

Abstract
We study the dynamics of the passage of a stiff chain through a pore into a cell containing particles that bind reversibly to it. Using Brownian molecular dynamics simulations we investigate the mean first-passage time as a function of the length of the chain inside for different concentrations of binding particles. As a consequence of the interactions with these particles, the chain experiences a net force along its length whose calculated value from the simulations accounts for the velocity at which it enters the cell. This force can in turn be obtained from the solution of a generalized diffusion equation incorporating an effective Langmuir adsorption free energy for the chain plus binding particles. These results suggest a role of binding particles in the translocation process that is in general quite different from that of a Brownian ratchet. Furthermore, nonequilibrium effects contribute significantly to the dynamics; e.g., the chain often enters the cell faster than particle binding can be saturated, resulting in a force several times smaller than the equilibrium value.
The transfer of genetic material through the membrane surrounding a cell nucleus is fundamental to the understanding of basic cell processes from gene therapy to viral infection. The motion of linear polymers through pores into confined volumes also arises in many other biological contexts (1), perhaps the most common examples of which include the translocation of proteins from the cytosol into the endoplasmic reticulum or into mitochondria or chloroplasts. The export of mRNA through nuclear pore complexes is still another example of great importance. Despite the longstanding and widespread interest in this process, however, our knowledge about it is still rudimentary.
The process of translocation under the influence of an external field or a chemical potential gradient has recently been studied extensively (2–5). There have also been several theoretical studies that have specifically investigated chain translocation in the presence of binding particles (6–9). The major role of these binding particles has been recognized as a “Brownian ratchet,” a mechanism that was introduced in pioneering work by Simon, Peskin, and Oster (6). They treated the case of protein translocation, but their arguments apply equally well to nucleic acids. To account for translocation rates fast compared with simple diffusion under a wide variety of conditions and circumstances, they proposed that nonspecific binding by globular proteins results in a “biased,” or “ratcheted,” motion of the chain. Many experiments confirm that efficient translocation can indeed take place without the involvement of motor proteins. For example, the entire length (≈40 μm) of the DNA that comprises the genome of T5 phage is observed to enter its bacterial cell host without requiring metabolic energy (10). The experiment of Salman et al. (11) on phage λ DNA shows similarly that translocation of a comparable length of chain can occur without the help of active processes.
Each of the mechanisms mentioned above for chain translocation, namely diffusion and ratcheting, corresponds to a different time scale and different physics. Simple diffusion requires a characteristic time td = L2/2D, where L is the total length of the polymer and D its diffusion coefficient. In the ratcheting scenario, as soon as a specific length, δ (the distance between binding sites), of the polymer enters, a protein binds to it and the chain is no longer able to diffuse backward because the pore size is too small for the DNA/protein complex to pass through. In this case, the chain simply diffuses from one binding site to the next, and the translocation time is equal to the product of the time it takes for the chain to diffuse the distance δ times the number of ratcheting sites M = L/δ, i.e., tratchet = Mδ2/2D = L2/(2MD) = td/M, corresponding to a speed-up of the translocation time by a factor of M over simple diffusion. As pointed out by Simon, Peskin, and Oster, this time represents an idealized limit in which the ratcheting mechanism functions “perfectly”; i.e., as each successive binding site passes into the cell it is bound irreversibly by a protein that prohibits the chain from diffusing backward. In actuality, however, an entering site is not necessarily bound immediately by a protein and/or the protein does not stay adsorbed long enough to act as a ratchet at that site; accordingly, the translocation time is increased beyond Lδ/2D by a factor that depends on the ratio of on and off rates for binding.
In this article we consider explicitly the effect of binding particles and show that, via a new mechanism, translocation can occur at rates significantly faster than that provided by the perfect ratcheting scenario described above. More specifically, we argue that the particles that bind reversibly to the chain give rise to a net force on the chain that pulls it into the cell. Furthermore, this force accounts fully for the translocation process and embraces the different mechanisms mentioned above, e.g., in a special limit the Brownian ratcheting appears as a particular idealization (and appealing simplification) of the effect of such a force. The magnitude of the force depends in a delicate way on the concentration of the binding particles and their diffusion coefficient relative to that of the chain. In the overdamped limit the translocation time in the presence of a force F is expected to take the form tF = L/v = Lζ/F, where ζ is the friction coefficient of the chain, related to its diffusion constant through the Einstein relation D = kBT/ζ.
Using Brownian molecular dynamics (BMD) simulations, we calculate separately the average force and the translocation time in the presence of binding particles and find that they do indeed obey the relation tF = Lζ/F. Consequently, the translocation process is force-driven, and the translocation time turns out to be longer or shorter than the ideal ratcheting time, depending on the concentration and diffusion coefficient of the binding particles. These results can be understood in the context of a generalized Fokker–Planck equation for the probability that at time t the chain will have entered the cell to a distance x and have n particles bound to it. The drift terms corresponding to chain entry and particle binding are obtained from derivatives with respect to entry distance and binding number of a Langmuir adsorption free energy for the overall system.§ We find that the binding process involves important nonequilibrium effects in general, on which depends the actual value of the force pulling the chain in, and hence the ratio tF/tratchet.
Simulation
BMD simulations of a stiff polymer translocating through a pore and into a cell are performed by using a coarse-grained model in which the chain is represented by a rigid rod of beads. The beads are rigidly linked to their nearest neighbors along the chain and do not interact with each other. In this way we model DNA as a perfectly straight (rather than the usual semiflexible) chain, because the focus of our work is on the entering segment of chain that is within a persistence length from the pore. The link between adjacent beads is rigid to avoid chain contraction and extension resulting from the binding of particles; we also neglect changes in the shape or twist of the chain caused by binding. The distance between monomers along the chain, σ, corresponds to the “footprint” of binding particles in that the center of each bead is considered as an absorbing site. Note that in this case δ = σ. The binding particles are modeled as spherical, interacting with each other through the repulsive part of a Lennard Jones (LJ) potential with diameter σ; the interaction between binding particles and chain monomers is treated by a full (12–6) LJ potential. Because the distance between the absorbing sites is equal to the diameter of the binding particles, each site can be surrounded by a maximum of six particles. The cell–particle interactions are taken to vanish for particles within the radius Rs of cell and to increase as (R – Rs)4 for particles at distances greater than Rs from the sphere center. This potential is simply a convenient way to describe an interior wall. Finally, we treat the pore itself as being completely “inert,” having no effect on the chain except to allow it to enter or leave the cell.
We focus on the dynamics of the chain once one end has been inserted. In our simulations, the initial condition corresponds to the first monomer being placed just at the entrance (x = 0) of the pore. Let x denote the length of chain inside the cell and ri the position of the ith binding particle (see Fig. 1). The time evolution of these coordinates is described by the overdamped Langevin equations¶
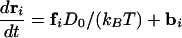 |
[1] |
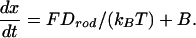 |
[2] |
Here fi and F are the deterministic forces acting on each particle and the rod, respectively, and bi and B are the corresponding random (Brownian) forces satisfying 〈bi(t)〉 = 0, 〈bi(t)·bj(t′)〉 = 6D0δ(t – t′)δij, 〈B(t)〉 = 0, and 〈B(t)B(t′)〉 = 2Drodδ(t – t′). D0 is the diffusion coefficient of an individual binding particle, related to its friction coefficient, ζ0, through the Einstein relation D0 = kBT/ζ0. As for the chain, we introduce an effective diffusion coefficient, Drod = kBT/ζrod, which in principle may include all of the pore–DNA interactions. Because so little is known about these complicated interactions, we have simply taken Drod = D0/(L/σ), consistent with the translational diffusion coefficient of a stiff chain being inversely proportional to its length. In all that follows we use σ, kBT, and σ2/D0 as the units of length, energy, and time, respectively. ε, the Lennard Jones binding energy between the particles and the monomers, is set equal to 5 kBT; the diameter of the spherical cell is 2 Rs = 24 σ, and the total length of the chain is L = 16 σ.
Fig. 1.

A schematic view of the translocation of the rigid chain into the spherical cell. The black spheres depict the monomers of the chain.
Fig. 1 is a schematic snapshot of the simulated system. The particles bind predominantly at the tip and then move back along the chain, with particles occasionally adding in empty spaces along the rod; at the same time, particles leave from other parts of the chain and allow for new particles to bind. Geometrically, up to six particles can sit around each monomer, but just as in the familiar Langmuir adsorption problem there are always empty sites on the chain caused by entropic factors. The fact that particles are added mostly at the tip is completely a dynamical effect. Under the influence of particle binding, the chain is in general moving too fast to allow for saturation of adsorption along its length. This results in less than the equilibrium number of occupied sites on the chain. In general, particles bind to the tip of the chain by pushing aside some already attached particles. However, the force pulling in the chain is exerted mostly at the entering positions where empty binding sites first appear.
Results
Fig. 2 shows the mean first-passage time (MFPT) versus the length of the chain inside the cell, x. MFPT is the average, over a large number of trajectories, of the time it takes for the front tip of the chain to first arrive to the position x. Each curve in Fig. 2 corresponds to a different value of N, the number of binding particles inside the cell.
Fig. 2.

MFPT as a function of entry distance x, for each of several different values of the number of binding particles N, as calculated from our BMD simulations. The thick solid curve describes the MFPT vs. x for simple diffusion of the chain into the cell. The dotted curve shows the MFPT for the case where translocation would occur via perfect ratcheting (see text).
As mentioned in the Introduction, different mechanisms for the translocation of the rod under the influence of the binding particles exist. If the chain simply diffused into the spherical cell, the MFPT would be equal to td = L2/(2Drod) = 2,048 σ2/D0 corresponding to the length L = 16 σ; see the quadratic function depicted by the heavy solid curve in Fig. 2. As shown in Fig. 2, td lies significantly above the translocation times we find in our simulations; even in the presence of only five binding particles, the MFPT is about three times shorter.
The dotted line in Fig. 2 represents the time that it would take for the entire chain to enter if there were ratchets functioning perfectly at every absorbing site. In this case, the chain simply undergoes successive and independent diffusions between neighboring sites, completing each in a time σ2/(2Drod); the MFPT is equal to the product of this time and the number of steps (x/σ) associated with the entry distance x. According to the ratcheting mechanism, then, the slope of time versus displacement is simply σ/(2Drod) = 8σ/D0, for Drod = D0/16.
The numerical results from our simulations, and the theory outlined in the next section, confirm the presence of a quite different translocation mechanism, namely, drift caused by a net force exerted on the chain by binding particles. Entry into the cell of successively longer portions of chain “feeds” new binding sites into the system; as each additional particle binds to the chain the free energy of the chain drops, and this reduction gives rise to a force pulling the chain into the cell. In the presence of a constant force, and in the overdamped limit, the corresponding translocation time is tF = L/v, where v = FDrod/kBT is the velocity of the chain.
With competing mechanisms operative, the MFPT will reflect predominantly the one with the smallest translocation time. As already remarked, the diffusion time is always much longer than those arising from the other two mechanisms (see Fig. 2). However, the ratcheting time could be longer or shorter than that of the force-driven process according to whether the deterministic force F is larger or smaller than the “effective” Brownian ratcheting force. Comparison of the ratcheting velocity of a chain, 2Drod/σ, with the usual expression for the velocity of a chain under a constant force, v = FratchetDrod/kBT, shows that the effective Brownian ratcheting force is 2 kBT/σ. Consequently, the ratcheting mechanism is expected to be dominant in the presence of weak enough driving forces. Nevertheless, even in this limit, as we shall see below, the attractive force is operative and completely determines the translocation velocity.
Several important observations can be extracted from Fig. 2. The first is that translocation times depend on the number (concentration) of binding particles. This is also true, of course, for the ratcheting mechanism, because higher concentrations of binding particles imply faster “on rates” (k+), hence higher translocation velocities. But, as Simon, Peskin, and Oster (6) have themselves emphasized, there is a maximum translocation rate corresponding to the limit of large k+/k–. This maximum rate, or minimum time, tratchet = Lδ/2Drod, also corresponds to the smallest distance (σ in the present model) between ratcheting (binding) sites. From Fig. 2 we see that the translocation occurs even faster than the limiting ratcheting prediction when the number of binding particles exceeds N = 100. This result suggests that an additional mechanism is operative, which we show is associated with a net force acting on the chain along its direction of motion.
More explicitly, the slope of MFPT vs. x plots reveals that the average velocity of the chain, v, remains almost constant throughout the translocation process (except right at the beginning and toward the end). From the relation F = ζrodv one expects that the average force on the chain also stays constant during this process, in which case the slope of time t versus 〈x〉 will be almost the same as that of MFPT versus x iff vx/Drod ≫ 1.∥ Using F = ζrodv, we calculated the effective force on the chain from the velocity obtained in our MFPT vs. length curves (Fig. 2). Specifically, we calculated the average velocity of the chain over the range x = 4 σ to x = 11 σ, i.e., in the region where the MFPT versus x curves are almost linear (see Fig. 2).
Alternatively, we can also determine the force acting on the chain directly from the simulations. The sum of the x components of the forces exerted by all binding proteins on each monomer of the chain provides the total instantaneous force on the chain. The squares in Fig. 3 represent the average force exerted on the chain by the binding particles (coarse-grained over σ), as a function of the length inside. This force is seen to be nearly constant; furthermore, it is found to agree within numerical uncertainty, as shown in Fig. 4, with the force calculated (as described above) from the average velocity (i.e., the inverse slope in Fig. 2) for all of the values of N (up to 300) that we treated.
Fig. 3.

The squares illustrate the force calculated directly in the simulation (coarse-grained over σ), as a function of the length of the chain inside, for the case N = 100. The circles show the force calculated from an identical BMD simulation but with a rod diffusion coefficient that is 16 times smaller. The dashed curve corresponds to the force derived from solution of the coupled entry/adsorption diffusion equation in the approximation of quasi-equilibrium. The solid curve is the solution of the full Fokker–Planck Eq. 4 by using a = 10.8 σ (see text).
Fig. 4.

Comparison of the force obtained directly in the simulation (○) with the one calculated by using the relation F = ζrodv (•), where v is the velocity corresponding to the inverse slope of MFPT vs. x data.
For N < 50, where the forces are of the order of unity or smaller (i.e., smaller than the effective Brownian ratchet force 2 kBT/σ), Fig. 2 shows that the translocation times begin to be significantly longer than the perfect ratcheting limit. This scenario is in principle embodied in the basic result of ref. 6, in which the translocation time is written as tratchet(1 + 2 K–1), where tratchet = Lδ/2 Drod is the ideal ratcheting time and K = k+/k– is the effective strength of binding, expressed as a ratio of on and off rates. K ≫ 1 corresponds to saturated binding and ideal ratcheting; otherwise, the ratchet mechanism becomes less efficient and the translocation times are longer than tratchet. It is notable that even in this weak force situation, where one expects the ratcheting mechanism to dominate, we still observe a force-controlled translocation (i.e., the translocation time is still determined by the force pulling the chain in, as shown in Fig. 4). The reason is that these weaker forces correspond to smaller numbers of less strongly bound particles, and hence also to less efficient ratcheting.
Dynamical Theory
The translocation process simulated above can be described theoretically by using a simplified dynamical model. The two relevant variables are the length of the chain x inside the cell and the number of particles n attached to the chain. As the chain enters a length x, the number of available absorbing sites increases. With ε the binding energy of a single particle we can write the (Langmuir adsorption) free energy, A, of the system as
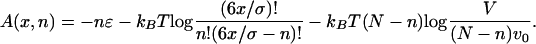 |
[3] |
Here N is the total number of particles, V is the volume of the spherical cell, and v0 is the volume of a single particle. The coefficient 6 in the second term of Eq. 3 is the number of particles that can interact attractively with a single binding site (chain monomer); accordingly, 6x/σ is the total number of available sites on a chain of length x. The first term in Eq. 3 is the energy gain caused by binding; the second is the entropic contribution associated with the total number of ways in which 6x/σ sites can be occupied by n particles; and the last term is the (ideal gas) contribution associated with the “free” particles, numbering N – n.
We consider the dynamics of translocation as a coupled diffusion process involving both the x and n degrees of freedom. Using mesoscopic nonequilibrium thermodynamics (12), one can derive the Fokker–Planck equation governing the time-dependent probability density ρ(x, n, t) that at time t a length x of the chain has passed through the hole and has n particles attached to it:
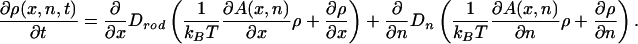 |
[4] |
Note that there is a drift and a diffusion term for each of the x and n variables. The force driving the translocation is F(x, n) = –∂A(x, n)/∂x, and we see from Eq. 3 that its origin is entropic, arising from the second (Langmuir) term in Eq. 3; the binding of the particles gives rise to a force that pulls the chain in. Similarly, the factor –∂A(x, n)/∂n is a “thermodynamic force” driving the particle binding. Drod is the spatial diffusion coefficient of the rod (with the usual dimensions of length2time–1), whereas Dn (which in general may depend on x and n) is the kinetic rate constant (with dimensions of time–1) for the process of particle binding and unbinding. A crude, but time-honored and physically reasonable, expression for Dn comes from the Smoluchowski theory of aggregation dynamics (13) for diffusing particles:
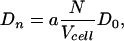 |
[5] |
where a is a length of order the particle size. Dn here is simply proportional to the concentration of binding particles N/V, and their spatial diffusion coefficient D0.
The Fokker–Planck Eq. 4 provides a complete description of the kinetics of both chain entry and particle binding. However, a simpler description can be achieved by considering the possibility of time-scale separation. The characteristic times for the entry and the binding processes scale as τx ≈ 1/Drod and τn ≈ 1/Dn, respectively. If the binding process is very fast compared with the chain entry (i.e., τn/τx ≈ Drod/Dn ≪ 1), it is reasonable to assume that the fast variable, here the number of attached particles, n, will decay very rapidly to its equilibrium distribution. In this case the process can be described by the evolution of the slow variable, the position x of the chain. Suppression of the fast variable can be carried out by using the standard adiabatic elimination technique (14), which is essentially equivalent to integration of the Fokker–Planck equation over the equilibrium distribution of the fast variable. The resulting 1D Fokker–Planck equation is
 |
[6] |
where
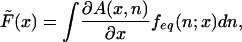 |
[7] |
is the average driving force. Here we have assumed that the spatial diffusion
coefficient of the rod is independent of n, and defined
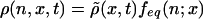 with feq(n; x) the local
equilibrium distribution,
with feq(n; x) the local
equilibrium distribution,
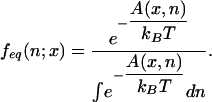 |
[8] |
In the particular case where feq(n; x) = δ(n –neq(x)), the force driving the translocation process becomes
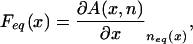 |
and the number of attached particles is equal to the equilibrium one, neq(x), given by the solution of
The above quasi-(or local) equilibrium approach basically assumes that, as soon as the chain's advance makes available new sites, particles bind to them immediately. Fig. 3 compares, for N = 100, the average force calculated in our simulation (squares) with the quasi-equilibrium force given by Eq. 7 (dashed curve). The comparison shows that the actual force that the binding particles exert on the chain is significantly smaller than the one that follows from the assumption that binding equilibration can keep up with chain entry (and this is true for all other N values considered). To check the limit in which the simple quasi-equilibrium 1D Fokker–Planck description, Eq. 6, provides an accurate description of the translocation process, we slowed down the entry of the rod by decreasing Drod. The circles in Fig. 3 shows the average force calculated via BMD simulation when Drod = 1/960 (vs. 1/16). Agreement with the quasi-equilibrium force evaluated from Eq. 7 is now excellent, confirming the validity of our model for the thermodynamic free energy A(x,n). Comparably good agreement is found between the number of attached particles calculated in the simulation and that predicted by the quasi-equilibrium theory. (Alternatively, we can increase the diffusion coefficient D0 of the binding particles, in which case the calculated force also approaches its quasi-equilibrium value, as we checked in the simulation.) The free energy given in Eq. 3 was written in the continuum limit under the assumption that the system is dilute. Considering its simplicity, the accuracy of the model in describing the system is surprisingly good.
In general, to compare simulation results with the predictions of our theory, we need to solve the full Fokker–Planck equation for the coupled x and n degrees of freedom. The numerical solution is obtained by converting Eq. 4 into its equivalent set of Langevin equations (14), and then solving these equations by using standard stochastic algorithms. From these numerical solutions the average force, the MFPT, and the average number of adsorbed monomers can be determined. Using the approximate expression for Dn, given by Eq. 5, with a = 10.8 σ (which in fact is very close to the value a = 4 πσ from the Smoluchowski theory) we have solved Eq. 4 for different values of N. The resulting MFPT values vs. x, and averaged pulling forces, compare well with those from the simulations. As an example, the resulting average force for N = 100 is represented by the solid line in Fig. 3, showing an excellent agreement with the results of the simulation.
Conclusion
The present work has attempted to provide a basic theoretical framework for treating the translocation dynamics of a stiff chain as it moves into a cell containing particles that interact attractively with it and bind to it. We conclude that this process is in general force-controlled. Obviously, pure diffusion occurs only in the absence of any binding particles; and the rectification of diffusion, which is the essence of the Brownian ratcheting mechanism, appears as a manifestation of the binding force in a special limit of interaction potentials and particle concentration.
To examine the extent to which the simple Brownian ratchet mechanism can account for chain translocation caused by binding particles, we have performed several simulations with different values for δ, the distance between binding sites. We varied δ from 1 to 4 σ, i.e., from δ smaller than to larger than the range of the Lennard–Jones attractive interaction (≈2 σ). When δ is sufficiently larger than the range of interaction, say for δ = 4 σ, we find that the chain performs a cycle of drift and diffusion movements. As an absorbing site enters the cell the chain is first pulled by the attractive force acting on the binding site, and then diffuses between adsorbing sites, during which time no net force is acting on the chain. In this case, the effect of the binding force is 2-fold: it pulls the chain in the region where it is acting and also impedes its backward diffusion. In the limit where the range of the force is small compared with the distance between sites, the contribution of the drift to the translocation is negligible, but nevertheless the force is still rectifying the diffusion. In these circumstances, the effect of the force can be most simply described by the Brownian ratchet idea. Specifically, the rectification arises from the free energy penalty for moving an absorbing site out of the cell (as provided approximately by our Langmuir adsorption model). The efficiency of the rectification depends on the ratio of free energy penalty to the thermal energy kBT.
We have also found that the effective force can be significantly (several times) smaller than the value one would estimate from a quasi-equilibrium treatment of the binding and entry dynamics, i.e., from assuming that the time scale for particles to diffuse and bind is much shorter than that for chain entry. More explicitly, only if we increase sufficiently the friction coefficient of the chain do we find agreement between the force calculated from the simulations and that obtained from the quasi-equilibrium solution to the coupled entry/binding Fokker–Planck equation. This result indicates that there can be significant differences between the force that would be measured by stalling a chain in single-molecule experiments (corresponding to the quasi-equilibrium value of the force) and the actual value of the force during the process of translocation, which depends on the kinetics of binding.
In summary, as established directly from our BMD simulations, we find that chain translocation can be understood in terms of the pulling force arising from particle binding. Moreover, this mechanism leads to translocation times that can become distinctly shorter than ratcheting. By decreasing the diffusion coefficient of the chain relative to that of the binding particles, the simulations give a still larger effective force pulling the chain along its length into the cell. The maximum value of this force, attainable in the limit N ≫ 1 and x → ∞ is precisely 6 ε/σ, i.e., the drop in free energy per unit length when all binding sites are occupied. These results are nicely confirmed by solving directly a coupled Fokker–Planck equation for the chain entry and particle binding dynamics. We conclude that physically realistic situations are in general more complicated than a ratcheting mechanism in which it is assumed that the only effect of particle binding at a chain site is to prohibit its diffusing back through the pore.
Acknowledgments
We acknowledge helpful discussions with Y. Kantor, M. Kardar, P. G. de Gennes, J. Widom, and M. Deserno. This research was supported by National Science Foundation Grants CHE99-88651 and CHE00-76384.
Abbreviations: BMD, Brownian molecular dynamics; MFPT, mean first-passage time.
Footnotes
In the simplest case of the fully equilibrated, fixed chemical potential Langmuir adsorption problem, the 1D pressure of the system corresponds to the force on the chain: P1D = f = (kBT/δ)ln(1 + ϕ exp(βε)), where ϕ is the volume fraction of binding particles, ε their binding energy, and δ the binding site size. In the limit of large binding energies (βε ≫ 1) one recovers the f ≈ ε/δ result discussed later; otherwise, entropic effects are important as well. We thank Pierre-Gilles de Gennes (College de France, Paris) for these clarifying observations.
We ignore the force on the chain in the y and z directions and let the rod move only in the x direction.
The translocation time of a polymer can be defined in terms of the MFPT τ(x), which satisfies the backward Fokker–Planck equation with a reflecting boundary condition at the hole and an absorbing boundary condition at x. The reflecting boundary condition is required because the first monomer is never allowed to move back out of the hole. The solution to the backward Fokker–Planck equation in the presence of a constant force F = ζv is τ(x) = x/v + D/v2(e–vx/D – 1). If v/D ≫ 1, then τ(x) = x/v. In the other limiting case, i.e., when v/D ≪ 1, then τ(x) = x2/2D.
References
- 1.Alberts, B., Bray, D., Lewis, J., Raff, M., Roberts, K. & Watson, J. D. (1994) Molecular Biology of the Cell (Garland, New York).
- 2.Lubensky, D. K. & Nelson, D. R. (1999) Biophys. J. 77, 1824–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muthukumar, M. (2001) Phys. Rev. Lett. 86, 3188–3191. [DOI] [PubMed] [Google Scholar]
- 4.Chern, S.-S., Cardenas, A. E. & Coalson, R. D. (2001) J. Chem. Phys. 115, 7772–7782. [Google Scholar]
- 5.Chuang, J., Kantor, Y. & Kardar, M. (2002) Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Top. 65, 0118021–0118028. [Google Scholar]
- 6.Simon, S. M., Peskin, C. S. & Oster, G. F. (1992) Proc. Natl. Acad. Sci. USA 89, 3770–3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sung, W. & Park, P. J. (1996) Phys. Rev. Lett. 77, 783–786. [DOI] [PubMed] [Google Scholar]
- 8.Elston, T. C. (2002) Biophys. J. 82, 1239–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liebermeister, W., Rapoport, T. A. & Heinrich, R. (2002) J. Mol. Biol. 305, 643–656. [DOI] [PubMed] [Google Scholar]
- 10.Maltouf, A. F. & Labedan, B. (1983) J. Bacteriol. 153, 124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salman, H., Zbaida, D. Rabin, Y., Chatenay, D. & Elbaum, M. (2001) Proc. Natl. Acad. Sci. USA 98, 7247–7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reguera, D. & Rubí, J. M. (2001) Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Top. 64, 0611061–0611068. [DOI] [PubMed] [Google Scholar]
- 13.Chandrasekhar, S. (1943) Rev. Mod. Phys. 15, 1–89. [Google Scholar]
- 14.Risken, H. (1984) The Fokker-Planck Equation (Springer, Berlin).