RBP-Jκ sites. Consistent with this finding, Gardella gel analysis of KSHV genomes 2 days post Ad-RTA induction reveals a striking failure of the mutant cells to accumulate progeny linear viral DNA, under conditions in which this process is readily observed in WT cells (Fig. 3B). This block to DNA synthesis also explains the impaired expression of K8.1, a late gene that is not directly regulated by RTA but whose expression, like that of many late genes, depends on viral DNA replication.
Fig. 4.

Lytic gene expression was defective in KSHV latently infected OT11 cells after RTA induction. Cells (OT13 or OT11) were infected by KSHV followed by Ad-RTA for lytic reactivation. At 4 days postinfection, the lytic markers KbZIP and ORF59 were detected by immunofluorescence assay. RTA staining was included to monitor the Ad-RTA infection efficiency.
To assess the stringency and biological significance of the block, we also examined the supernatants of Ad-RTA-induced WT and RBP-Jκ null cells for infectious KSHV, by inoculation of TIME cells with said supernatants. Two days after inoculation, TIME cells were stained for KSHV LANA. As shown in Fig. 5A, peak virus production from OT13 cells occurs on day 4 post Ad-RTA induction, and reaches titers sufficient to infect nearly 40% of TIME cells in the recipient monolayer. [Although this is less virus than is produced by Ad-RTA induction of KSHV-infected HFF cells (Fig. 5A, open circles), it is substantially more than is produced by another human cell line, SLK, an endothelial line derived from a KS tumor.] Importantly, no infectivity was produced by RBP-Jκ-/- cells at any time point. Fig. 5B shows representative immunofluorescence images of the recipient HFF monolayer infected with culture supernatants from KSHV-infected OT13 (Left) or OT11 (Right). No detectable infectivity was released from latently infected cells (Middle), but Ad-RTA induction resulted in virus production in WT but not mutant cells (Bottom).
Fig. 5.

Comparison of virus release from induced cells. (A) Time-course analysis of virus release from induced human and mouse cell lines. HFF, SLK, OT13, and OT11 were KSHV-infected for 2 h, followed by Ad-RTA superinfection. Supernatants were collected every day and concentrated by ultracentrifugation. The concentrated virus was then used to infect TIME cells, which were stained for LANA at 48 h postinfection. Serial 2-fold dilutions of the concentrated virus were used in the infectivity assay to ensure that the virus titer is in linear range. The percentage of LANA-positive cells obtained with the undiluted stock from each cell type is plotted against time (days postinfection). ○, HEF; •, SLK; ▪, OT13; □, OT11. (B) No infectious KSHV particles were released after RTA induction in KSHV-infected OT11 cells. OT13 (Left)or OT11 (Right) cells were infected with Ad-RTA alone (Top), KSHV alone (Middle), or KSHV plus Ad-RTA (Bottom). Six days postinfection, supernatants were collected from each culture, concentrated equivalently, and used to infect HFF cells. Two days postinfection, HFFs were stained for LANA expression by immunofluorescence assay.
To conclusively demonstrate that the defect of RTA-induced lytic reactivation is due to the absence of RBP-Jκ and not to some secondary defect of OT11 cells, we used a retroviral expression system to reintroduce RBP-Jκ into OT11 cells and selected cells stably expressing RBP-Jκ (designated OT11-RBPJκ). As in the case of OT13 cells, nuclear extracts of this OT11-RBPJκ were able to specifically supershift an RBP-Jκ-site-containing oligonucleotide (Fig. 1 A, lane 10). Furthermore, by transient transfection, the RTA-mediated activation of an RBPJκ-driven LUC reporter was restored in OT11-RBPJκ, indicating the presence of functional RBP-Jκ protein (Fig. 6A). We then infected OT11-RBPJκ with KSHV, which, as expected, resulted in the efficient establishment of latency, as judged by LANA staining (Fig. 6B). When these cells were superinfected with Ad-RTA, KSHV lytic cycle genes (KbZIP and ORF59) were readily activated (Fig. 6B). The fact that OT11-RBPJκ restored RTA-induced lytic cycle gene expression provides further evidence that RBP-Jκ is essential for RTA-induced KSHV reactivation program.
Fig. 6.
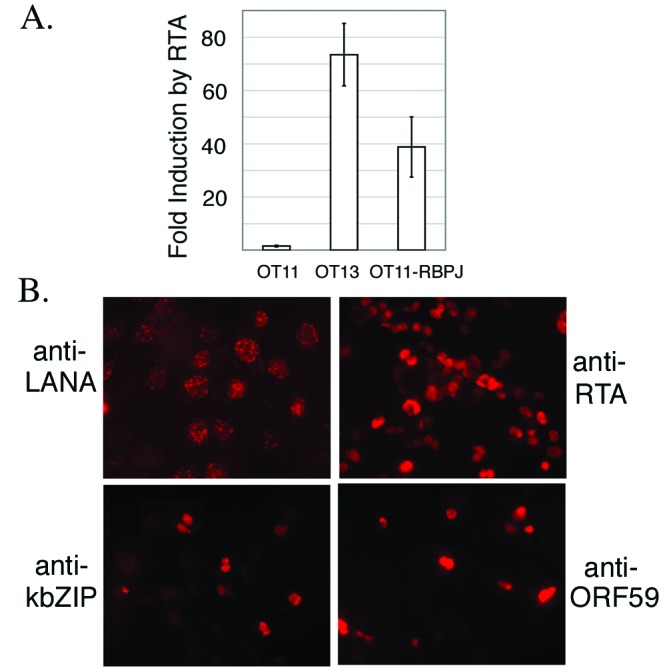
Stable cells established from OT11 that express RBP-Jκ restore functional RBP-Jκ activity and KSHV lytic gene expression after induction. OT11-RBP-Jκ cells were stable cells selected from OT11 cells after infection by a retrovirus expressing RBP-Jκ.(A) Cells were cotransfected with template DNA 4×RBPJrev-Luc and effector DNA pcDNA3-RTA. LUC assay was performed as in Materials and Methods. The fold induction by RTA was plotted, with the error bars representing SDs of the results from at least two independent experiments. (B) OT11-RBP-Jκ cells were infected by KSHV, followed by Ad-RTA for lytic induction. Latency marker (LANA) and lytic genes (KbZIP and ORF59) were analyzed by immunofluorescence assay. RTA staining was included to monitor Ad-RTA infection efficiency.
Discussion
These experiments use murine cells to explore the role of RBP-Jκ in KSHV infection. Although mice are not natural hosts of KSHV in vivo, results presented here and elsewhere (18) indicate that cultured murine cells can correctly support all phases of the KSHV life cycle. KSHV latency in OT13 cells meets all known criteria of latent herpesviral infection: expression of latency-specific genes, extinction of lytic-cycle markers, circularization of viral DNA, and ability to support lytic reactivation on induction by RTA. Virus produced by lytic reactivation in mouse cells retains infectivity for human cells. We therefore believe that mouse cells represent a biologically relevant system in which to explore the latent-lytic switch of KSHV. The fact that RBP-Jκ is one of the most conserved transcription factors in vertebrate biology (its human and mouse homologs share 92% amino acid identity) suggests that its functional roles are likely to be conserved in both species. This finding further supports the use of murine cells for the study of this specific question.
Our results serve to delineate the role of the host transcriptional repressor RBP-Jκ in the biology of the KSHV life cycle. Despite the presence of functional RBP-Jκ sites in the promoter of the key regulator of the latent-lytic switch of KSHV, we could not demonstrate active repression of the locus by endogenous RBP-Jκ by using transiently expressed reporter genes (Fig. 1). Consistent with this result, we find that latency establishment is normal in RBP-Jκ-/- cells (Figs. 2 and 3A), indicating that RTA expression is extinguished normally (Fig. 2) even in the absence of this known repressor. We think it likely that RBP-Jκ-based repressive complexes do form on the viral RTA promoter at these sites, because the sites can clearly support RBP-Jκ interactions during the lytic cycle (Fig. 1D). However, our results indicate that such repressive complexes cannot be sufficient for effective extinction of RTA expression, and point to the existence of additional layers of control of RTA, either the presence of redundant negative regulators or the absence of additionally required positive signals. We do not yet know the identities of these additional regulators.
Despite its dispensability for the establishment of latency, RBP-Jκ still plays a key role in KSHV biology, namely, to allow positive regulation by RTA of genes essential for the lytic cycle. When RBP-Jκ is absent, even deliberate overexpression of RTA cannot trigger replication, indicating that it plays crucial roles downstream of RTA expression. Although it is formally possible that RBP-Jκ may also have functions in lytic replication that are RTA-independent, the entire phenotype of the RBP-Jκ null cells can be explained on the basis of a requirement for RTA-RBP-Jκ interactions in the transcription of delayed-early genes. Numerous important DE genes harbor RBP-Jκ sites that are critical for their RTA responsiveness in transient transfection assays, e.g., the regulator MTA and the major single-stranded DNA-binding protein (SSB) (28). We presume that the multiple defects in DE gene expression we have observed (Fig. 4) account for the failure of viral DNA replication, and that the latter is the principal determinant of the failure to activate late gene expression. As a result, no infectivity can be generated in the absence of RBP-Jκ.
Clearly, however, there is much complexity in lytic gene expression in vivo that cannot be simply explained by the results of transient transfection assays. For example, the ORF59 gene, a DE gene encoding a polymerase processivity factor, has predicted RBP-Jκ sites in its promoter but is still activated strongly by RTA in both OT11 and OT13 cells (unpublished results), implying that it has RBP-Jκ-independent pathways of RTA responsiveness. This situation recalls that of the KSHV early gene polyadenylated nuclear RNA (PAN), which has RBP-Jκ sites but also contains a high-affinity RTA-binding site whose impact dwarfs that of the RBP-Jκ site (28). Nonetheless, when the ORF59 gene is in the context of the authentic viral genome, it is completely unresponsive to exogenous RTA in OT11 cells (Fig. 4). This result suggests that ORF59 may be being controlled more indirectly by RBP-Jκ in vivo, e.g., by other viral regulators (like MTA) which are themselves directly influenced by RBP-Jκ, or by cis-acting changes in the template DNA or chromatin that are not recapitulated by transfected plasmids.
Finally, we note that, despite RBP-Jκ's essential role in lytic reactivation, not every stimulus that acts through RBP-Jκ sites can induce lytic KSHV growth. Ectopic expression of Notch intracellular domain or Epstein–Barr virus-encoded nuclear antigen 2 (32, 34, 35, 47–50), two proteins that displace repressive complexes from RBP-Jκ and replace them with activators, fails to induce lytic KSHV gene expression (unpublished results). This finding suggests that activation through the RBP-Jκ site, although necessary for lytic induction, is not sufficient for this process, and supports the inference that the non-RBP-Jκ-dependent functions of RTA (for example, its direct binding to high-affinity sites in the genome) also play critical roles in lytic reactivation.
Acknowledgments
We thank Dr. T. Honjo for providing OT13 and OT11 cells, Dr. J. Bechtel and Dr. A. Grundhoff for technical support, and all of the members of Ganem laboratory for advice. D.G. is an investigator of the Howard Hughes Medical Institute.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: RBP-Jκ, recombination signal sequence-binding protein-Jκ; KS, Kaposi's sarcoma; KSHV, KS-associated herpesvirus; RTA, replication and transcription activator; TIME, telomerase-immortalized microvascular endothelial; LUC, luciferase; LANA, latency-associated nuclear antigen; Ad, adenovirus; HFF, human foreskin fibroblast.
References
- 1.Beral, V. (1991) in Cancer, HIV and AIDS, eds. Beral, V., Jaffe, H. W. & Weiss, R. (Cold Spring Harbor Lab. Press, Plainview, NY), Vol. 10, pp. 5-22. [Google Scholar]
- 2.Ensoli, B., Barillari, G. & Gallo, R. C. (1991) Hematol. Oncol. Clin. North Am. 5 281-295. [PubMed] [Google Scholar]
- 3.Chang, Y., Cesarman, E., Pessin, M. S., Lee, F., Culpepper, J., Knowles, D. M. & Moore, P. S. (1994) Science 266 1865-1869. [DOI] [PubMed] [Google Scholar]
- 4.Ganem, D. (1997) Cell 91 157-160. [DOI] [PubMed] [Google Scholar]
- 5.Schulz, T. F. (1999) Adv. Cancer Res. 76 121-160. [DOI] [PubMed] [Google Scholar]
- 6.Boshoff, C., Schulz, T. F., Kennedy, M. M., Graham, A. K., Fisher, C., Thomas, A., McGee, J. O., Weiss, R. A. & O'Leary, J. J. (1995) Nat. Med. 1 1274-1278. [DOI] [PubMed] [Google Scholar]
- 7.Staskus, K. A., Zhong, W., Gebhard, K., Herndier, B., Wang, H., Renne, R., Beneke, J., Pudney, J., Anderson, D. J., Ganem, D. & Haase, A. T. (1997) J. Virol. 71 715-719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Radkov, S. A., Kellam, P. & Boshoff, C. (2000) Nat. Med. 6 1121-1127. [DOI] [PubMed] [Google Scholar]
- 9.Rivas, C., Thlick, A. E., Parravicini, C., Moore, P. S. & Chang, Y. (2001) J. Virol. 75 429-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharp, T. V., Wang, H. W., Koumi, A., Hollyman, D., Endo, Y., Ye, H., Du, M. Q. & Boshoff, C. (2002) J. Virol. 76 802-816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friborg, J., Jr., Kong, W., Hottiger, M. O. & Nabel, G. J. (1999) Nature 402 889-894. [DOI] [PubMed] [Google Scholar]
- 12.Sturzl, M., Hohenadl, C., Zietz, C., Castanos-Velez, E., Wunderlich, A., Ascherl, G., Biberfeld, P., Monini, P., Browning, P. J. & Ensoli, B. (1999) J. Natl. Cancer Inst. 91 1725-1733. [DOI] [PubMed] [Google Scholar]
- 13.Moore, P. S., Boshoff, C., Weiss, R. A. & Chang, Y. (1996) Science 274 1739-1744. [DOI] [PubMed] [Google Scholar]
- 14.Boshoff, C., Endo, Y., Collins, P. D., Takeuchi, Y., Reeves, J. D., Schweickart, V. L., Siani, M. A., Sasaki, T., Williams, T. J., Gray, P. W., et al. (1997) Science 278 290-294. [DOI] [PubMed] [Google Scholar]
- 15.Nicholas, J., Ruvolo, V. R., Burns, W. H., Sandford, G., Wan, X., Ciufo, D., Hendrickson, S. B., Guo, H. G., Hayward, G. S. & Reitz, M. S. (1997) Nat. Med. 3 287-292. [DOI] [PubMed] [Google Scholar]
- 16.Neipel, F., Albrecht, J. C., Ensser, A., Huang, Y. Q., Li, J. J., Friedman-Kien, A. E. & Fleckenstein, B. (1997) J. Virol. 71 839-842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bais, C., Santomasso, B., Coso, O., Arvanitakis, L., Raaka, E. G., Gutkind, J. S., Asch, A. S., Cesarman, E., Gershengorn, M. C., Mesri, E. A. & Gerhengorn, M. C. (1998) Nature 391 86-89. [DOI] [PubMed] [Google Scholar]
- 18.Bechtel, J., Liang, Y., Hvidding, J. & Ganem, D. (2003) J. Virol. 77 6474-6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vieira, J., O'Hearn, P., Kimball, L., Chandran, B. & Corey, L. (2001) J. Virol. 75 1378-1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun, R., Lin, S. F., Gradoville, L., Yuan, Y., Zhu, F. & Miller, G. (1998) Proc. Natl. Acad. Sci. USA 95 10866-10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lukac, D. M., Renne, R., Kirshner, J. R. & Ganem, D. (1998) Virology 252 304-312. [DOI] [PubMed] [Google Scholar]
- 22.Lukac, D. M., Kirshner, J. R. & Ganem, D. (1999) J. Virol. 73 9348-9361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gradoville, L., Gerlach, J., Grogan, E., Shedd, D., Nikiforow, S., Metroka, C. & Miller, G. (2000) J. Virol. 74 6207-6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seaman, W. T., Ye, D., Wang, R. X., Hale, E. E., Weisse, M. & Quinlivan, E. B. (1999) Virology 263 436-449. [DOI] [PubMed] [Google Scholar]
- 25.Song, M. J., Li, X., Brown, H. J. & Sun, R. (2002) J. Virol. 76 5000-5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lukac, D. M., Garibyan, L., Kirshner, J. R., Palmeri, D. & Ganem, D. (2001) J. Virol. 75 6786-6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang, P. J., Shedd, D., Gradoville, L., Cho, M. S., Chen, L. W., Chang, J. & Miller, G. (2002) J. Virol. 76 3168-3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang, Y., Chang, J., Lynch, S. J., Lukac, D. M. & Ganem, D. (2002) Genes Dev. 16 1977-1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kao, H. Y., Ordentlich, P., Koyano-Nakagawa, N., Tang, Z., Downes, M., Kintner, C. R., Evans, R. M. & Kadesch, T. (1998) Genes Dev. 12 2269-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsieh, J. J., Zhou, S., Chen, L., Young, D. B. & Hayward, S. D. (1999) Proc. Natl. Acad. Sci. USA 96 23-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oswald, F., Kostezka, U., Astrahantseff, K., Bourteele, S., Dillinger, K., Zechner, U., Ludwig, L., Wilda, M., Hameister, H., Knochel, W., et al. (2002) EMBO J. 21 5417-5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Waltzer, L., Logeat, F., Brou, C., Israel, A., Sergeant, A. & Manet, E. (1994) EMBO J. 13 5633-5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Waltzer, L., Perricaudet, M., Sergeant, A. & Manet, E. (1996) J. Virol. 70 5909-5915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsieh, J. J. & Hayward, S. D. (1995) Science 268 560-563. [DOI] [PubMed] [Google Scholar]
- 35.Grossman, S. R., Johannsen, E., Tong, X., Yalamanchili, R. & Kieff, E. (1994) Proc. Natl. Acad. Sci. USA 91 7568-7572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oka, C., Nakano, T., Wakeham, A., de la Pompa, J. L., Mori, C., Sakai, T., Okazaki, S., Kawaichi, M., Shiota, K., Mak, T. W. & Honjo, T. (1995) Development 121 3291-3301. [DOI] [PubMed] [Google Scholar]
- 37.Polson, A. G., Huang, L., Lukac, D. M., Blethrow, J. D., Morgan, D. O., Burlingame, A. L. & Ganem, D. (2001) J. Virol. 75 3175-3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kedes, D. H., Lagunoff, M., Renne, R. & Ganem, D. (1997) J. Clin. Invest. 100 2606-2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gardella, T., Medveczky, P., Sairenji, T. & Mulder, C. (1984) J. Virol. 50 248-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Renne, R., Lagunoff, M., Zhong, W. & Ganem, D. (1996) J. Virol. 70 8151-8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mavromara-Nazos, P. & Roizman, B. (1987) Virology 161 593-598. [DOI] [PubMed] [Google Scholar]
- 42.Silver, S. & Roizman, B. (1985) Mol. Cell. Biol. 5 518-528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meier, J. L. & Stinski, M. F. (1997) J. Virol. 71 1246-1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meier, J. L. & Pruessner, J. A. (2000) J. Virol. 74 1602-1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deng, H., Young, A. & Sun, R. (2000) J. Gen. Virol. 81 3043-3048. [DOI] [PubMed] [Google Scholar]
- 46.Sakakibara, S., Ueda, K., Chen, J., Okuno, T. & Yamanishi, K. (2001) J. Virol. 75 6894-6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jarriault, S., Brou, C., Logeat, F., Schroeter, E. H., Kopan, R. & Israel, A. (1995) Nature 377 355-358. [DOI] [PubMed] [Google Scholar]
- 48.Tamura, K., Taniguchi, Y., Minoguchi, S., Sakai, T., Tun, T., Furukawa, T. & Honjo, T. (1995) Curr. Biol. 5 1416-1423. [DOI] [PubMed] [Google Scholar]
- 49.Fortini, M. E. & Artavanis-Tsakonas, S. (1994) Cell 79 273-282. [DOI] [PubMed] [Google Scholar]
- 50.Henkel, T., Ling, P. D., Hayward, S. D. & Peterson, M. G. (1994) Science 265 92-95. [DOI] [PubMed] [Google Scholar]