

Abstract
Traditional approaches to protein profiling were built around the concept of investigating one protein at a time and have long since reached their limits of throughput. Here we present a completely new approach for comprehensive compositional analysis of complex protein mixtures, capable of overcoming the deficiencies of current proteomics techniques. The Combinatorial methodology utilises the peptidomics approach, in which protein samples are proteolytically digested using one or a combination of proteases prior to any assay being carried out. The second fundamental principle is the combinatorial depletion of the crude protein digest (i.e. of the peptide pool) by chemical crosslinking through amino acid side chains. Our approach relies on the chemical reactivities of the amino acids and therefore the amino acid content of the peptides (i.e. their information content) rather than their physical properties. Combinatorial peptidomics does not use affinity reagents and relies on neither chromatography nor electrophoretic separation techniques. It is the first generic methodology applicable to protein expression profiling, that is independent of the physical properties of proteins and does not require any prior knowledge of the proteins. Alternatively, a specific combinatorial strategy may be designed to analyse a particular known protein on the basis of that protein sequence alone or, in the absence of reliable protein sequence, even the predicted amino acid translation of an EST sequence. Combinatorial peptidomics is especially suitable for use with high throughput micro- and nano-fluidic platforms capable of running multiple depletion reactions in a single disposable chip.
Keywords: peptidomics, combinatorial peptidomics, proteomics, biotechnology, mass spectrometry, proteins, peptides
Background
Gerardus Mulder, a Dutch chemist who was the first to purify proteins in the middle of the 19th century, defined them to be "without doubt the most important of all substances of the organic kingdom, and without it life on our planet would probably not exist". However, despite more than one and a half centuries of scientific effort, proteins are routinely being studied using traditional technologies, which have long since reached their limits of throughput. Techniques such as 2D gel electrophoresis, chromatography or a combination of the two are now widely available, but have a number of disadvantages in that they do not allow a highly parallel approach due to their physical limitations, large sample consumption and high costs. Protein staining on gels is biased towards highly abundant proteins and yields only qualitative information. In addition, in all proteomics applications based on electrophoretic or chromatographic separation of complex protein mixtures, the purity of final preparations is inversely proportional to the quantity of the materials obtained. This means that larger amounts of highly complex protein mixtures and more purification steps (or separation dimensions) are required in order to yield enough material of sufficient purity for subsequent mass spectrometry (MS) or other applications. Most chromatography based techniques suffer from poor reproducibility. An alternative approach using isotope-coated affinity tags (ICAT) has been developed to allow relative quantitation of proteins by MS [1]. ICAT utilises isotope coding to quantitate differential protein expression, but the peptide pools obtained are too complex for convenient resolution by MS. More recently another completely different technology has been applied for proteomics research. This technology employs arrays of affinity ligands (antibodies or other agents) immobilised on a variety of solid supports [2,3]. Using arrayed affinity ligands avoids the need for protein separation, as all of the spotted reagents are spatially separated and their positions known. The use of fluorescently labelled protein mixtures further simplifies protein detection. Additional increases in protein array sensitivity and signal-to-noise ratio were reported using time resolved fluorescence [4] and planar waveguides as protein immobilisation substrates [5,6]. However, unlike DNA chips, protein chip based proteomics faces significant difficulties due to the much more heterogeneous character of proteins compared to nucleic acids. A significant improvement in protein microarray technology has been achieved through the use of competitive displacement strategies [7]. However, a whole cell protein repertoire is extremely complex and different proteins may require different solubilisation and separation techniques. The less than reproducible character of the protein sample preparation is capable of compromising any protein assay which follows. Current state-of-the-art in protein biochemistry has not yielded universal solubilisation and affinity assay conditions applicable to all cellular proteins, e.g., small and large, hydrophobic and hydrophilic, soluble and membrane associated, basic and acidic proteins. Protein heterogeneity significantly limits the applicability of affinity-based systems to small subsets of proteins having very similar physical characteristics.
Peptidomics
We have previously shown that the composition of a protein mixture can be determined by directly assaying the peptides from crude tryptic or otherwise digested protein preparations using immobilised antibodies. The Peptidomics approach [8] resolves many of the problems associated with multiplex affinity assays (e.g. arrays of antibodies) by allowing single optimised reaction conditions to be used irrespective of the starting material (small soluble proteins or large transmembrane receptors). This has been achieved through proteolytic digestion of the protein sample, for example with Trypsin. Since protein samples are to be digested, protein solubilisation is less of an issue and may be omitted altogether. Peptidomics enables the high throughput screening of proteins in a microarray format and has several advantages over affinity capture of intact proteins. These include, the homogeneity of digested proteins (typically in the form of tryptic peptides), which results in a more uniform pool of target species, allowing more regular quantitation. As peptides are much more stable and robust than proteins, protein degradation is not an issue. Also, antibody reagents can be more easily generated, such as by chemical synthesis of in silico predicted peptides against which antibodies are raised, e.g. by phage display techniques [9-12] or using in-vitro evolution [13-15] and DNA-protein fusions [16-18]. Such affinity reagents can be obtained in a truly high throughput manner and their specificities and affinities can be more easily controlled. In Peptidomics, each protein is broken down into many smaller components, resulting in the availability of a range of peptides thus allowing multiple independent assays for the same "original" protein to be performed using most antigenic species. Peptides are also particularly suited to detection by mass spectrometric techniques, such as MALDI-TOF-MS for direct analysis of samples on a solid substrate such as microarrays. The peptide mass range is such that isotopic resolution is easily achieved and hence their masses can be accurately determined, allowing for mass matching database searches to be performed to confirm the specificity of the affinity capture.
Digestion of cellular fractions or even intact tissues results in the release of peptides, which will be mostly hydrophilic, thus further improving the assay. In contrast to a traditional affinity assay, Peptidomics allows multiple antibody-peptide pairs to be used to assay the same protein target (similarly to Affymetrix DNA oligonucleotide arrays, where up to 20 oligonucleotides may be generated against the same mRNA sequence http://www.affymetrix.com), thus increasing the reliability of the assay. One of the major drawbacks of any affinity assay-based technique, including Peptidomics, is the availability and the cost of capture agents. Unlike nucleic acids, which are both information carriers and perfect affinity ligands, every protein requires the production of its own unique affinity reagent (e.g. an antibody) the development of which, unlike the synthesis of an oligonucleotide or purification of a PCR product, may require significant amounts of time and resources.
Principles behind combinatorial peptidomics
We have substituted the affinity purification step of the peptidomics approach with quantitative depletion of the peptide pools through chemical crosslinking of a subset of peptides (through their amino acid side chains) to a solid support. Chemical depletion reduces the complexity of a peptide pool to a required degree to make it compatible with direct MS detection. Because only those peptides that do not contain an amino acid recognized by the amino acid filter(s) remain in the mixture, the depleted peptide pools contain peptides of reduced amino acid compositional complexity. This further facilitates the analysis of mass spectra produced by MALDI-TOF mass spectrometry and permits a greater number of peptide peaks to be identified from a mass spectrum. Unmodified peptides as well as proteins generally contain up to 8 reactive groups. These include six amino acid specific groups: Sulfhydryl groups of Cysteines, Thioether groups of Methionines, Imidazolyl groups of Histidines, Guanidinyl groups of Arginines, Phenolic groups of Tyrosines and Indolyl groups of Tryptophans (Figure 1). Any of these groups or combinations thereof can be used to covalently immobilise respective amino acids (and peptides which contain them) in a specific and fully predictable manner with respect to amino acid content. The remaining two reactive groups – amino groups (H2N-) and carboxyl groups (HOOC-) are present on every peptide as N- and C-terminal groups or epsilon amino groups of Lysines or as parts or Aspartic acid and Glutamic acid side chains. These groups may be used for amino acid sequence- and content-independent manipulations, for instance through chemical, radioactive, fluorescent or isotopic labelling. Chemically reactive surfaces (derivatised beads, for example) which covalently bind amino acids in a side chain specific manner are referred to as amino acid filters. Any combination of amino acid filters of various specificities or reactivities is possible. The amino acid filtering (depletion) step may be repeated using combinations of up to 6 filters (equivalent to a six-dimensional separation, see Figure 2) or until the complexity of the peptide pool and the amino acid complexity of the remaining peptides is decreased to the desired level (i.e. suitable for direct MS detection). A quantitative chemical depletion principle can only be applied to peptides since proteins are mostly globular folded molecules, having a large fraction of the chemically reactive amino acid residues buried deep in the protein globule, thus preventing quantitative interactions. Combinatorial peptidomics relies on neither affinity reagents of any kind (whether antibodies, antibody-mimics, their fragments etc.) nor other chromatographic or electrophoretic separation techniques. Combinatorial peptidomics is the first generic methodology applicable to protein expression profiling, which is independent of the physical properties of proteins and does not require any prior knowledge of the proteins. Alternatively, a specific strategy based on combinatorial depletion, may be calculated and predicted for analysis of a known protein on the basis of that protein sequence alone or, in the absence of a reliable protein sequence, even the predicted amino acid translation of an EST sequence.
Figure 1.
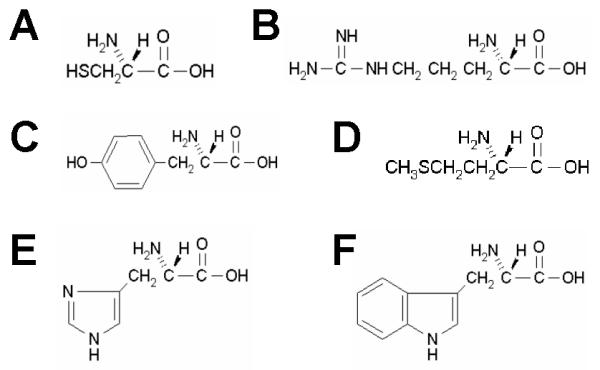
Six amino acids which contain chemically reactive side chains: Sulfhydryl groups in Cysteines (A), Guanidinyl groups in Arginines (B), Phenolic groups in Tyrosines (C), Thioether groups in Methionines (D), Imidazolyl groups in Histidines (E) and Indolyl groups of Tryptophans (F).
Figure 2.
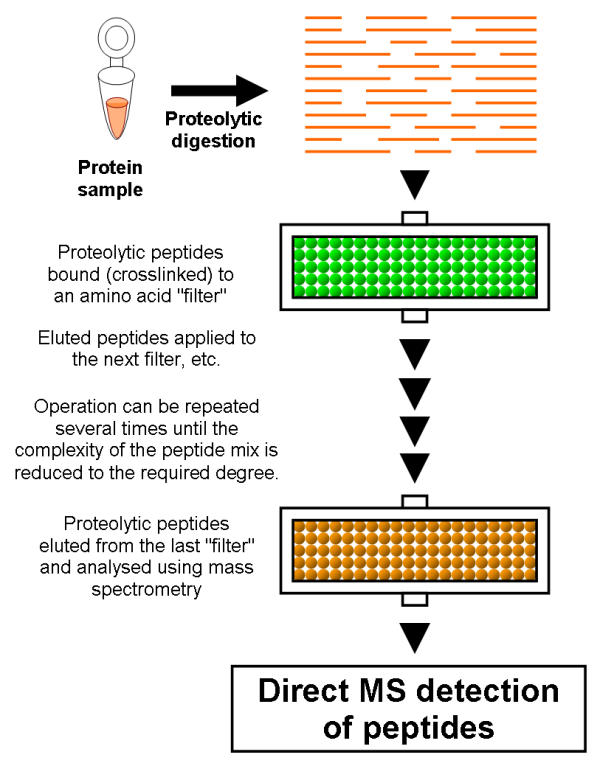
Principles behind combinatorial peptidomics. Sample is proteolytically digested, but the affinity purification step of the peptidomics approach [8] is substituted by quantitative depletion of the peptide pools through chemical crosslinking of a subset of peptides (through their amino acid side chains) to a solid support (e.g. derivatised beads, derivatised capillaries, etc). Sulfhydryl groups of Cysteines, Thioether groups of Methionines, Imidazolyl groups of Histidines, Guanidinyl groups of Arginines, Phenolic groups of Tyrosines and Indolyl groups of Tryptophans can be used to covalently immobilise respective amino acids (and peptides which contain them) in a specific and fully predictable manner with respect to amino acid content. Any combination of such amino acid "filters" of various specificities or reactivities is possible (can be used sequentially or as a single "filter" with mixed specificity). Chemical depletion reduces the complexity of a peptide pool to a required degree to make it compatible with direct MS detection.
Combinatorial approach
The Combinatorial approach includes two key stages, which are described here in more detail. The first stage is protein digestion. This can be achieved using a variety of proteases or alternatively, chemical cleavage could be used. Table 1 lists some of the most frequently used protein cleavage reagents. The second stage includes one or more depletion steps. A number of amino acid side-chain specific chemistries are available for use at this stage. Table 2 gives a few examples of the suitable amino acid side chain-specific reagents. Some individual applications of amino acid side chain specific chemistries can be found in the literature. For example, the use of acetylimidazole as Tyr- selective reagent [19-21], mercurial reagents or N-ethylmaleimide as Cys- selective reagents [22-25], diketones and phenylglyoxal as Arg- selective reagents [26,27], diethylpyrocarbonate as a selective His-specific compound [28]. Specific reaction of iodoacetate with methionine was described in [29] and bromoacetyl compounds for selective immobilisation of met-containing proteins have been used by The Nest Group, Inc. (USA) in their commercially available "Pi3-Met" reagent http://www.nestgrp.com. Specific chemical crosslinking of the tryptophan residues has been previously achieved using 2-hydroxy-5-nitrobenzyl bromide [30].
Table 1.
Frequently used protein cleavage reagents
| Enzymes | Preferred cleavage site | Approximate frequency |
| Ancrod | Arg-X, Arg-Gly | |
| Bromelain | C-terminal to Lys, Ala and Tyr | |
| Chymotrypsin | C-terminal to hydrophobic residues, e.g., Phe, Tyr, Trp. Less sensitive with Leu, Met, Ala | |
| Clostripain | C-terminal to Arg residues | 20 |
| Collagenase | N-terminal to Gly (X-Gly) in Pro-X-Gly-Pro | |
| Elastase | C-terminal to amino acids with small hydrophobic side chains | |
| Endoproteinase Arg-C | C-terminal to Arg residues | 20 |
| Endoproteinase Asp-N | N-terminal to Asp and Cys | 10 |
| Endoproteinase Glu-C | C-terminal to Asp and Glu | 10 |
| Endoproteinase Lys-C | C-terminal to Lys | 20 |
| Factor Xa | C-terminal to Arg in Gly-Arg-X | |
| Ficin | uncharged or aromatic amino acids | |
| Follipsin | Arg-X | 20 |
| Kallikrein | C-terminal to Arg in (Phe-Arg-X or Leu-Arg-X) | |
| Pepsin | Broad specificity; preference for cleavage C-terminal to Phe, Leu, and Glu | 7 |
| Thermolysin | N-terminal to amino acids with bulky hydrophobic side chains, e.g., Ile, Leu, Val, and Phe 5 | |
| Thrombin | C-terminal to Arg | |
| Trypsin | C-terminal to Lys and Arg | 10 |
| V8 protease | C-terminal to Glu, less active with Asp | |
| Chemistry | Preferred cleavage site | |
| Cyanogen bromide | Trp, (Met) | |
| Formic acid | Asp – Pro | |
| HCl | Asp-X, X-Asp | |
| Hydroxylamine (alkaline pH) | Asn – Gly | |
| N-bromosuccinimide (NBS) or N- chlorosuccinimide | Trp | |
| 2-Nitro-5-thiocyanobenzoate (NTCB) | Cys | |
Table 2.
Examples of amino acid side-chain specific chemistries
| Reagents | Group specificity | Crossreactivity |
| α Haloacetyl compounds: Iodoacetate; α haloacetamides; bromotrifluoroacetone; N chloroacetyliodotyramine | Cys, His, Met, Tyr | NH2 groups (slow at low pH) |
| N Maleimide derivatives: N ethylmaleimide (at pH < = 7) | Cys | NH2 groups (slow at low pH) |
| Mercurial compounds (most specific): p chloromercuribenzoate(PCMB)/p hydroxymercuribenzoate(PHMB) in H2O (optimum at pH 5, competitive displacement possible) | Cys | |
| Disulphide reagents (reversible): 5,5 dithiobis (2 nitrobenzoic acid) (DTNB); 4,4 dithiodipyridine; methyl 3 nitro 2 pyridyl disulphide; methyl 2 pyridyl disulphide | Cys | |
| N acetylimidazole | Tyr | NH2 groups (slow) |
| Diazonium compounds (optimum at pH9, unstable) | Tyr, His | NH2, Trp, Cys and Arg (slow) |
| Dicarbonyl compounds (pH > = 7): glyoxal; phenylglyoxal; 2,3 butanedione; 1,2 cyclohexanedione | Arg | Lys at pH < = 7 |
| p toluenesulphonylphenyl alaninechloromethyl ketone (TPCK); p toluenesulphonyllysine chloromethyl ketone (TLCK); Methyl-p-nitrobenzenesulphonate | His | Cys |
| Diethylpyrocarbonate (reversible at pH > = 7) | His (at pH4) | NH2 |
| 2 hydroxy 5 nitrobenzyl bromide (HNBB) | Trp | |
| p nitrophenylsulphenyl chloride | Trp, Cys | |
| α Haloacetyl compounds | Met at pH3; also Cys, His, Tyr | NH2 groups (slow at low pH) |
The choice of proteases, crosslinking chemistries and of their combinations is important and is determined by the degree of depletion required and the frequency of the amino acids being targeted. The frequency (F) with which any amino acid occurs in proteins varies, but could approximately be taken as 1/20 to illustrate the principle:
F = 1/20
Number of chances (C) to find any particular amino acid "1" in the peptide containing n amino acids will therefore be approximately equal:
C1 = n × F1 = n/20 (approx)
If our assumption (F = 1/20) is correct, each 20 amino acid long peptide has on average one chance of being covalently linked to any single "filter". If two filters are used (in parallel or consecutively), then the number of chances (C) to precipitate a peptide containing n amino acids, i.e. to find any two amino acids "1" and "2" in such peptide will be equal approximately:
C1+2 = n × F1 + n × F2 = n/10 (approx)
For any 3 amino acid filtering steps:
C1+2+3 = n × F1 + n × F2 + n × F 3 = n/7 (approx)
Any 4 amino acids:
C1+2+3+4 = n × F1 + n × F2 + n × F3 + n × F4 = n/5 (approx)
Any 5 amino acids:
C1+2+3+4+5 = n × F1 + n × F2 + n × F3 + n × F4 + n × F5 = n/4 (approx)
Any 6 amino acids:
C1+2+3+4+5+6 = n × F1 + n × F2 + n × F3 + n × F4 + n × F5 + n × F6 = n/3.5 (approx)
To deplete a complex peptide mixture by amino acid-specific sorption a different number of filters may be needed, depending on the range of peptide lengths, which depends on the cleavage technique. Using our assumption that F = 1/20, a 3 – 4 amino acid long peptide may on average be crosslinked once (i.e. has on average one chance to be removed from the sample) through one of its amino acid side chains using all six filters. The degree of the depletion also depends on the average peptide length (see Table 1). The degree of depletion needs to be adjusted such as to yield a sufficiently depleted peptide pool suitable for direct analysis by a mass spectrometer, i.e. having ~1000 peptides in the sample. Generally speaking, the shorter the range of peptide fragment lengths, the greater the number of filters required for the same degree of depletion. The origin of the protein sample (whether whole cellular proteome or partially purified narrow subfraction, containing only few proteins) is another key factor determining the required degree of peptide depletion.
Results
Peptide depletion using Methionine-reactive amino acid filter
To illustrate the depletion principle we have used a mixture of synthetic peptides (See Table 3) and Methionine-reactive beads. Five of the ten peptides used contained Methionine in different positions along their sequences. The MALDI MS spectrum of the original peptide mixture (incubated with no beads) is shown on Figure 3A. The ten peaks corresponding to the peptides present in the mixture are indicated. The same peptide mixture was incubated with Methionine-reactive beads for depletion of all Met-containing peptides by their irreversible cross-linking to beads.
Table 3.
Synthetic peptides used in this study
| Peptide sequence* | Methionine present | m/z | Seq. ID |
| RPPQTLSR | no | 1293.56 | 1 |
| NLSPDGQYVPR | no | 1584.83 | 6 |
| SANAEDAQEFSDVER | no | 2007.13 | 9 |
| NFHQYSVEGGK | no | 1604.82 | 11 |
| LERPVR | no | 1108.38 | 16 |
| VFAQNEEIQEMAQNK | yes | 2118.43 | 4 |
| DLPLLIENMK | yes | 1524.92 | 8 |
| ETYGEMADCCAK | yes | 1659.95 | 19 |
| FIMLNLMHETTDK | yes | 1932.37 | 5 |
| DLVTQQLPHLMPSNCGLEEK | yes | 2592.07 | 13 |
* All peptides were biotinylated at their N termini.
Figure 3.
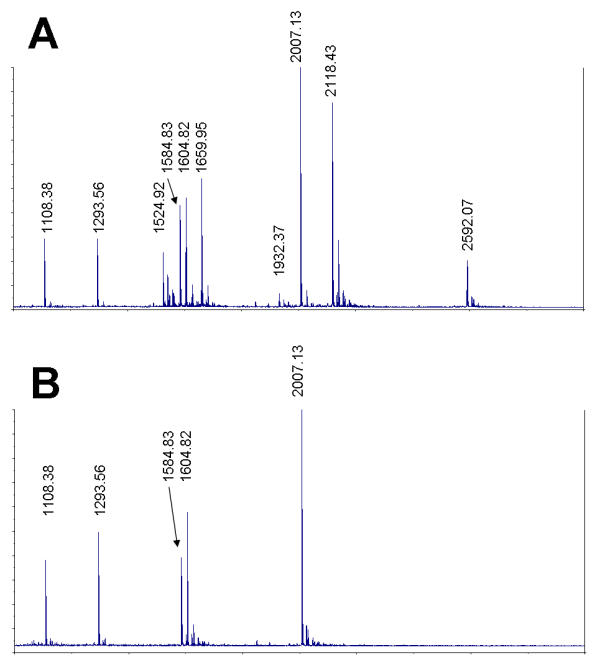
Depletion of peptides using Methionine-reactive amino acid filter. A – Untreated mixture containing 10 synthetic peptides (see Table 3). B – the same mixture following a Met-reactive chemistry mediated depletion (see Methods).
In affinity based systems the equilibrium state always includes both free and bound analyte (i.e. peptide or protein), with their ratio being dependant on the dissociation constant KD. Unlike an affinity recognition event, the chemical reaction can be brought to completion more easily. Because of its quantitative character, the depletion by irreversible chemical cross-linking is preferred to any affinity-based separation because of the inherently incomplete character of the latter. Accordingly, the MS spectrum of the depleted mixture, shown on Figure 3B, reveals no Met-containing peptides. Thus the single depletion step has reduced the complexity of this model peptide mixture two fold.
Relative quantitation of depleted peptide mixtures
We have further investigated, using the same model peptides and the Met-reactive bead system, whether the depletion preserves quantitative differences between different peptides (i.e. their concentrations). Three separate samples were prepared from the original and the depleted peptide mixtures. Each such aliquot was treated and analysed in parallel using identical conditions and MS settings. Three separate spectra from each of the samples were obtained by accumulation of data from 400 laser shots. Peak areas were measured for each peak on each spectrum and average values were expressed as means. The results obtained for the five peptides (without Methionine) from the original and the depleted samples are shown on Figure 4. The error bars indicate standard deviations. The Figure 4 indicates that relative peptide abundance for each individual peptide within each sample remains very similar between separate MS measurements (with standard deviation mostly within 20% of relative peak values). Figure 4 also demonstrates that the chemical depletion step does not lead to major changes in relative peak values of the remaining peptides (compare open and filled bars on Figures 4A). This means that relative quantitation using mass spectrometry in conjunction with the combinatorial approach is achievable without a need for differential isotope labelling of peptides as in ICAT [1] and that the depletion approach described can be used for relative quantitation of protein expression levels and other proteomic measurements.
Figure 4.
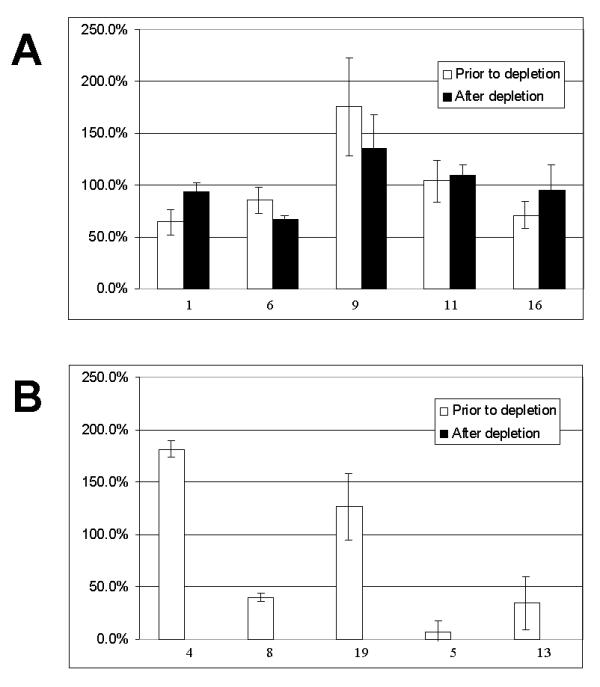
Relative quantitation of peptides. A – Five peptides containing no Methionines (see Table 3) prior to (open bars) and after the depletion using Met-reactive beads (filled bars). B – Five peptides containing Methionines (see Table 3) prior to (open bars) and after the depletion using Met-reactive beads. No Met-containing peptides were detected in the depleted samples. Bar heights (both panels) represent averaged peak values detected (+/- STDEV, n = 9). Peptides are identified by their Seq IDs below each bar (on both panels).
Peptide labelling
Two or more samples originating from different tissues/cells/etc can be subject to mass spectrometry at the same time. To do this one must be able to distinguish the peptide peaks which come from the different samples used. Isotope labelling has been used before [1] for this purpose. However, such labelling can also be achieved by tagging peptides (or peptide pools) using non-identical but similar chemical entities, having identical or closely matching chemical and physical properties, but different molecular masses. The Combinatorial approach allows such labelling to be done through amino groups, preferably alpha-amino groups, or through carboxyl groups, preferably alpha-carboxyl groups. The use of other reactive groups is also possible, but less preferable as other reactive side chains are more useful for combinatorial depletion, whilst amino- or carboxyl- groups are present on every peptide/protein and therefore represent the best target for sequence-independent labelling. Unlike isotopic labelling, which is severely limited to a very few suitable isotopes, there is a vast choice of commercially available materials for use as "chemical" labels. Mass differences may be introduced into peptide pools either by using the same amino- or carboxyl- group modifying chemistry (but with varying side chains on the reagent used), or using slightly different reagents whilst keeping their side chains the same, or varying both. Examples of such suitable amino-group reactive chemistries include: aryl halides, aldehydes, ketones, alpha-haloacetyl (used at pH > 7), N-maleimide (used at pH > 7) and derivatives, as well as acylating reagents. Examples of carboxy-group reactive chemistries include: diazoacetate esters, diazoacetamides and carbodiimides. Figure 5A shows the mass spectrum of the NFHQYSVEGGK peptide, used for differential labelling. The peptide was chemically labelled with either 4-fluorophenyl-isothiocyanate (introduced Δ m/z = 153, Figure 5B) or with 3,5-difluorophenyl-isocyanate (introduced Δ m/z = 155, Figure 5C). The fluorophenyl-isocyanates and fluorophenyl-isothiocyanates are just two of the numerous examples of acylating reagents with mass differences introduced through modifying the reagents or their own side chain modifications. The peptides could alternatively be labelled through their carboxyl groups using a large variety of reactive chemicals available. The Discussion section further exemplifies available alternatives.
Figure 5.
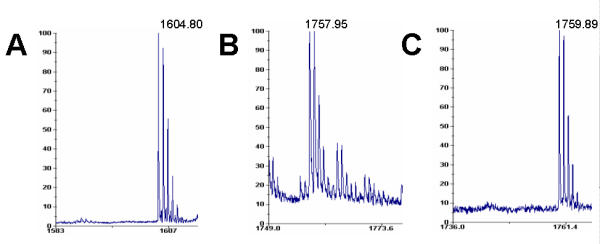
Differential mass labelling of the NFHQYSVEGGK peptide. A – mass spectrum of the unmodified peptide. B – mass spectrum of the same peptide modified with 4-fluorophenyl-isothiocyanate (introduced Δ m/z = 153). C – mass spectrum of the same peptide modified with 3,5-difluorophenyl-isocyanate (introduced Δ m/z = 155).
Discussion
Combinatorial approach
Characterization of the complement of expressed proteins from a single genome is a central focus of the evolving field of proteomics and can only be accomplished using a high-throughput, generic process. The number of expressed genes in a cell is estimated to be of the order of 10,000, resulting in up to 100,000 proteins, including splice forms and post-translational modifications. Any single protein could theoretically be identified by a single peptide using a TOF or TOF/TOF MS, meaning that an order of 100,000 non-identical "random" peptides may be required to cover a complete cellular proteome. A single mass spectrum is capable of resolving approximately 1000 different peptide peaks within the mass range of 500 to 3500 Da, corresponding to 5 to ~35 amino acid long peptides (significantly larger numbers of peptides cannot be resolved due to the resolution capabilities of TOF mass spectrometry). Therefore only 10–20 such non-degenerate spectra (from non-identically depleted samples) may be sufficient to reveal on average a single peptide from each cellular protein, not including isoforms. Statistically significant results, or detailed isoform analysis may require more different spectra, e.g. a 96-well plate worth of non-degenerate low-complexity samples. If splicing and PTMs are of no concern and for pre-fractionated proteomes, the number of representative spectra (whichever way arising) may be much lower. Recent developments in the field of the FT-MS capable of sub-ppm resolution (e.g. the APEX series of machines developed at Bruker-Daltonics, http://www.daltonics.bruker.com) may further reduce the number of representative spectra required, ultimately down to just few or one per proteome.
So far two main ideologies have been followed to decrease the complexity of samples submitted to MS. One relied on protein fractionation, followed by digestion and MS. In another approach, samples are digested first, followed by peptide fractionation. All fractionation techniques to date have utilised physical properties of proteins/peptides (i.e. size, charge, hydrophilicity/hydrophobicity, affinity interactions, etc.) resulting in poorly reproducible, not very quantitative techniques and expensive affinity reagents.
In the combinatorial approach presented here, the peptide mixture is depleted in a quantitative and reproducible manner by passage through one or more of the amino acid side chain specific "filters". The depleted peptide pools contain much fewer peptides, i.e. only those which do not contain the targeted amino acids (i.e. not crosslinked by the "filters") and can therefore be directly subjected to TOF MS and TOF/TOF MS analysis. These peptides are also of reduced amino acid complexity (e.g. would only contain 14 amino acids if all six filters had been used) thus facilitating peptide mass matching. The existence of the six reactive groups provides for the use of up to six independent amino acid covalent filters (with many more different chemistries available), thus bringing the total number of different filter combinations to 63. Table 4 lists possible combinations of such filters. Using all such filters simultaneously will result in a maximum possible depletion and should preferably be used for the most complex peptide mixtures, e.g., whole cell proteomes. Using individual amino acid "filters" or subsets of "filters" may be preferable for low-complexity protein mixtures, containing fewer individual proteins, e.g., simple micro-organism proteomes, or protein fractions resulting from pre-fractionating of more complex proteomes. The depletion degree may be varied either by changing the number of filters used or through the length of peptide fragments (longer fragments are more likely to be crosslinked due to higher probabilities of amino acid occurrences in long peptides). Nearly 20 different digestion specificities available (see Table 1) combined with up to 63 crosslinker combinations will result in hundreds of potential combinations, providing a sufficient number of non-identical depleted peptide pools of the low complexity to achieve the required total number of peptides. The combinatorial approach may be further streamlined by predicting the optimal combinations (digestion + crosslinker) if protein sequence information is available. The Combinatorial approach is inherently tolerant to experimental artefacts arising from the incomplete digestion (usually the enzymatic and therefore least predictable step of the technique, and a course of complications in other peptidomics applications). The products of incomplete digestion will be mostly eliminated from the samples, because for longer protein fragments, the chances of any single reactive amino acid being present in their sequences is much higher. This feature further reduces the complexity of the Mass Spectra and improves signal/noise ratio.
Table 4.
Amino acid side chain specific "filters"
| Average Peptide length in mixture | Number of amino acid filters required for near complete depletion | Maximum number of possible combinations of amino acid filters |
| 20 | 1 | 6 (i.e. either"1" or "2" etc., etc.) |
| 10 | 2 | 15 (i.e. either "1+2" or "1+3" etc.) |
| 7 | 3 | 20 (i.e. either "1+2+3" or "1+2+4" etc.) |
| 5 | 4 | 15 (i.e. "1+2+3+4" or "1+2+3+5" etc.) |
| 4 | 5 | 6 (i.e. "all but 1" or "all but 2" etc.) |
| 3 | 6 | 1 (i.e. "1+2+3+4+5+6") |
Depletion versus enrichment
The combinatorial peptidomics approach described so far has relied on depletion as the means to reduce complexity of the peptide mixture. As opposite to depletion, the combinatorial approach can also be carried out using enrichment selection techniques (i.e. by retaining peptides containing the required amino acids, i.e. Cysteines, Methionines, Histidines, Arginines, Tyrosines and/or Tryptophans, individually or in combinations). In such an embodiment, the peptide mixture is passed through the selected amino acid filter which crosslinks or binds peptides containing the respective amino acid. The filter assembly is then washed to remove unbound peptides and the chemical crosslink is cleaved in order to free the bound peptides. The protocol can be repeated using one or more of the other amino acid filters. Both depletion and enrichment reduce the complexity of the sample and will result in complementary non-degenerate peptide pools. However these approaches have a few characteristic differences (summarised in Table 5). Most significantly, the depletion approach allows for a more straightforward and faster route, suitable for peptides of 10 amino acids and longer, while the slower enrichment approach provides a better alternative for shorter peptides (e.g. 10 amino acids or less). Depletion is advantageous over enrichment in that it also decreases the compositional complexity of the remaining peptide pool, thus further facilitating analysis, such as the analysis of the mass spectra. Another important difference includes the resistance of the depletion approach towards incompletely digested peptides. Since these will generally be of increased lengths, the products of incomplete digestion (a major course of complications in other peptidomics applications) will be mostly eliminated from the depleted samples (due to longer peptides having higher chance of being crosslinked).
Table 5.
Comparison of the depletion and enrichment approaches to combinatorial peptidomics
| Depletion approach | Enrichment approach | |
| 1. Individual steps per each "filtering" stage | 1 step process, i.e. bind to beads (beads discarded) | 3 step process, i.e. bind to beads, wash and elute |
| 2. Number of combinations using 6 amino acid filters | 63 | 63 |
| 3. Range of suitable peptide lengths | Longer peptides require less "filtering" stages (i.e. 10 or more amino acid residues preferred) | Shorter peptides require less "filtering" stages (i.e. 10 or less amino acid residues preferred) |
| 4. Complexity of peptide mixtures | Decreased | Decreased |
| 5. Amino acid compositional complexity of the remaining peptides | Decreased (by the number of "filters" used) | Not changed (20 amino acids) |
| 6. Quantitative analysis | A single-stage depletion is more straightforward and quantitative than a triple-stage enrichment | Enrichment approach is less straightforward and robust than the depletion |
| 7. Scaling up | Possible (larger "filters" or consecutive stages) | Possible (larger "filters" or parallel reactions) |
| 8. Scaling down | Possible (low fmol level MS sensitivity requires high pmol filter binding capacities) | Especially suitable : low fmol level MS sensitivity requires fmol binding capacities |
| 9A. Limitations (overloading) | Large binding capacity of the "filters" is crucial – overloading will allow all peptides to pass the "filter" | Overloading of the "filters" is not an issue, excess of sample may be applied |
| 9A. Limitations (incompletely digestion) | Products of incomplete digestion will be mostly eliminated | Products of incomplete digestion will be mostly retained and may interfere with the downstream purification and analysis steps |
| 10. Nano-applications | Problematic due to limitation (see above) – excess of binding sites required to maintain efficient separation. Suitable for micro-fluidic applications | Suitable for nano-applications, since smaller number of binding sites required (compared to depletion strategy) |
Labelling
Differential labelling may be very useful if absolute quantitation is required (as opposed to relative quantitation). This can be exemplified further as follows. The acylating reagents, such as described in Section 2.3 above, could, for example, be modified with either Fluorine, Chlorine, Bromine or Iodine (i.e. use bromine-isothiocyanate versus iodine-isothiocyanate). Alternatively these could be "mono-", "di-" or "tri-" etc. modifications (i.e. use fluorophenyl-isothiocyanate vs. difluorophenyl-isothiocyanate) or the amino-reactive chemistry could be modified itself (i.e. use isocyanate vs. isothiocyanate), or the isocyanates could be derivatised differently (i.e. isocyanate vs. phenyl-isocyanates etc). A combination of the above could lead to 100s of differential molecular tags (far exceeding the capabilities of isotope labelling) by using isocyanates or derivatives alone. The Δ m/z mass difference introduced could be made as low as 2 (1 is less preferable due to potential difficulties in resolving naturally occurring isotopes of the peptides), as in the case with 4-fluorophenyl-isothiocyanate (Δ m/z = 153) and 3,5-difluorophenyl-isocyanate (Δ m/z = 155), and there is practically no limit on the upper range of the mass differences. However, larger mass differences will complicate the resolution of corresponding peptides in complex mixtures and are therefore less preferable. The use of chemical labelling allows much smaller Δ m/z to be used and the number of chemical labels is not limited by the number of available isotopes (as in ICAT method, known in the field). Also more than two complex peptide mixtures could be differentially labelled and analysed simultaneously unlike the ICAT method [1] or D0-/D3-per-methyl esterification approach [31] or acrylamide/deuterated acrylamide labelling method [32] (two samples only in each method). In addition to the ability to easily choose the desired Δ m/z, the chemical modifications allow the mass range of the analysed peptides to be shifted upwards. So the low molecular weight peptides, otherwise subject to interference from MALDI matrix related species, could be shifted into high MW range (in addition to, or instead of being differentially labelled).
High throughput and nano-applications
Combinatorial peptidomics is suitable for use with a variety of platforms, including traditional systems (column-based co-IP), arrayed affinity reagents (antibody microarrays, e.g. on MALDI plates) and a variety of micro- and nano-fluidic applications. Except for a few obvious and easily accepted reasons for miniaturisation, such as increasing the throughput of an assay by packing more reaction chambers into the same volume, and decreasing the assays cost, miniaturisation is especially suitable if combinatorial peptidomics approaches are sought. Miniaturisation has the potential to increase the reaction kinetics due to much smaller reaction volumes (and therefore faster reagent diffusion times) and significantly increased surface to volume ratios. This is especially applicable to combinatorial peptidomics, since both depletion and enrichment involves immobilising amino acids and peptides onto the solid surfaces. Truly miniature applications do not require the use of porous resins and careful selection of pore sizes, since a high surface to volume ratio of a small capillary channel, which is also easier to control, may be sufficient.
The Enrichment strategy, as opposed to Depletion, would better suit nano-fluidic or other applications where the size of reaction chambers matters (e.g. chips, micro- or nano-fluidics devices, etc). Since no excess of binding sites is required and because of its stability towards sample overloading, the overall size of such a device could be made smaller if the enrichment approach is used. For example, if only a single enrichment step is used, and peptide capture is totally reversible, then as few as low femtomole amounts of crosslinking chemistries may be required to retain peptide amounts sufficient for MS detection. Overloading of such a "filter" should not lead to an increase in the retained and released peptides, since the binding capacity of such a "filter" is limited. Although depletion may be performed faster, the overall binding capacity of a single "depletion filter" may need to be order(s) of magnitude larger to secure the reaction kinetics and to secure stability against sample overloading.
Conclusions
Modern proteomics aims to systematically analyse proteins for their identity, quantity and function and therefore requires new more generic and truly multiplex approaches for protein research. The combinatorial peptidomics approaches presented in this manuscript rely on the quantitative depletion or enrichment of peptide pools by chemical crosslinking through amino acid side chains. Chemical depletion reduces both the complexity of a peptide pool and the amino acid compositional complexity to a degree required to allow direct MS analysis. The combinatorial approach utilises commonly used proteolytic digestion techniques and widely available chemistries, many of them being well documented. The combinatorial peptidomics approach allows the determining of the composition of a protein mixture by assaying peptides directly from crude tryptic digests without using antibodies or any other affinity selection and therefore enables protein identification on a proteome-wide scale. Using all possible combinations of enzymatic/chemical protein digestion methods plus all combinations of the adsorption chemistries may allow thousands of peptides to be identified. The method presented greatly advances the effort of identifying all cellular proteins in "one go". The simplicity and predictability of the combinatorial approach (i.e. on the basis of protein sequence alone) provides for optimization of strategies for quantitative analysis of known proteins by using calculated and predicted combinations of digestion and separation features. Combinatorial peptidomics is suitable for studying both novel protein targets and for routine diagnostic applications. Combinatorial peptidomics is the first generic proteomics technology, reliant on the information content of proteins and peptides for their separation and analysis. The Combinatorial peptidomics approach compares to existing affinity based separation systems just as digital signal processing compares to analogue systems – it forms the basis of the high throughput proteomics technologies of the future.
Materials and Methods
Peptide depletion using Methionine-reactive amino acid filter
All peptides were obtained from SIGMA-Genosys. The Met-reactive beads (obtained from The Nest Group, Southborough, MA, USA) were activated as follows: beads from one "Pi3" isolation pack (approx 10 ul dry settled volume) were washed ×5 times with ×400 ul Methanol, followed by ×3 washes with 10% Acetic Acid using a spin column. Following washing, the beads were resuspended in 400 ul 10% Acetic Acid and transferred to a 1.5 ml microcentrifuge tube. Beads were precipitated by centrifugation and the supernatant (Acetic Acid) was removed. Peptide samples were prepared as follows: 75 ul of a peptide mixture (Table 3), containing approximately 75 ug peptides in total, was mixed with 25 ul of glacial Acetic Acid. The peptide mixture was divided into two 50 ul aliquots. One aliquot was transferred to the microcentrifuge tubes with the activated Met-reactive beads, whilst another aliquot was incubated without beads. Samples were left at 22°C for 18 hours. Following the incubation tube with beads was spun for 1 min at 10,000 RPM in a microcentrifuge and supernatant was transferred to a fresh tube.
Mass spectrometric analysis was performed as described previously [7,8]. Briefly, 5 ul aliquots were taken from each sample. The volume was then made up to 10 ul in 0.1 % TFA and the overall amount of TFA adjusted to 0.1 %. Each sample was bound to a Zip Tip, washed in 0.1 % TFA and eluted in 1 ul of a solution containing alpha-cyano-4-hydroxycinnamic acid (~2.5 mg/ml in 3:2 methanol/0.1% TFA) and deposited directly onto a target for MALDI-TOF-MS. All spectra were acquired in the standard reflector mode of a Voyager DE STR (Applied Biosystems, Foster City, CA). Four hundred laser shots were fired and the resulting mass spectra were averaged to produce each final trace.
For the relative quantitation study each graph was obtained from 3 separate mass spectra taken from three separately prepared samples (9 spectra altogether) for peptide mixtures both prior to depletion and after depletion with Met-reactive beads. All spectra were obtained under identical MS settings and all other details were the same. Values for each individual peptide were calculated as individual peak areas taken relative to the average peak areas from the 5 peptides (not-containing Met) for each separate spectrum. The normalised values were then plotted as means for each peptide (see above, +/- STDEV, n = 9) on both plots.
Labelling
Figure 5A shows the mass spectrum of the NFHQYSVEGGK peptide, used for differential labelling. The labelling was done with either 4-fluorophenyl-isothiocyanate or 3,5-difluorophenyl-isocyanate. Labelling was carried out at 4°C. The peptide solution (5 ul) was added to 85 ul of H2O and the buffer concentration was adjusted to 20 mM Na-CO3 (pH9.5). Following that 10 ul of 10% solution of acylating reagents in DMSO was added to the peptide, mixed and incubated overnight. The peptide was chemically labelled with 4-fluorophenyl-isothiocyanate (introduced Δ m/z = 153), see Figure 5B and with 3,5-difluorophenyl-isocyanate (introduced Δ m/z = 155), see Figure 5C.
Contributor Information
Mikhail Soloviev, Email: Mikhail.Soloviev@ogs.co.uk.
Richard Barry, Email: Richard.Barry@ogs.co.uk.
Elaine Scrivener, Email: Elaine.Scrivener@ogs.co.uk.
Jonathan Terrett, Email: Jon.Terrett@ogs.co.uk.
References
- Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold RR. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- Barry R, Scrivener E, Soloviev M, Terrett J. Chip-Based Proteomics Technologies. Int Genomic Proteomic Technology. 2002. pp. 14–22.
- Soloviev M. EuroBiochips: spot the difference. Drug Discov Today. 2001;6:775–777. doi: 10.1016/S1359-6446(01)01919-5. [DOI] [PubMed] [Google Scholar]
- Luo LY, Diamandis EP. Preliminary examination of time-resolved fluorometry for protein array applications. Luminescence. 2000;15:409–413. doi: 10.1002/1522-7243(200011/12)15:6<409::AID-BIO628>3.3.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Weinberger SR, Morris TS, Pawlak M. Recent trends in protein biochip technology. Pharmacogenomics. 2000;1:395–416. doi: 10.1517/14622416.1.4.395. [DOI] [PubMed] [Google Scholar]
- Pawlak M, Grell E, Schick E, Anselmetti D, Ehrat M. Functional immobilization of biomembrane fragments on planar waveguides for the investigation of side-directed ligand binding by surface-confined fluorescence. Faraday Discuss. 1998;111:273–288. doi: 10.1039/a806704j. [DOI] [PubMed] [Google Scholar]
- Barry R, Diggle T, Terrett J, Soloviev M. Competitive Assay Formats for High-Throughput Affinity Arrays. J Biomol Screen. [DOI] [PubMed]
- Scrivener E, Barry R, Platt A, Calvert R, Masih G, Hextall P, Soloviev M, Terrett J. Peptidomics: A new approach to affinity protein microarrays. Proteomics. 2003;3:122–128. doi: 10.1002/pmic.200390020. [DOI] [PubMed] [Google Scholar]
- Chiswell DJ, McCafferty J. Phage antibodies: will new 'coliclonal' antibodies replace monoclonal antibodies? Trends Biotechnol. 1992;10:80–84. doi: 10.1016/0167-7799(92)90178-X. [DOI] [PubMed] [Google Scholar]
- Thompson J, Pope T, Tung JS, Chan C, Hollis G, Mark G, Johnson KS. Affinity maturation of a high-affinity human monoclonal antibody against the third hypervariable loop of human immunodeficiency virus: use of phage display to improve affinity and broaden strain reactivity. J Mol Biol. 1996;256:77–88. doi: 10.1006/jmbi.1996.0069. [DOI] [PubMed] [Google Scholar]
- Parsons HL, Earnshaw JC, Wilton J, Johnson KS, Schueler PA, Mahoney W, McCafferty J. Directing phage selections towards specific epitopes. Protein Eng. 1996;9:1043–1049. doi: 10.1093/protein/9.11.1043. [DOI] [PubMed] [Google Scholar]
- Vaughan TJ, Williams AJ, Pritchard K, Osbourn JK, Pope AR, Earnshaw JC, McCafferty J, Hodits RA, Wilton J, Johnson KS. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nat Biotechnol. 1996;14:309–314. doi: 10.1038/nbt0396-309. [DOI] [PubMed] [Google Scholar]
- He M, Taussig MJ. Antibody-ribosome-mRNA (ARM) complexes as efficient selection particles for in vitro display and evolution of antibody combining sites. Nucleic Acids Res. 1997;25:5132–5134. doi: 10.1093/nar/25.24.5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Menges M, Groves MA, Corps E, Liu H, Bruggemann M, Taussig MJ. Selection of a human anti-progesterone antibody fragment from a transgenic mouse library by ARM ribosome display. J Immunol Methods. 1999;231:105–117. doi: 10.1016/S0022-1759(99)00144-1. [DOI] [PubMed] [Google Scholar]
- Xu L, Aha P, Gu K, Kuimelis R, Kurz M, Lam T, Lim A, Liu H, Lohse P, Sun L, Weng S, Wagner R, Lipovsek D. Directed evolution of high-affinity antibody mimics using mRNA display. Chem Biol. 2002;9:933. doi: 10.1016/S1074-5521(02)00187-4. [DOI] [PubMed] [Google Scholar]
- Roberts RW, Szostak JW. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc Natl Acad Sci U S A. 1997;94:12297–12302. doi: 10.1073/pnas.94.23.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Barrick JE, Szostak JW, Roberts RW. Optimized synthesis of RNA-protein fusions for in vitro protein selection. Methods Enzymol. 2000;318:268–293. doi: 10.1016/s0076-6879(00)18058-9. [DOI] [PubMed] [Google Scholar]
- Fletcher G, Mason S, Terrett J, Soloviev M. Self-assembly of proteins and their nucleic acids. Journal of Nanobiotechnology. 2003;1:1. doi: 10.1186/1477-3155-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong KH, Nishida M, Inoue H, Takahashi K. Tyrosine-7 is an essential residue for the catalytic activity of human class PI glutathione S-transferase: chemical modification and site-directed mutagenesis studies. Biochem Biophys Res Commun. 1992;182:1122–1129. doi: 10.1016/0006-291x(92)91848-k. [DOI] [PubMed] [Google Scholar]
- Kiss L, Korodi I, Nanasi P. Study on the role of tyrosine side-chains at the active centre of emulsin beta-D-glucosidase. Biochim Biophys Acta. 1981;662:308–311. doi: 10.1016/0005-2744(81)90043-7. [DOI] [PubMed] [Google Scholar]
- Heller J, Horwitz J. Involvement of tyrosine and lysine residues of retinol-binding protein in the interaction between retinol and retinol-binding protein and between retinol-binding protein and prealbumin. Acetylation with N-acetylimidazole and alkaline titration. J Biol Chem. 1975;250:3019–3023. [PubMed] [Google Scholar]
- van Berkel WJ, Weijer WJ, Muller F, Jekel PA, Beintema JJ. Chemical modification of sulfhydryl groups in p-hydroxybenzoate hydroxylase from Pseudomonas fluorescens. Involvement in catalysis and assignment in the sequence. Eur J Biochem. 1984;145:245–256. doi: 10.1111/j.1432-1033.1984.tb08545.x. [DOI] [PubMed] [Google Scholar]
- Werner PK, Lieberman DM, Reithmeier RA. Accessibility of the N-ethylmaleimide-unreactive sulfhydryl of human erythrocyte Band 3. Biochim Biophys Acta. 1989;982:309–315. doi: 10.1016/0005-2736(89)90071-0. [DOI] [PubMed] [Google Scholar]
- Smyth DG, Blumenfeld OO, Konigsberg W. Reactions of N-ethylmaleimide with peptides and amino acids. Biochem J. 1964;91:589–595. doi: 10.1042/bj0910589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring H, Christen P. A diagonal procedure for isolating sulfhydryl peptides alkylated with N-ethylmaleimide. Anal Biochem. 1980;107:358–361. doi: 10.1016/0003-2697(80)90396-6. [DOI] [PubMed] [Google Scholar]
- Yankeelov J. In Methods Enzymology. Academic Press. 1972;25:566. doi: 10.1016/S0076-6879(72)25056-X. [DOI] [PubMed] [Google Scholar]
- Takahashi K. The reaction of phenylglyoxal with arginine residues in proteins. J Biol Chem. 1968;243:6171–6179. [PubMed] [Google Scholar]
- Miles EW. Modification of histidyl residues in proteins by diethylpyrocarbonate. Methods Enzymol. 1977;47:431–442. doi: 10.1016/0076-6879(77)47043-5. [DOI] [PubMed] [Google Scholar]
- Cheng KW. Carboxymethylation of methionine residues in bovine pituitary luteinizing hormone and its subunits. Location of specifically modified methionine residues. Biochem J. 1976;159:79–87. doi: 10.1042/bj1590079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loudon GM, Koshland DE. The chemistry of a reporter group: 2-hydroxy-5-nitrobenzyl bromide. J Biol Chem. 1970;245:2247–2254. [PubMed] [Google Scholar]
- Goodlett DR, Keller A, Watts JD, Newitt R, Yi EC, Purvine S, Eng JK, von Haller P, Aebersold R, Kolker E. Differential stable isotope labeling of peptides for quantitation and de novo sequence derivation. Rapid Commun Mass Spectrom. 2001;15:1214–1221. doi: 10.1002/rcm.362. [DOI] [PubMed] [Google Scholar]
- Sechi S. A method to identify and simultaneously determine the relative quantities of proteins isolated by gel electrophoresis. Rapid Commun Mass Spectrom. 2002;16:1416–1424. doi: 10.1002/rcm.734. [DOI] [PubMed] [Google Scholar]