

Abstract
Recent results from x-ray crystallography and molecular dynamics free-energy simulations have revealed the existence of a number of specific cation-binding sites disposed along the narrow pore of the K+ channel from Streptomyces lividans (KcsA), suggesting that K+ ions might literally “hop” in single file from one binding site to the next as permeation proceeds. In support of this view, it was found that the ion configurations correspond to energy wells of similar depth and that ion translocation is opposed only by small energy barriers. Although such features of the multiion potential energy surface are certainly essential for achieving a high throughput rate, diffusional and dissipative dynamical factors must also be taken into consideration to understand how rapid conduction of K+ is possible. To elucidate the mechanism of ion conduction, we established a framework theory enabling the direct simulation of nonequilibrium fluxes by extending the results of molecular dynamics over macroscopically long times. In good accord with experimental measurements, the simulated maximum conductance of the channel at saturating concentration is on the order of 550 and 360 pS for outward and inward ions flux, respectively, with a unidirectional flux-ratio exponent of 3. Analysis of the ion-conduction process reveals a lack of equivalence between the cation-binding sites in the selectivity filter.
Keywords: molecular dynamics, Brownian dynamics, potential of mean force, membrane potential, Poisson–Boltzmann equation
Potassium channels are transmembrane proteins that have the ability to conduct K+ ions at nearly the diffusion limit while remaining very selective (1). The availability of high-resolution crystallographic structures (2–4), together with the development of sophisticated computer models (5–10), is providing us with a unique opportunity to refine our understanding of these systems at an unprecedented level. Although the complexity of these channels does present a formidable challenge to biomolecular modeling, it is particularly encouraging to note that many of the recent results from molecular dynamics (MD) simulations based on realistic all-atom models have been consistent with the information emerging from higher-resolution structural data (for a recent review, see ref. 11). In particular, the free-energy profile, or potential of mean force (PMF), associated with the position of three K+ along the axis of the pore was calculated with umbrella sampling simulations of the K+ channel from Streptomyces lividans (KcsA) embedded in a phospholipid membrane with explicit solvent (5). This MD calculation reproduced the four cation-binding sites (S1–S4) located in the narrow selectivity filter that were already known from x-ray diffraction data at 3.2-Å resolution (3) and also anticipated the existence of two additional sites (Sext and S0) located on the extracellular side of the channel, which were observed independently in diffraction data at 2.0-Å resolution (2). This result increased the confidence in the ability of such simulations to provide meaningful information about the ion-conduction process that is not available from the crystallographic structure (12).
One of the most striking features of the calculated multiion PMF was the absence of significant activation free-energy barriers opposing concerted translocation of the ions along the channel axis according to a process reminiscent of the “knock-on” mechanism originally proposed by Hodgkin and Keynes nearly 50 years ago (13). The absence of large energy barriers is traditionally interpreted as strong evidence that the ion-conduction process ought to be essentially diffusion-limited. Nonetheless, ion fluxes were not actually simulated, and the channel conductance was not calculated. Ultimately, for a complete description of ion permeation it is of fundamental importance to establish a direct link between those observable quantities that constitute the primary source of information about ion channels and the crystallographic protein structure. At the present time, this cannot be done by using simple “brute-force” MD simulations (11). According to single-channel measurements, the ionic flux across the KcsA under a transmembrane potential of 100 mV corresponds to the net translocation of approximately one ion in 10–20 ns (14), which is on the order of a typical MD trajectory (6–10). Despite the steady increase in computer power, the direct simulation of ionic fluxes across a selective biological channel with all-atom MD remains computationally prohibitive. Designing a theoretical framework able to rigorously extend this time scale to calculate ion fluxes is one of the most important goals in computational studies of ion channels (11).
A very attractive computational approach for simulating ion permeation over long time scales without having to treat a system in all atomic details explicitly is Brownian dynamics (BD). In its simplest 1D form, the random BD trajectory of the permeating ions along the pore axis is generated by integrating the stochastic equations of motion (15, 16)
 |
[1] |
where Zi and Żi are the position and velocity of the ith ion along the channel axis, respectively, D is the diffusion constant, ζi(t) is a random Gaussian noise, and Wtot is an effective potential energy function. Such BD have been used to simulate multiion conduction through 1D (17, 18) and in 3D (19, 20) models of K+ channels. As embodied by Eq. 1, the construction of a BD model requires very specific input quantities such as the ion diffusion constant (D) and the total PMF (Wtot), which serve as the fundamental “microscopic ingredients” of the theory. If the input quantities are treated as free adjustable parameters to fit experimental data, then BD can be used as a phenomenological framework to analyze experimental data. Alternatively, the theory may be anchored tightly to the molecular reality of ion channels if all the fundamental input quantities are rigorously extracted from calculations based on detailed atomic models. Such a strategy finds its roots in statistical mechanical theories of nonequilibrium transport phenomena in which dissipative equations of motion are derived for a reduced set of degrees of freedom while rigorously projecting out the dynamics of the rest of the system (see ref. 21 and references therein). Investigations based on a similar approach have provided much insight into the detailed mechanism of numerous dynamical processes in dense liquids (22). Although there are some similarities, the approach differs essentially from previous BD simulations of ion channels (18–20).
Theory and Method
Total PMF and Diffusion Constant. The total PMF, Wtot, is expressed (23) as
 |
[2] |
where Weq is the equilibrium PMF and ϕmp(Z) is the transmembrane potential profile along the Z axis. This decomposition is obtained by rigorously separating the microscopic system into a channel region and intra- and extracellular bulk regions (23). The equilibrium PMF is dominated by the strong local molecular interactions (in the absence of any transmembrane potential), whereas the transmembrane potential arises from a very small ionic charge imbalance widely distributed at the membrane–solution interface away from the pore region (23). The equilibrium PMF was calculated previously from all-atom umbrella sampling MD free-energy simulations (5) generated from the x-ray structure of the KcsA (PDB ID code 1BL8) (3). All equilibrium properties are rigorously included in Weq(Z1, Z2, Z3), including the explicit influence of water molecules and a flexible protein. Although this structure corresponds to a closed state of the channel [the passage of cations through the tight bundle formed by the four inner helices on the intracellular side is energetically unfavorable (24)], the PMF can be used in a model designed to simulate the movements of K+ through the selectivity filter (from the center of the intracellular cavity and up to the extracellular side).
The transmembrane potential needed to simulate ion fluxes was calculated for a model of the KcsA in the open state constructed on the basis of the 3.4-Å resolution x-ray structure of the calcium-activated K+ channel from Methanobacterium thermoautrophicum (4). The result is shown in Fig. 1. The voltage profile was calculated by using a modified Poisson–Boltzmann theory, in which the intra- and extracellular bulk regions are kept in equilibrium with electrodes at a potential difference of Vmp (the Poisson–Boltzmann voltage equation) (23, 24). The PBEQ module (25) implemented in the charmm program (26) was used with the optimized atomic Born radii for amino acids (27). It should be noted that the protein and ion charges must be formally turned off in the calculation of ϕmp (see Supporting Text, which is published as supporting information on the PNAS web site, www.pnas.org, for further details).
Fig. 1.

Transmembrane potential profile along the pore of an open-state model of the KcsA (3) generated from the structure of the calcium-activated K+ channel from M. thermoautrophicum (4). The curve is drawn assuming a positive unitary value of the intracellular potential. The potential at the position of the five most important cation-binding sites, S0–S4, is highlighted (filled dots). For comparison, the profile calculated for the closed state of KcsA is also shown (24).
The diffusion-constant profile shown in Fig. 5, which is published as supporting information on the PNAS web site, was extracted from the velocity autocorrelation function of the ions calculated by using an analysis based on the generalized Langevin equation for nonuniform systems (22, 28, 29) (see Supporting Text for further details).
BD Algorithm. The stochastic Brownian motion of the multiion system was implemented as a continuous-time Markov chain with discrete states corresponding to the ion positions, and the state-to-state random walk was constructed by generating exponentially distributed random survival times. Such a Markov random walk satisfies the condition of detailed balance under equilibrium conditions in the absence of net flux, and the stochastic evolution of the system obeys a multidimensional Smulochowsky (Nernst–Planck) diffusion equation as δZ becomes increasingly small (30–32). The forward and backward transition rates are given by (e.g., for ion 1)
 |
[3] |
where D(Z) is the space-dependent diffusion constant of the ion in the channel, and δZ is the grid spacing. Similar expressions can be written for the forward and backward transitions involving the other ions in doubly or triply occupied channels. Only the states with two or three K+ are being considered. Entry and exit of ions into the simulation region are represented as a first-order process. In a three-ion occupied channel, an ion can attempt to exit the system as soon as it reaches the ends of the pore, at Zmin or at Zmax. The exit rate of the innermost ion on the intracellular side is
 |
[4] |
(the ions are numbered in increasing order starting from the extracellular side). An ion can attempt to enter a two-ion occupied channel at any time with a rate (e.g., on the intracellular side) of
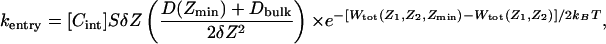 |
[5] |
where [Cint] is the ion concentration on the intracellular side and S is the cross-sectional area of the vestibule. Similar expression holds for the entry and exit on the extracellular side. The equilibrium PMF of the two-ion occupied channel, W(Z1, Z2), was calculated from the three-ion PMF with the third ion as far as possible from the selectivity filter, i.e., W(Z1, Z2) ≈ W(Z1, Z2, Zmin) and W(Z1, Z2) ≈ W(Zmax, Z1, Z2); in practice a Boltzmann average of the two limiting values was used. The cross-sectional area was set to 30 Å2, which corresponds roughly to an entrance vestibule of 3-Å radius. To focus on the multiion conduction through the selectivity filter, the value of the entry (kentry) and exit (kexit) rates were scaled by a dimensionless factor λ, which was set to a large value to impose equilibrium conditions of concentration exactly at the extremities of the pore and removes all time scales associated with entry and exit processes.
The probability of finding three ions in the pore follows a first-order saturation, [C]/(KD + [C]), where KD is the equilibrium dissociation constant determined by the relative free energy of a third ion entering a two-ion occupied channel: [Wtot(Z1, Z2, Z3) – Wtot(Z1, Z2)]. Both MD free-energy perturbation calculations and continuum electrostatic calculations indicate that the relative free energy of a third ion entering the cavity is on the order of 0 to –4 kcal/mol, which corresponds to a considerable uncertainty on the association constant and, consequently, the dependence of the channel conductance after ion concentration. In the model, the free-energy difference was empirically set to –1.4 kcal/mol to yield a channel that is occupied by three K+ 50% of the time at a concentration of 800 mM, in accord with experimental measurements (14). All the results were obtained from BD trajectories of ≈3 μs, corresponding to ≈109 Markovian transitions. Each BD simulation took ≈2 h on a single Pentium 800-MHz CPU.
Results and Discussion
The total free-energy landscape of the K+ across the selectivity filter, Wtot, is the most important ingredient of the present theory. As an illustration, we consider the total PMF in the case of membrane potential differences of ±150 mV. The transmembrane potential along the axis of the channel in the open state, obtained by solving the Poisson–Boltzmann voltage equation (23, 24), is shown in Fig. 1. For comparison, the voltage profile along the axis of the channel in the closed state is shown also (24). In accord with previous results (24, 33), the variation in the transmembrane potential difference becomes increasingly localized across the selectivity filter as the channel opens. The main cation-binding sites (S0–S4) are located in the region where variation in the transmembrane potential is the steepest, whereas the potential difference across the wide aqueous intracellular vestibule is very small. The resulting total PMF, Wtot, is shown in Fig. 2. The results are presented as 2D topographic maps obtained from a projection along the reduced reaction coordinates (see legend for further details). The concerted multiion processes corresponding to the dominant mechanism for outward (+150 mV) and inward (–150 mV) ion movements are indicated with black arrows in the 2D maps. Comparison of the results for the two voltages shows that the total PMF, and presumably the ion-conduction mechanism, may be affected significantly by the applied transmembrane potential (see also ref. 5).
Fig. 2.

The total multiion free-energy profile Wtot(Z1, Z2, Z3) including the equilibrium PMF calculated from MD and a transmembrane voltage of ±150 mV. Wtot(Z1, Z2, Z3) is obtained from Eq. 2 by using the transmembrane potential profile shown in Fig. 1. The 2D projection maps were calculated as a function of the reduced reaction coordinates (Z1+Z2)/2 and Z3 (the ions are numbered from 1 to 3 starting from the extracellular side; see ref. 5). Each color contour corresponds to 1 kcal/mol. The dominant mechanism for outward (+150 mV) (a) and inward (–150 mV) (b) ion movements are indicated with dashed arrows. The principal ionic configurations along the conduction pathways are illustrated: A, [Cavity, S3, S1]; B, [S4, S3, S1]; C, [S4, S2, S0].
The ions in the channel undergo a random walk on the total free-energy surface with the space-dependent diffusion constant D(Z) (see Fig. 5 and Supporting Text). The calculated diffusion constant varies weakly throughout the entire permeation pathway, decreasing to ≈70% of its bulk value in the selectivity filter region. Further analysis of the magnitude and fast decay of the friction kernel shows that dynamical memory and inertial effects are negligible and that using a noninertial Markovian dynamics approximation (i.e., BD) is physically justified. For these reasons, the average structural character of the random ion movements governing the conduction mechanism (i.e., single-file diffusion of the ions with water in between) is determined largely by the multiion free-energy surface Wtot rather than the dissipative and frictional forces.
Several BD trajectories on the microsecond time scale were generated to explore the average ion fluxes under various nonequilibrium conditions of ion concentration and transmembrane voltage. As an illustration, a 100-ns segment from a typical BD trajectory corresponding to the evolution of the position Z(t) of the ions along the pore is shown in Fig. 3. During the BD trajectories, the ions stay in well defined states for some time, followed by sudden concerted “hopping” transitions occurring in a random fashion. The relation of the BD trajectory to the cation-binding sites and the average density of ions along the pore axis is illustrated schematically in Fig. 3 Lower Right. Because the trajectories were generated by using the total PMF calculated from Eq. 2, the dominant positions of the K+ during the trajectories are consistent with the cation-binding sites observed in the pore (2, 5).
Fig. 3.

Typical BD trajectory generated with an applied membrane potential of +50 mV and under symmetric conditions of K+ concentration chosen to yield a channel occupied by three ions 50% of the time. The Z(t) of the two or three ions in the system is alternatively plotted in blue, red, and green for the sake of clarity. The relative ion density along the pore is shown in relation to the different binding sites. Many outward translocation events can be observed; for example, the red curve shows some of these events between 5 and 10, 25 and 30, 35 and 45, and 90 and 95 ns. A reentry of a translocating ion in the direction opposite to the transmembrane potential can be observed between 45 and 68 ns. Binding of an ion from the extracellular solution briefly to site S0 stabilizes ions in sites S2 and S4 (e.g., around 48 ns).
A few typical events resulting in the net translocation of an ion can be observed in Fig. 3 (see legend). Such “productive” translocation events normally occur according to a particular pattern during which the five cation-binding sites (S0–S4) are occupied by three ions for a very brief period. The elementary microscopic events leading to outward ion conduction can be summarized as follows. For extensive periods of time, the selectivity filter is occupied by two K+ ions located alternatively in the S1 and S3 sites or the S2 and S4 sites; a third ion frequently attempts to enter the channel, either on the intra- or extracellular side. The exchange between these two states, which occurs on a time scale of ≈1–2 ns, corresponds to a rapid but nonproductive “back-and-forth shuttling” of the two K+ in the selectivity filter. While the two ions are located in sites S1 and S3, a third ion hops from the intracellular vestibule into site S4 and collides with the ion in S3. This induces a concerted transition to a state with the three ions in the S4, S2, and S0 binding sites, which then is followed by the rapid dissociation and departure of the outermost ions in S0 on the extracellular side, yielding the conduction of one net charge. This process is qualitatively similar to the knock-on mechanism proposed by Hodgkin and Keynes (13). A typical conduction event can be observed in Fig. 3 between 26 and 35 ns. Such a microscopic process is possible because simultaneous occupancy of the adjacent sites S3 and S4 is not energetically prohibitive while the dissociation of the outermost ion in S0 is accelerated by ion–ion repulsion (5). Similar collisions between two ions occupying adjacent sites can also briefly occur in the selectivity filter. For example, the back-and-forth shuttling process of the two ions in the binding sites is typically initiated by a brief transition to a state with two ions in adjacent sites S2 and S3. Simultaneous occupancy of the adjacent sites S1 and S2 is energetically unfavorable and is not observed. Such rapid collisions suggest that the effective electrostatic repulsion between the ions in the filter is manifested at short distances.
In the trajectories shown in Fig. 3, one ion enters the channel from the extracellular side at 45 ns, in the direction opposite to the applied transmembrane potential, and stays in the selectivity filter for an extended period before leaving the channel at 68 ns. At such a high potential, no ion succeeds in completing a translocation across the selectivity filter against the applied potential, but such events can be observed at lower potentials. At ±25 mV the net current is ≈5–6 pA, and ≈5% of the ions actually can translocate in the direction opposite to the applied transmembrane potential during the BD trajectories. The ratio of unidirectional outward and inward fluxes as a function of the applied potential is an important characteristic of ion conduction in long narrow pores (13). If all ions were moving independently, the unidirectional flux ratio would follow the simple Boltzmann rule, exp[–eVmp/kBT], as a function of applied membrane potential. In a concerted multiion-conduction mechanism, this rule is violated and the unidirectional flux ratio varies as exp[–neVmp/kBT], where n is called the flux-ratio exponent (1). Calculating unidirectional fluxes from the BD trajectories, it is found that the value of n is 3, reflecting the fact that the ions cannot exchange their position inside the pore during conduction. Experimentally, values of n, from 2.4 to 3.4 with a slight voltage dependence, have been measured for the voltage-gated Shaker channel, which has a high sequence similarity to KcsA (34).
The current–voltage relation (I–V curve) at 400 mM of K+ is shown in Fig. 4a, and the saturation curve of the conductance estimated at ±50 mV is shown in Fig. 4b. Experimental results from LeMasurier et al. (14) are shown also. The I–V is well reproduced at small and moderate voltages, but nonlinearity becomes too pronounced at large voltages because access resistance is not included in the model. The calculated conductance G exhibits a typical first-order saturation, which is expected because the model is limited to two- and three-ion occupied states. The observed deviations from the first-order behavior in the experimental data at low concentration could reflect the influence of unscreened electrostatic interactions or perhaps the onset of a different ion-conduction mechanism with less than three K+. The values of Gmax, the maximum conductance of the channel at saturating concentration, estimated from Fig. 4b is on the order of 550 and 360 pS for outward and inward ions flux, respectively. These values are in remarkable agreement with the experimental measurements (14). The calculated Gmax values correspond to the maximum K+ throughput of the pore, extending from the center of the intracellular cavity to the external binding site, Sext. Incorporating diffusion-limited access resistances at the ends of the pore (not taken into account in the current simulations) would only decrease Gmax. It should be noted that the variation of the conductance as a function of the ion concentration depends sensitively on the equilibrium factors that govern the occupancy of the pore. In the model, the free-energy difference between two and three K+ in the pore was empirically set to yield a pore occupied by three K+ ions 50% of the time at 800 mM, in accord with experimental observations (14). Nonetheless, we emphasize that no adjustable parameter affects the calculated Gmax value.
Fig. 4.

(a) I–V relation calculated from BD simulations under symmetric conditions and K+ concentration of 400 mM. (b) Conductance of the KcsA at ±50 mV as a function of permeant ion concentration. The variation of the channel conductance as a function of K+ concentration follows a first-order saturation with KD values of 740 and 640 mM for +50 and –50 mV, respectively. Experimental data from ref. 14 (open symbols and gray lines) were taken from www.jgp.org/cgi/content/full/118/3/303/DC1/1.
Interestingly, the calculated I–V curve exhibits a slight outward rectification that is also observed in single-channel experiments with the KcsA (14). One naive interpretation could be that the small fraction of the transmembrane potential in the wide cavity gives rise to an increased voltage-driven entry rate on the intracellular side. To test this hypothesis, the I–V curve was recalculated by using a simplified transmembrane field that is constant across the selectivity filter and zero everywhere else. As shown in Fig. 4 (dashed line), the constant field approximation yields essentially the same I–V curve, ruling out this possibility. This computational experiment suggests that the outward rectification is an intrinsic property of the selectivity filter. Detailed analysis reveals that the outward rectification arises from a subtle difference between the five cation-binding sites (S0–S4), which play a dominant role in ion conduction. Outward ion conduction proceeds mainly according to the sequence of states [S3, S1] → [S4, S3, S1] → [S4, S2, S0] → [S4, S2] → [S3, S1], whereas inward conduction takes place according to the same sequence of states occurring in the opposite order. Notably, the process corresponding to the equivalent inward knock-on mechanism does not occur because simultaneous occupancy of adjacent sites S0 and S1 or S1 and S2 is energetically unfavorable (5). The concerted transition, [S4, S3, S1] → [S4, S2, S0], plays a critical role for both outward and inward ion conduction. As observed in Fig. 2b, a small barrier opposing the concerted transition remains even at moderately high negative voltage. In contrast, Fig. 2a shows that the concerted transition becomes essentially barrierless at moderately high positive value of the transmembrane potential, yielding a significant increase in the outward flux (a second ion-conduction mechanism corresponding to a vacancy-diffusion process could become possible at very high positive transmembrane potential, although it does not contribute significantly at potentials <150 mV). The forward and backward transition rates differ because the multiion configuration [S4,S3,S1] is less stable than the configuration [S4, S2, S0] due to the repulsion between the ion occupying adjacent cation-binding sites. The observed difference challenges the traditional view that rapid conduction is made possible because the cations can hop from one equivalent binding site to the next along the selectivity filter. One should note that the slight outward rectification exhibited by KcsA is most likely not a universal property of all K+ channels, because even small changes in the amino acid sequence would be expected to cause small but significant alterations in the atomic structure of the pore (e.g., see ref. 35) and also in the multiion free-energy surface governing conduction.
Conclusion
A rigorous computational strategy has enabled us to simulate ion fluxes for various conditions of permeant ion concentration and transmembrane potential. Although no parameters were specifically adjusted to reproduce the value of the maximum conductance, the results are in excellent accord with available observations. In view of the extreme sensitivity of calculations based on atomic models (a small increase of approximately kBT in the central energy barrier in the ion-conduction mechanism is sufficient to decrease the ion flux by a factor three), even a semiquantitative agreement with experimental results is very satisfying. The present effort demonstrates that the calculation of the conductance characteristics of a selective ion channel from first principles is possible.
Beyond the specifics of the actual numerical results, we would like to emphasize that an important strength of the present strategy is to help provide a rigorous conceptual framework to represent the mechanism of ion conduction through K+ channels at the microscopic level. Ion conduction is shown here to proceed from the random motion taking place on a total multiion free-energy surface, where the ions in the pore undergo sudden concerted hopping transitions between well defined states. In particular, specific concerted transitions involving three K+ simultaneously were identified as the key microscopic events governing the magnitude of the conductance.
It is heartening that the current results are, in fact, remarkably coherent with classical theoretical models of ion permeation widely used by electrophysiologists (see ref. 1 and references therein). The present framework thus enables a rigorous interpretation of the basic elements entering into such models (i.e., the multiion equilibrium free-energy profiles, transmembrane voltage profile, and diffusion-constant profile) without altering the fundamental structure that these models provide.
It is likely that a similar approach could be generally useful in studies of slow biomolecular processes whenever there is a need to extend the information extracted from all-atom MD trajectories to long time scales. Future extensions of the current model may be aimed at characterizing the properties of intracellular and extracellular blockers of K+ channels as well as the punch-through phenomenon exhibited by Na+ (36).
Supplementary Material
Acknowledgments
Useful discussions with Fred Sigworth, Olaf Andersen, Toby Allen, Mark Schumaker, Lise Heginbotham, and Chris Miller are gratefully acknowledged. The idea of a MD/BD hierarchy for computing ion fluxes was suggested by Kent Wilson during a private conversation at the Biophysical Discussions on Ionic Channels in Membranes (October 2–5, 1983, Airlie, VA). This work was supported by National Institutes of Health Grant GM62342-01.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: MD, molecular dynamics; PMF, potential of mean force; KcsA, Streptomyces lividans K+ channel; BD, Brownian dynamics; I–V, current–voltage.
References
- 1.Hille, B. (2001) Ionic Channels of Excitable Membranes (Sinauer, Sunderland, MA), 3rd Ed.
- 2.Zhou, Y., Morais-Cabral, J. H., Kaufman, A. & MacKinnon, R. (2001) Nature 414, 43–48. [DOI] [PubMed] [Google Scholar]
- 3.Doyle, D. A., Cabral, J. M., Pfuetzner, R. A., Kuo, A., Gulbis, J. M., Cohen, S. L., Chait, B. T. & MacKinnon, R. (1998) Science 280, 69–77. [DOI] [PubMed] [Google Scholar]
- 4.Jiang, Y., Lee, A., Chen, J., Cadene, M., Chait, B. T. & MacKinnon, R. (2002) Nature 417, 515–522. [DOI] [PubMed] [Google Scholar]
- 5.Bernèche, S. & Roux, B. (2001) Nature 414, 73–77. [DOI] [PubMed] [Google Scholar]
- 6.Guidoni, L., Torre, V. & Carloni, P. (1999) Biochemistry 38, 8599–8604. [DOI] [PubMed] [Google Scholar]
- 7.Allen, T. W., Bliznyuk, A., Rendell, A. P., Kuyucak, S. & Chung, S. H. (2000) J. Chem. Phys. 112, 8191–8204. [Google Scholar]
- 8.Åqvist, J. & Luzhkov, V. (2000) Nature 404, 881–884. [DOI] [PubMed] [Google Scholar]
- 9.Shrivastava, I. H. & Sansom, M. S. (2000) Biophys. J. 78, 557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bernèche, S. & Roux, B. (2000) Biophys. J. 78, 2900–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roux, B. (2002) Curr. Opin. Struct. Biol. 12, 182–189. [DOI] [PubMed] [Google Scholar]
- 12.Miller, C. (2001) Nature 414, 23–24. [DOI] [PubMed] [Google Scholar]
- 13.Hodgkin, A. L. & Keynes, R. D. (1955) J. Physiol. (London) 128, 61–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.LeMasurier, M., Heginbotham, L. & Miller, C. (2001) J. Gen. Physiol. 118, 303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ermak, D. L. & McCammon, J. A. (1978) J. Chem. Phys. 69, 1352–1360. [Google Scholar]
- 16.Cooper, K. E., Jakobsson, E. & Wolynes, P. G. (1985) Prog. Biophys. Mol. Biol. 46, 51–96. [DOI] [PubMed] [Google Scholar]
- 17.Bek, S. & Jakobsson, E. (1994) Biophys. J. 66, 1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mashl, R. J., Tang, Y., Schnitzer, J. & Jakobsson, E. (2001) Biophys. J. 81, 2473–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chung, S. H., Allen, T., Hoyles, M. & Kuyucak, S. (1999) Biophys. J. 77, 2517–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Allen, T. & Chung, S. H. (2001) Biophys. Biochim. Acta 155, 83–91. [DOI] [PubMed] [Google Scholar]
- 21.Zwanzig, R. W. (2001) Nonequilibrium Statistical Mechanics (Oxford Univ. Press, New York).
- 22.Berne, B. J., Borkovec, M. & Straub, J. E. (1988) J. Phys. Chem. 92, 3711–3725. [Google Scholar]
- 23.Roux, B. (1999) Biophys. J. 77, 139–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roux, B., Bernèche, S. & Im, W. (2000) Biochemistry 39, 13295–13306. [DOI] [PubMed] [Google Scholar]
- 25.Im, W., Beglov, D. & Roux, B. (1998) Comput. Phys. Commun. 111, 59–75. [Google Scholar]
- 26.Brooks, B. R., Bruccoleri, R. E., Olafson, B. D., States, D. J., Swaminathan, S. & Karplus, M. (1983) J. Comput. Chem. 4, 187–217. [Google Scholar]
- 27.Nina, M., Beglov, D. & Roux, B. (1997) J. Phys. Chem. B 101, 5239–5248. [Google Scholar]
- 28.Straub, J. E., Berne, B. J. & Roux, B. (1990) J. Phys. Chem. 93, 6804–6812. [Google Scholar]
- 29.Crouzy, S., Woolf, T. B. & Roux, B. (1994) Biophys. J. 67, 1370–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McGill, P. & Schumaker, M. F. (1996) Biophys. J. 71, 1723–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schumaker, M. F., Pomes, R. & Roux, B. (2000) Biophys. J. 79, 2840–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schumaker, M. F., Pomes, R. & Roux, B. (2001) Biophys. J. 80, 12–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang, Y., Lee, A., Chen, J., Cadene, M., Chait, B. T. & MacKinnon, R. (2002) Nature 417, 523–526. [DOI] [PubMed] [Google Scholar]
- 34.Stampe, P. & Begenisich, T. (1996) J. Gen. Physiol. 107, 449–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuo, A., Gulbis, J. M., Antcliff, J. F., Rahman, T., Lowe, E. D., Zimmer, J., Cuthbertson, J., Ashcroft, F. M., Ezaki, T. & Doyle, D. A. (2003) Science 300, 1922–1926. [DOI] [PubMed] [Google Scholar]
- 36.Nimigean, C. M. & Miller, C. (2002) J. Gen. Physiol. 120, 323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.