

Abstract
Distamycin A is a well known polyamide antibiotic that can bind in the minor groove of duplex DNA primarily at AT-rich sequences both as a monomer or as a side-by-side antiparallel dimer. The association phase of the distamycin binding reaction has not been studied in either of its binding modes, because of the lack of an adequate UV or CD signal at the low concentrations needed to monitor the fast bimolecular reaction. We report a significant increase in fluorescence amplitude, accompanied by a small red shift, on binding distamycin to its specific target sites. This signal can be used to monitor drug binding in steady-state and time-resolved processes. Distamycin shows extremely fast association with the 1:1 binding site, with a bimolecular rate of 7 × 107 M−1⋅s−1 and also fairly rapid dissociation (≈3 s−1). When DNA is in excess, there is a slow component in the association reaction whose rate decreases strongly with increasing DNA concentration. Binding of the drug to the 2:1 site occurs in two distinct steps: fast, sequential binding of each drug molecule to the DNA with a bimolecular rate comparable to that at the 1:1 site, followed by a slow (≈4 s−1) equilibration to the final population. Dissociation from the 2:1 site is ≈40-fold slower than from the 1:1 site. This study provides the groundwork for analysis of the binding kinetics of longer polyamides and covalently linked polyamides that have recently been shown to inhibit transcription in vivo.
Keywords: fluorescence, stopped-flow kinetics
Distamycin A (Dst), a naturally occurring polyamide antibiotic, is one of the most extensively studied members of a class of molecules that bind in the minor groove of duplex DNA. Its binding affinity derives from specific hydrogen bonding contacts between the amide protons and the O2 or N3 of pyrimidines and purines, respectively, electrostatic interactions with the backbone, and van der Waals contacts with the walls of the minor groove. Two distinct modes of binding have been observed. In the 1:1 motif, a single drug molecule binds in the minor groove, either making contacts with one of the two strands, or forming bifurcated hydrogen bonds involving bases on both strands of the duplex (1). In the 2:1 binding mode, each drug molecule makes contacts with bases on only one of the two strands(2, 3). In both modes, binding of these compounds causes a small change in UV absorbance (≈10 mAU⋅μM−1⋅ cm−1) as well as a small change in molar ellipticity (≈0.1 mdeg⋅μM−1⋅ cm−1). The 1:1 and 2:1 binding modes are distinguishable by features in the CD spectrum in the region from 240 to 280 nm (4). Despite these spectroscopic changes on binding, the kinetics of formation of specific complexes with DNA have not been well studied, primarily because, at the concentrations needed to obtain an adequate signal, the association rates are too fast to measure by using commercially available stopped-flow instruments (5). Thus, whereas there is a wealth of information regarding the thermodynamics (6, 7) and structures, both in crystals (1, 2, 8) and in solution (3, 9, 10), for each type of complex, very little is known about the kinetic routes that result in their formation.
Due to the greater sensitivity of fluorescence-based techniques in comparison with UV absorbance or CD, we turned to fluorescence measurements as a handle for observing the binding of Dst to its target sites. Fluorescence-based approaches have been used in the past to measure indirectly the binding of drugs in the minor groove of DNA. Binding of the related polyamide drug netropsin has been monitored in steady-state measurements by quenching of fluorescence of the base 2-aminopurine incorporated at the target site (11). Quantitative measurement of the binding of minor groove binders has also been reported in a system where a dansyl “semantophore” was tethered to a base in the major groove and its fluorescence enhancement was monitored as a function of concentration of Dst (12). Direct measurement of the change in fluorescence spectra on binding to DNA has been reported for some of the more fluorescent compounds of this family, such as Hoechst 33258 and 4′,6-diamidino-2-phenylindole (DAPI), as well as a few others (13, 14).
It has been known for some time that Dst itself is weakly fluorescent and that its fluorescence is enhanced on binding to poly(dA-dT) (15). However, the fluorescence change on binding to specific sites has not been characterized or used in quantitative steady-state measurements or for monitoring the kinetics of association at discrete binding sites. We report that the fluorescence changes on binding can be used to monitor the association of Dst at both its 1:1 and 2:1 binding sites in steady-state and stopped-flow measurements. These measurements have, for the first time, enabled us to estimate the Arrhenius activation energy and the entropy of activation involved in the interaction of these drugs with their specific binding sites. Our studies provide a potentially useful assay for studying the binding kinetics of longer polyamides (16) that have recently been shown to possess extremely high affinity and specificity for larger binding sites as well as promising effects in vivo.
Materials and Methods
Oligonucleotide Target Sites and Dst.
Oligonucleotides were purchased from the Keck oligonucleotide synthesis facility at Yale University. Oligonucleotides were purified by reversed-phase HPLC on a Pharmacia ProRPC column using a triethylamine acetate/acetonitrile buffer system followed by lyophilization and dialysis against water. Concentrations were estimated by using the pairwise extinction coefficient approximation. Duplexes were made by annealing stoichiometric amounts of each strand with its respective complement. Serial dilutions of each duplex were made from the same stock solutions, which were stored frozen at −20°C until use. The sequences of oligonucleotides used in these studies are illustrated in Fig. 1. Clearly, in the absence of a “placeholder” residue such as imidazole, several binding modes are possible at any given 4-bp sequence. Our choice for the target sequences was governed by the availability of structural and thermodynamic data indicating the preponderance of one mode over the other (17). To minimize the possibility of multiple binding modes at the same sequence, we chose the sequence 5′-GCGATTAGCG-3′ for the 1:1 binding site and the sequence 5′-GCGAAGTTGCG-3′ for the 2:1 binding site. Dst was purchased from Sigma and used without further purification. Analysis by HPLC showed the purity to be greater than 95%. Due to the slow hydrolysis of Dst in aqueous solutions (18), only fresh solutions (<2 days after preparation) were used in all experiments.
Figure 1.

Structure of Dst (A) and the representative 1:1 and 2:1 binding sites (B).
Steady-State Fluorescence Measurements.
Steady-state fluorescence measurements were obtained on a Hitachi (Tokyo) F4500 fluorescence spectrophotometer by irradiation at 300 nm with excitation slit width at 5 nm and emission slit width at 10 nm. These large slit widths were used to maximize the observed fluorescence signals from Dst and its complexes with DNA. Each spectrum in Fig. 2A is an average of five measurements at the rate of 240 nm/min. Spectra are shown after correction by subtraction of the buffer scattering contribution. All steady-state measurements were performed at 25°C.
Figure 2.

(A) Steady-state fluorescence change on Dst binding to the 1:1 sites, shown here for a solution containing 2.5 μM each of Dst and DNA. Samples were irradiated at 300 nm with excitation slit at 5 nm and emission slit at 10 nm. (B) Representative fluorescence-detected stopped-flow trace of Dst binding to its 1:1 binding site under excess Dst conditions. The trace shown here corresponds to a reaction in which the final concentration of Dst and 1:1 site was 1.25 μM and 500 nM. (C) Representative stopped-flow trace of the slow phase observable under excess DNA conditions. Shown here [1:1DNA] = 15 μM and [Dst] = 2.5 μM.
Fluorescence Detected Stopped-Flow Kinetics.
Stopped-flow measurements were performed in an Applied Photophysics (Surrey, U.K.) DX 17 MV single-mixing stopped-flow microvolume reaction analyzer. Observations were conducted in the fluorescence mode by using excitation light set at 300 nm and both slits of the excitation monochromator set to between 0.5 and 2 mm (≈2.5 to 10 nm). A 360-nm longpass filter (Schott, Duryea, PA) was used to prevent any excitation light from reaching the emission photomultiplier. The photomultiplier voltage was set to between 900 and 1,000 V, and appropriate offset voltage was applied to center the observable signal at 0 mV. Sensitivity was set to between 0.2 and 1.0 mV for full scale. Reactions were monitored for a time corresponding to at least six t1/2s, and 1,000 data points were collected for each shot. The split time-base feature of the instrument was used to collect more data points in the initial phase of the reaction and enable observation of any slow phase wherever necessary. At least three and up to 10 shots were used for each experiment using 50 μl of each reactant per shot in 1:1 mixing mode. The dead-time of the instrument is less than 2 ms. Temperature was varied (within ±0.1°C) by using an attached circulating water bath and monitored by using an internal thermistor in the instrument. Unless specified, all experiments were performed at 25°C. Data from stopped-flow experiments were processed by using the software origin (Microcal, Northampton, MA) and fitted to appropriate equations by using the Levenberg Marquardt non-linear-least-squares fitting subroutine.
Results and Discussion
Dst Fluorescence Is Enhanced on Binding to Both Its 1:1 and 2:1 Binding Sites.
Dst has an extended conjugated system consisting of aromatic N-methyl pyrrole rings linked by carboxamide groups. It is not surprising that such an extended conjugated system is fluorescent. However, the fluorescent properties of Dst have long been ignored. Indeed, Dst is commonly used in histochemical protocols as a negative stain to counteract the bright fluorescence of minor groove binding ligands such as 4′,6-diamidino-2-phenylindole (DAPI) (19). At least one study has reported the use of a DAPI displacement assay to study the binding of “nonfluorescent” minor groove binding molecules (20). The fluorescence of Dst in aqueous solutions and its enhancement on binding to DNA were first reported by Stockert and coworkers (15). The authors demonstrated that there is a sequence-dependent enhancement of Dst fluorescence on binding DNA. The order of relative increase in fluorescence observed was poly(dA-dT) > calf thymus DNA >> poly (dG-dC). The increase in fluorescence was ascribed to both an increase in rigidity of the Dst chromophore on binding into the minor groove, and also to a process of fluorescence resonance energy transfer between base excimers and Dst, resulting in enhancement of drug fluorescence with a corresponding decrease in DNA fluorescence. The fluorescence of Dst has, however, not been used in studying its interactions with specific DNA sequences, primarily because it is very small in magnitude and is quenched strongly in aqueous solution. We observe a significant enhancement of fluorescence on binding at both the 1:1 (Fig. 2A) and 2:1 sites.
The Bimolecular Association Rate of Dst at Its 1:1 Binding Site Is Close to Diffusion Limited.
The process of association of Dst with DNA is likely to be complex, consisting of individual steps during which the drug samples the many available sites in the DNA minor groove, before establishing a complex with the maximum number of favorable interactions. However, the observed bimolecular rate will depend only on the rate of formation of a metastable complex (the nucleation unit), for which addition of a favorable interaction is faster than dissociation of the complex. Several previous studies (4, 21) have concluded that the association kinetics of Dst are too fast to measure by using stopped-flow methods . However, the concentrations of Dst and DNA used in these studies were in the range of tens to hundreds of micromolar, and absorbance was used as the mode of detection.
By using the more sensitive fluorescence detection, we have been able to measure the extremely fast association of Dst with its 1:1 site. Under conditions of excess Dst, the association signal is monophasic (Fig. 2B), and the apparent rates show a linear dependence on concentration of Dst. The bimolecular association rate constant obtained from the slope of a plot of the observed rate vs. Dst concentration is 6.6 × 107 M−1⋅s−1 (Table 1). Therefore, compared with intercalating drugs such as actinomycin, which have association rates in the order of 105 M−1⋅s−1, and triplex-forming oligonucleotides, which have association rates in the range of 103-104 s−1, Dst is able to search for and bind its target site very rapidly.
Table 1.
Kinetic parameters for distamycin binding to its 1∶1 and 2∶1 sites
| Site | Phase | k1 | Ea1, kcal⋅mol−1 | ΔS1, cal⋅K−1⋅mol−1 | k2, s−1 | Ea2, kcal⋅mol−1 | ΔS2, cal⋅K−1⋅mol−1 |
|---|---|---|---|---|---|---|---|
| 1∶1 | On | 66.4 (3.8) μm−1⋅s−1 | 2.3 | −16.6 | 15.1 (0.3) | 19.7 | 16 |
| Off | 3.1 (0.08) s−1 | 23.7 | 33.7 | — | — | — | |
| 2∶1 | On | 74.5 (3.3) μm−1⋅s−1 | — | — | 5.1 (0.2) | 19.5 | 11 |
| Off | 0.08 (0.004) s−1 | 23.2 | 9.4 | — | — | — |
Values of bimolecular association constant for both sites were derived from plots of apparent rate vs. [Dst] at 25°C. Figures in parentheses indicate SD about the mean. The values of Ea and ΔS were derived from Arrhenius plots with temperatures ranging from 10°C to 35°C. The values of k2 indicated were obtained at [DNA] = 5 μM and [Dst] = 10 μM for the 1∶1 site and [DNA] = 2.5 μM and [Dst] = 25 μM for the 2∶1 site.
By studying the temperature dependence of the fast association rate, we can estimate the enthalpy and the entropy of activation of the association reaction. A plot of ln (kon) vs. 1/T gives a straight line from which the energy of activation and entropy of activation can be determined by using the expressions
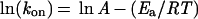 |
and
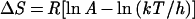 |
where kon is the observed association rate constant, A is the Arrhenius preexponential factor, Ea is the energy of activation of the process, R is the gas constant, k is Boltzman's constant, h is Planck's constant, and T the temperature in Kelvin. The values of energy and entropy of activation for the fast bimolecular step are 2.3 kcal⋅mol−1 and −16.6 cal⋅K−1⋅mol−1, consistent with expectation for a diffusion-limited bimolecular reaction.
A Slow Phase Is Observable in the Association Kinetics Under Excess DNA Conditions.
Whereas the binding kinetics of Dst at the 1:1 site are essentially monophasic under excess Dst conditions, the behavior under excess DNA is markedly different. A slower phase is clearly discernible (Fig. 2C); the rate of this phase slows down with increasing DNA concentration (Fig. 3A), with a linear dependence on reciprocal DNA concentration (Fig. 3B). This property is unusual in DNA–ligand reaction kinetics. One previous example is the reaction of RNA polymerase with DNA (22), for which the authors proposed that polymerase exists in reactive and nonreactive states. The slow component was assigned to equilibration over the polymerase states as binding occurs.
Figure 3.

(A) [Dst] dependence of slow phase in association. (B) Variation of slow phase in association reaction with 1/[1:1DNA]. The values of slope and intercept are 7.9(0.3) s−1 and 176.6(3.0) M⋅s−1 respectively.
It is plausible that Dst could have nonreactive conformations because of the potential for 180° bond rotations between the pyrrole rings, which would disrupt the crescent shape of the molecule and prevent insertion into the minor groove (23, 24). The ≈20 kcal/mol activation energy for the slow signal (Table 1) would be a consequence of the partial double bond character of the bonds in question. This model would explain the observation that no slow signal is seen when Dst is in excess, because all of the DNA molecules would find reaction-competent conformers in that case. The slow signal would appear only in DNA excess, when the entire Dst population must react. However, standard relaxation kinetic analysis shows that the high concentration limit of the rate corresponds to the rate constant for conversion of the inactive to the active form, and the low concentration limit to the sum of the forward and reverse rate constants for isomerization. Thus, the data in Fig. 3 imply that the equilibrium constant for the isomerization should favor the unreactive over the reactive form by at least 5-fold. This conclusion is not consistent with the observation that most of the drug reacts rapidly, and must therefore be in the reactive form.
We have also considered models in which the drug molecules are uniform in conformation, but they can bind transiently to incorrect sites. If the correct and incorrect sites do not overlap (noncompetitive binding), such models can show the inverse concentration dependence documented in Fig. 3, although, for many values of the rate constants, the predicted slope of Fig. 3B is smaller than observed. Relaxation kinetic analysis of this case shows that the high concentration limit for the rate is proportional to the rate of drug dissociation. The similarity between the activation energies for dissociation (see below, Table 1) and the slow phase of the association reaction is a supportive factor for this model.
The actual circumstance is likely to be more complicated than these limiting models. For example, Dst conformers unsuited for the minor groove may bind noncompetitively to DNA, for example in the major groove, slowing their rate of isomerization at high DNA concentrations. Solution of this problem will require transient analysis of Dst structure during the binding process.
Dissociation of Dst from Its 1:1 Site Is Fast and Monophasic.
A commonly used technique for measuring the dissociation rates of drugs from DNA is the observation of a spectroscopic signal on mixing with solutions of detergents such as SDS, which can sequester dissociated drug molecules and prevent their rebinding to DNA (25). Dissociation of Dst from its 1:1 complex was observed by mixing a preequilibrated complex of target duplex and a 10-fold excess of Dst with 2% SDS. Dissociation is essentially monophasic (see Fig. 5A), with an apparent dissociation rate of ≈4 s−1 at 25°C. The measured dissociation rates are independent of SDS concentration between 1% and 10% SDS (see Fig. 6, which is published as supplementary data on the PNAS website, www.pnas.org). Again, the temperature dependence of the dissociation rates can be used to calculate the energy and entropy of activation for the dissociation reaction (Table 1).
Figure 5.

(A) Representative stopped-flow trace for SDS-assisted dissociation of Dst from its target 1:1 site. Dissociation experiments carried out here used equilibrated complex formed with [DNA] = 5 μM, [Dst] = 10 μM, and mixed with an equal volume of 2% SDS. (B) Representative stopped-flow trace of dissociation of Dst from its 2:1 site, shown here for an equilibrated complex formed by incubating [DNA] = 2.5 μM and [Dst] = 25 μM and injecting against an equal volume of 2% SDS.
Association Kinetics at the 2:1 Site Show Biphasic Behavior.
As in the case of the 1:1 site, enhancement of Dst fluorescence is seen on binding to the 2:1 site. In contrast to the uniform direction of change in fluorescence observable when Dst binds at the 1:1 site, the association reaction at the 2:1 site shows (Fig. 4) a rapid increase in fluorescence, followed by a decrease, with a time constant 50- to 100-fold slower than the first process. The rate of the first process is linearly dependent on concentration of Dst, which rules out a rate-limiting step involving both Dst monomers at the target site. The bimolecular rate constant calculated from a plot of observed rate for this phase vs. Dst concentration yields a value very close to that observed for binding of a single Dst molecule at the 1:1 site (Table 1). The linear dependence of rate on Dst concentration implies that the second Dst binds more rapidly than the first dissociates. We were not able to measure at low enough Dst concentrations to reverse this inequality.
Figure 4.

Representative stopped-flow trace for Dst binding to its 2:1 site under excess Dst conditions. Shown here for [DNA] = 2.5 μM, [Dst] = 5 μM.
By using excess Dst, the first phase can be accelerated to a point where it is faster than the dead time of the instrument, allowing observation of the second phase alone and measurement of its variation with concentration of Dst. The rate of the second process, calculated from fitting to a single exponential, increases only slightly with Dst concentration (Fig. 7 in supplementary data). This observation is a common feature of drug-DNA reaction kinetics. For example, for the proflavine–DNA reaction the slow reaction phase was proposed to correspond to conversion from an initial nonintercalated to a final intercalated complex (26), and the slow phase in the reaction of echinomycin with DNA was assigned to dissociation from transiently occupied, suboptimal sites (27).
One general model to explain our observations invokes sequential binding of two molecules of Dst to the target site, followed by their rearrangement in the minor groove to give the optimal 2:1 complex. The rearrangement could be accompanied by the large negative fluorescence signal observed in the second phase if the fluorescence because of two Dst molecules binding in a suboptimal way were greater than that of the final 2:1 complex. Such “sliding” rearrangements have been observed in NMR studies of 2:1 complexes by Wemmer and coworkers (10, 28). Another possible model proposes that much of the drug binds initially in the 1:1 mode, and the slow phase corresponds to dissociation of the 1:1 complex in favor of rebinding at another 1:1 complex to form the final 2:1 state. This model is supported by the similarity between the activation energies for the second reaction phase and the dissociation reaction (Table 1).
To gain further insight into the process, we carried out double mixing experiments, in which dissociation of the transient complexes is observed at different aging times after mixing Dst and the 2:1 site. The observed dissociation rates were the same over a range of mixing times from 3 ms to 1 s (Fig. 8 in supplementary data; see below). Given the much more rapid dissociation expected for the 1:1 complex, this result is not consistent with equating the slow phase with conversion of 1:1 to 2:1 complexes. Whatever the nature of the “annealing” process, the transient intermediates do not differ strongly in dissociation rate.
Dissociation of Dst from the 2:1 Site Displays Biphasic Behavior.
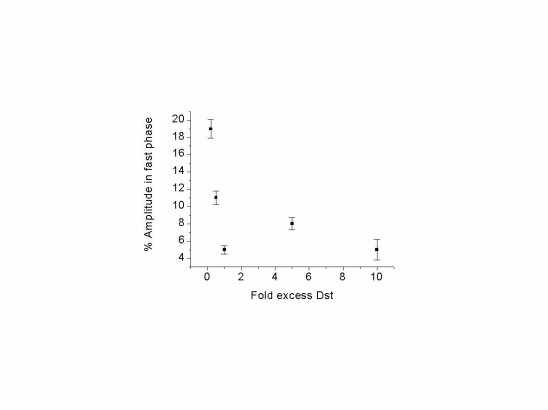
The dissociation reaction from 2:1 sites is markedly slower than that from 1:1 sites (Fig. 5B), confirming the observation by Sha and Chen (4) using absorbance-detected stopped-flow measurements. Also, a fast phase is observed in the dissociation reaction at early time points. This signal represents only a small fraction of the total amplitude observed in the dissociation reaction and probably reflects dissociation of drug molecules bound at the target site in the 1:1 mode because of incomplete saturation of the 2:1 mode. Consistent with this model is the observed decrease in relative amplitude of the first (fast) phase with increasing excess of drug over DNA (Fig. 9 in supplementary data). The variation of dissociation rate with temperature under large excess of drug can be used to estimate the activation enthalpy and entropy of dissociation from the 2:1 mode (Table 1).
Conclusions
We have used fluorescence-detected stopped-flow studies to measure the rates of association and dissociation of Dst from sites representing its two distinctive binding motifs. We show the existence of slower phases in the association reactions at both the 1:1 and 2:1 sites. Dissociation of “incorrect” complexes, possibly involving nonreactive isomers of the drug, is likely to play a role in the observed phenomena. In more complex mixtures, in which stronger competing sites are available, the apparent association rates will depend on the dissociation rates from these mismatched sites. Covalently linked polyamide dimers (29) should have simpler reaction pathways than unlinked dimers such as Dst in its 2:1 site, because of the many possible ways that unlinked dimers can form 1:1 complexes. Therefore, we predict that covalently linked dimers will show faster association rates in comparison with unlinked monomers, contributing to their enhanced binding affinity.
A large body of recent work in this area has demonstrated the enormous potential of polyamide drugs for extremely tight binding (30) to specified sites of arbitrary sequence and for inhibition of transcription by binding to regulatory elements (31). Whereas there is a sizable database of structural and thermodynamic data on these complexes, little was understood about their binding kinetics. Our studies with Dst demonstrate the utility of fluorescence-based stopped-flow studies for measuring the kinetic parameters of all these compounds and could prove useful for guiding future design of polyamide drugs.
Supplementary Material
Acknowledgments
We thank Professors Peter Dervan and Michael Waring for their comments, Professors Axel Brunger, Allana Schepartz, Lila Gierasch, and Nicholas Geacintov for allowing use of their instruments, and the National Institutes of Health for funding this work (GM21966).
Abbreviation
- Dst
distamycin A
References
- 1.Coll M, Frederick C A, Wang A H J, Rich A. Proc Natl Acad Sci USA. 1987;84:8385–8389. doi: 10.1073/pnas.84.23.8385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen X, Ramakrishnan B, Sundaralingam M. J Mol Biol. 1997;267:1157–1170. doi: 10.1006/jmbi.1997.0941. [DOI] [PubMed] [Google Scholar]
- 3.Pelton J G, Wemmer D E. Proc Natl Acad Sci USA. 1989;86:5723–5727. doi: 10.1073/pnas.86.15.5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen F M, Sha F. Biochemistry. 1998;37:11143–11151. doi: 10.1021/bi980950l. [DOI] [PubMed] [Google Scholar]
- 5.Smirnov I V, Poletaev A I. Biofizika. 1978;23:384–386. [PubMed] [Google Scholar]
- 6.Rentzeperis D, Marky L A, Dwyer T J, Geierstanger B H, Pelton J G, Wemmer D E. Biochemistry. 1995;34:2937–2945. doi: 10.1021/bi00009a025. [DOI] [PubMed] [Google Scholar]
- 7.Rentzeperis D, Marky L A, Kupke D W. J Phys Chem. 1992;96:9612–9613. [Google Scholar]
- 8.Chen X, Ramakrishnan B, Rao S T, Sundaralingam M. Nat Struct Biol. 1994;1:169–175. doi: 10.1038/nsb0394-169. [DOI] [PubMed] [Google Scholar]
- 9.Pelton J G, Wemmer D E. J Am Chem Soc. 1990;112:1393–1399. [Google Scholar]
- 10.Pelton J G, Wemmer D E. J Biomol Struct Dyn. 1990;8:81–97. doi: 10.1080/07391102.1990.10507791. [DOI] [PubMed] [Google Scholar]
- 11.Patel N, Berglund H, Nilsson L, Rigler R, McLaughlin L W, Graslund A. Eur J Biochem. 1992;203:361–366. doi: 10.1111/j.1432-1033.1992.tb16558.x. [DOI] [PubMed] [Google Scholar]
- 12.Barawkar D A, Ganesh K N. Nucleic Acids Res. 1995;23:159–164. doi: 10.1093/nar/23.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bostock-Smith C E, Searle M S. Nucleic Acids Res. 1999;27:1619–1624. doi: 10.1093/nar/27.7.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wiederholt K, Rajur S B, Giuliano J, Odonnell M J, McLaughlin L W. J Am Chem Soc. 1996;118:7055–7062. [Google Scholar]
- 15.Stockert J C, Delcastillo P, Bella J L. Histochemistry. 1990;94:45–47. doi: 10.1007/BF00266788. [DOI] [PubMed] [Google Scholar]
- 16.Dervan P B, Burli R W. Curr Opin Chem Biol. 1999;3:688–693. doi: 10.1016/s1367-5931(99)00027-7. [DOI] [PubMed] [Google Scholar]
- 17.Fagan P, Wemmer D E. J Am Chem Soc. 1992;114:1080–1081. [Google Scholar]
- 18.Zajac M, Nogowska M, Jatczak D, Rewekant R. Pharmazie. 1993;48:531–534. [Google Scholar]
- 19.Miyoshi N, Noriki S, Ozaki M, Kitatani M, Chiyo Y, Fukuda M. Jpn J Hum Genet. 1987;32:199–200. [Google Scholar]
- 20.Browne K A, He G X, Bruice T C. J Am Chem Soc. 1993;115:7072–7079. [Google Scholar]
- 21.Bourdouxhe C, Colson P, Houssier C, Sun J S, Montenaygarestier T, Helene C, Rivalle C, Bisagni E, Waring M J, Henichart J P, Bailly C. Biochemistry. 1992;31:12385–12396. doi: 10.1021/bi00164a013. [DOI] [PubMed] [Google Scholar]
- 22.Jia Y P, Kumar A, Patel S S. J Biol Chem. 1996;271:30451–30458. doi: 10.1074/jbc.271.48.30451. [DOI] [PubMed] [Google Scholar]
- 23.Berman H M, Neidle S, Zimmer C, Thrum H. Biochim Biophys Acta. 1979;561:124–131. doi: 10.1016/0005-2787(79)90496-9. [DOI] [PubMed] [Google Scholar]
- 24.Gurskaia G V, Grokhovski S L, Zhuze A L, Gottikh B P. Dokl Akad Nauk SSSR. 1978;243:645–648. [Google Scholar]
- 25.Muller W, Crothers D. J Mol Biol. 1968;35:251–290. doi: 10.1016/s0022-2836(68)80024-5. [DOI] [PubMed] [Google Scholar]
- 26.Li H J, Crothers D M. J Mol Biol. 1969;39:461–477. doi: 10.1016/0022-2836(69)90138-7. [DOI] [PubMed] [Google Scholar]
- 27.Fox K R, Wakelin L P G, Waring M J. Biochemistry. 1981;20:5768–5779. doi: 10.1021/bi00523a020. [DOI] [PubMed] [Google Scholar]
- 28.Geierstanger B H, Jacobsen J P, Mrksich M, Dervan P B, Wemmer D E. Biochemistry. 1994;33:3055–3062. doi: 10.1021/bi00176a039. [DOI] [PubMed] [Google Scholar]
- 29.Mrksich M, Parks M E, Dervan P B. J Am Chem Soc. 1994;116:7983–7988. [Google Scholar]
- 30.Trauger J W, Baird E E, Dervan P B. Nature (London) 1996;382:559–561. doi: 10.1038/382559a0. [DOI] [PubMed] [Google Scholar]
- 31.Dickinson L A, Gulizia R J, Trauger J W, Baird E E, Mosier D E, Gottesfeld J M, Dervan P B. Proc Natl Acad Sci USA. 1998;95:12890–12895. doi: 10.1073/pnas.95.22.12890. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}