

Abstract
We have developed a semiempirical algorithm called Family Values (FamVal), which identifies residues that encode functional specificity in a protein sequence. Given a multiple sequence alignment (MSA) grouped into functionally distinct subfamilies, FamVal calculates a specificity score for each subfamily at every amino acid position of an MSA. This algorithm was used to predict specificity-encoding positions within the tetramerization assembly (T1) domain of voltage-gated potassium (Kv) channel subfamilies Kv3 and Kv4. The importance of one such position (Arg to Ala at MSA position 93) was confirmed by in vitro pull-down assays. The structural basis of this assembly discrimination was elucidated by determining the crystal structure of the Kv4 T1 domain and comparing it to the Kv3 T1 domain.
Functional information about proteins can be extracted from the growing collection of genome sequences by using computational methods. Sequence homology provides a starting point for the determination of certain structural and functional properties. By comparing the sequences of proteins of unknown function with the sequence of a protein of known function, it is possible to obtain information about the function of the unknown protein. In a similar manner, structural scaffolds can be deduced if structural templates exist among homologous proteins. The diversity of structural scaffolds for a given protein family, however, is smaller than the diversity of protein sequences and, thus, proteins with similar scaffolds can exhibit a variety of functions. In other words, a family of structurally similar proteins can be segregated into different groups (subfamilies) with different biological functionalities. For a particular protein fold, structurally important positions can be classified as those contributing to the fold stability itself, or to the specific functional features of a particular subfamily. This study is concerned with the development of a method to identify the latter class of amino acid positions.
Previously, a number of approaches have been used to identify functionally important positions in a common protein fold. In particular, Shannon entropy, set theory, the evolutionary trace method, and principal-component analysis have been used effectively (1–4). However, no currently available algorithms identify specificity-encoding residues based on the amino acid frequency in a multiple sequence alignment (MSA) in combination with a chemical profile of each subfamily. To achieve this goal, we have developed a nonprobabilistic semiempirical algorithm called Family Values (FamVal). The predictive power of the FamVal algorithm was tested by using the tetramerization assembly (T1) domain of voltage-gated potassium (Kv) channels as a system. The T1 domain is known to govern assembly specificity among related subfamily members of Kv channels. We aimed to use FamVal to determine the structural basis of subfamily-specific assembly. The T1 domain is particularly attractive for testing the algorithm, because (i) its biological function as a domain mediating tetramerization among Kv channels is well established, (ii) there are many protein sequences that have been separated distinctly into four major subfamilies, and (iii) significant structural knowledge about the T1 domain is available. We used FamVal to analyze T1 sequences in public sequence databases. Residues identified by FamVal were mutated and tested biochemically. Finally, by determining and comparing three-dimensional structures of T1 from two related subfamilies (Kv3 and Kv4), we have provided a structural explanation for the functional assembly specificity that FamVal identified.
Materials and Methods
FamVal Algorithm. FamVal requires as input an MSA template grouped into distinct subfamilies. It is assumed that the MSA template is as accurate as possible and, if available, structural alignments can be used. Conventional MSA algorithms based entirely on primary sequence, such as CLUSTALW (5), have been used for this study. At each horizontal MSA position, an n-dimensional profile is calculated first for each subfamily (subfamily vector) and then for the entire family of all sequences (overall vector). The n-dimensional vector is given by
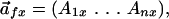 |
where 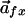 is the vector
representing the chemical profile at an MSA position x for subfamily
f, n is the number of chemical properties and
Anx is a scalar representing the chemical
identity at a particular MSA position. Anx is
given by
is the vector
representing the chemical profile at an MSA position x for subfamily
f, n is the number of chemical properties and
Anx is a scalar representing the chemical
identity at a particular MSA position. Anx is
given by
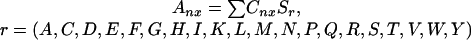 |
where r is the amino acid, cn is the number of residues of type r at an MSA position x, and sr is the score assigned for residue type r. Each subfamily vector is compared with the overall vector by a scoring function that returns a scalar “specificity” value.
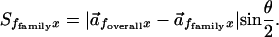 |
Sffamily x is the scalar
score for subfamily f at position x in the MSA. θ is
the angle between the two composite vectors
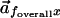 and
and
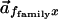 .
.
Because FamVal was intended to work for n chemical properties an n-dimensional matrix formulation is necessary. In matrix terms, the scoring function is
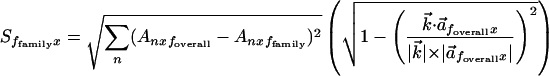 |
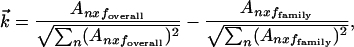 |
where 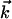 is a difference vector between
the subfamily and overall vectors.
is a difference vector between
the subfamily and overall vectors.
Algorithm Training and Interface Language. FamVal is written in perl and has a Common Gateway Interface-based web interface for data entry and processing. A test-bed program that assembles all permutations of a list of chemical-property scales and runs FamVal on each biochemical test case was written. Data output was evaluated at positions that were known to be biochemically important or unimportant and then evaluated qualitatively to confirm the usefulness of a given set of chemical-property files.
Structure Determination. The protein-expression vector contains residues 40–146 of rat Kv channel 4.2 (rKv4.2 T1) that were inserted into the coding region of a modified pET28 vector. BL21(DE3) cells were used for protein expression. This T1 segment of rKv4.2 is equivalent to residues 66–173 for the T1 domain of Aplysia Kv1.1 (aKv1 T1). Protein expression was performed for pull down assays as described below. Isolated protein was concentrated to 20 mg/ml in 5 mM Hepes, pH 7.5, and stored at –80°C. Crystals were obtained in 0.1 M Tris·HCl, pH 8.5/0.2 M MgCl2/5% polyethylene glycol 4000, at 23°C by hanging-drop vapor diffusion. Diffraction data were collected on a rotating-anode source on flash-frozen crystals by using 10% polyethylene glycol 8000/0.1 M Tris·HCl, pH 8.5/0.2 M MgCl2 as a cryoprotectant (Table 1). The composition of the cryoprotectant is different from that of the crystallization solution; the cryoprotectant is essential to preserve the crystals upon freezing. The phases were determined by evolution program molecular replacement methods, by using a polyserine model of Aplysia Kv3.1 T1 (6) as a search model.
Table 1. Crystallographic data collection and structure refinement statistics.
| Data set | Kv4.2 T1 |
| Wavelength, Å | 1.54 |
| Cell a, b, c, Å | a = 60.0, b = 60.0, c = 61.6 |
| Space group | P4212 |
| Resolution, Å | 2.1 |
| Completeness, % | 99.5 |
| Rmerge,* % | 5.5 |
| Rcryst,† % | 23.04 |
| Rfree,‡ % | 27.7 |
| No. reflections | 163,537 |
| No. unique reflections | 7,071 |
| Reflections in test set, % | 10.8 |
| No. protein atoms | 885 |
| No. water molecules | 57 |
| Avg. B factor, Å2 | 30 |
Rmerge = Σ|li — 〈l〉|/Σ li.
Rcryst = Σ|Fo — Fc|/Σ|Fo|.
Rfree = Σ|Fo — Fc|/Σ|Fo| for a 10.8% subset of diffraction data.
T1 Mutagenesis and Protein Expression. The Ala-to-Arg mutation in aKv3.1 T1 (referred to as Kv3 A63R) was made by using a pET16b expression vector (residues 1–115) (6) as a template for site-specific mutagenesis. Mutations were made in rKv4.2 at Arg-93, to Ala, and at Leu-66, to Arg [referred to as Kv4(R93A) and Kv4(L66R), respectively], by using a modified pET28 vector containing the rKv4.2 T1 (residues 40–146) as a template. The rKv4.2 T1 long form (residues 40–162), which is referred to as rKv4.2 T1(L), has an additional 16-aa C-terminal extension relative to rKv4.2 T1 (residues 40–146). All mutations were confirmed by DNA sequencing. All proteins were expressed and purified as described (6). Histidine tags were removed by thrombin as needed. Thrombin and uncleaved protein were completely removed on a benzamidine-Sepharose column and then cleaned by an additional run on a Ni·nitrilotriacetic acid (NTA) column. rKv4.2 T1 (R93A) produced greatly diminished yields of protein relative to the WT. rKv4.2 T1 (L66R) did not produce any useful amount of protein, even from large-scale cultures. Reasons for diminished expression levels are unknown; the diminished expression levels could, however, be due to the misfolding of proteins followed by degradation. All protein samples were concentrated to 150 μM and frozen at –70°C.
T1 Pull-Down Assays. Pull-down assays were performed by using a 4:1 ratio of mutant to His-tagged protein. Seven and a half micromoles of His-tagged protein was added to 30 μmol of Kv4 T1 protein. This reaction was brought up to 2 ml in 50 mM Tris·HCl, pH 8.0/500 mM NaCl/20 mM 2-mercaptoethanol/10 mM imidazole/10% glycerol. EDTA was then added to 1 mM. This reaction was incubated for 12 h with agitation at 4°C. The reaction mixture was then dialyzed into 50 mM Tris·HCl, pH 8.0/500 mM NaCl/20 mM 2-mercaptoethanol/10 mM imidazole/100 μM ZnSO4/10% glycerol and agitated at 4°C for another 12 h. This reaction was then applied to a 100-μl Ni·NTA agarose column and washed with 50 column volumes of the dialysis buffer. Ni·NTA agarose was then removed from the column and 20 μl of the slurry was analyzed by a Tricine-based SDS/PAGE system.
Results
A Vector Representation of Chemical Properties. FamVal first creates a representation of the chemical identity for every position in a functionally grouped MSA. For each MSA position, an n-dimensional vector represents the chemical identity, for which n is the total number of chemical-property scales used. Each element of the vector is a sum of the chemical properties of all of the amino acids at that MSA position (see Materials and Methods for formulation). Chemical properties are described by numerical values for each amino acid, normalized between –1 and 1, and assembled into a table of amino acid-value pairs called a “chemical-property scale” (Fig. 1). Chemical-property vectors are calculated for the entire family (the “overall” vector), as well as for each functional subfamily (the “subfamily” vector). The vector difference between them is then calculated. The magnitude of the difference vector is weighted by multiplying by sin (θ/2) where θ is the smaller angle between the two vectors. θ ranges from 0° to 180°, thus sin (θ/2) ranges from 0 to 1 for parallel and antiparallel vectors, respectively. This scalar quantity (Sf, x) is referred to as the FamVal score, which provides a quantitative measure of the uniqueness of a subfamily f compared with all other family members at position x in the MSA. FamVal scores for each position x in each subfamily are then expressed in units of standard deviation above the mean score.
Fig. 1.

Schematic representation of chemical-property vectors. Vectors are constructed for each MSA position independently for a given subfamily and again for the entire family. Here, an example MSA containing six sequences from two subfamilies is plotted in two dimensions (hydrophobicity on the y and polarity on the x axis). Red and blue vectors represent subfamily vectors for subfamilies A and B, respectively. In the leftmost plot, they represent the sums of three Ds for subfamily A, or three Es for subfamily B. A purple vector represents the overall vector at the MSA position. The subfamily's FamVal scores are computed by comparing the distances (green dotted lines) between the subfamily-vector endpoints and the overall-vector endpoints, weighted by multiplying by sin (θ/2). A table containing normalized chemical-property scales used for FamVal scores in this study is shown at the bottom.
Selection of Chemical-Property Scales. Because a variety of chemical-property scales are available, an objective method for selecting the most useful scales is important. Three test cases were used based on the availability of accurate MSAs, biochemical data clearly identifying specific subfamilies, and information on which specific amino acid positions are important or unimportant for their functional identity. The test cases used were the interaction of intracellular messenger proteins with kinase domains of type-β transforming growth factor and bone morphogenetic protein receptors (7), the conversion between platelet-derived growth factors types A and B (8), the host specificity of Listeria monocytogenes for E-cadherin (9), and the conversion of fibroblast growth factor types 1 and 2 (10). For each case, MSA positions known for their subfamily specificity or known not to play a role in subfamily specificity (by biochemical methods) were identified as benchmark positions.
To calculate FamVal scores, an initial set of eight chemical properties was assembled, which represented a cross section of chemical properties. These were amino acid frequency in the Swiss-Prot Protein Knowledgebase (11), mutability (12), polarity (13), accessibility (14), molecular weight (15), fractional area loss upon unfolding (16), hydrophobicity (17), and steric bulk (18). Sets containing all 255 possible combinations (28 – 1) of these scales were created and FamVal scores were calculated by using each test set. These FamVal scores were then compared with benchmark positions to identify sets that predicted the biochemical data. It was found that a set of scales composed of relative mutability (12), the average surface area buried upon folding (16), and the hydrophobicity (17) (Fig. 1) provided the most consistent agreement between high (1.5σ or greater than the average) FamVal scores and the biochemical data.
Every possible combination of the eight property files was created, which resulted in 255 unique sets containing from one to eight properties. By searching for the best set from these parameter sets, three main test cases were used. In the most basic training case [E-cadherin specificity for internalin (9)], a single position (Pro-16) was found to be responsible for host specificity. Pro-16 of human E-cadherin sequences and Glu of mouse and rat E-cadherin sequences switches E-cadherin's binding specificity to internalin. Thus, in analyzing the training results, we looked for positions that scored high for both sequence groups. With the final parameter set, MSA position 16 had high FamVal scores for both binding (1.72σ) and nonbinding (5.49σ) groups.
Type-β transforming growth factor–Smad specificity was the next training case. Among four positions prescreened as benchmark positions, two (Asn-267 and Asp-269 of type-β transforming growth factor receptor 1, National Center for Biotechnology Information/Entrez code NP_033396) of four scored high, with the final parameter set. Specifically, MSA position 267 had a FamVal score of 1.59 σ for Smad2/3 and 4.28 σ for Smad1 binding. Asp-269 had 2.17σ for Smad2/3 and 3.96σ for Smad1 binding, respectively.
The final training set was the conversion of human platelet-derived growth factors (PDGFs) from PDGF-AA to PDGF-BB (8). Wild-type PDGF-AA only binds the α receptor, whereas wild-type PDGF-BB binds both α and β receptors. Therefore, PDGF-BB broadens the receptor-binding specificity. As a result, we looked for MSA positions that scored high for PDGF-BB. Two sets of positions are known to be important. A mutant at position 67 and a double mutant containing positions 26 and 28 both were shown to be biochemically important for PDGF-BB specificity. MSA positions 26 and 67 had high FamVal scores of 2.34σ and 2.52σ, respectively.
Specificity-Encoding Positions for Channel Tetramerization. Eukaryotic Kv channels are assembled as tetramers; the genes of the subunit are segregated into four main subfamilies (Kv1, Kv2, Kv3, and Kv4) (19, 20). The N-terminal T1 domain that precedes the first transmembrane helix S1 of Kv channels is known to govern the specificity of assembly (19, 20). Within the T1 domain, there is a high level of sequence conservation (≈70%) within each subfamily (21), but significantly less at ≈40% between them. This high level of sequence conservation emphasizes an evolutionary conservation of subfamily-specific functionality. The amino acids contributing to the subfamily-specific assembly can be broadly viewed as (i) those providing energetically favorable affinity interactions and (ii) those providing energetically unfavorable interactions with other subfamily sequences to prevent intersubfamily association.
Because public sequence databases make available a large number of T1 sequences that are clearly distinguishable among the four subfamilies, T1 is an ideal candidate to test the FamVal algorithm. We have shown the biochemical feasibility of altering the subfamily specificity of Kv1 and Kv3 T1 domains by swapping a 14-aa segment within T1 (6). Here we focused on analyzing the specificity-encoding amino acids of Kv3 and Kv4 by FamVal. All positions with high FamVal scores are candidates for encoding subfamily specificity. We were particularly interested, however, in positions that scored high for both Kv3 and Kv4 because mutations at these positions might not only disrupt self-assembly but also switch the specificity of subfamily assembly between them. Seventy-one Kv4 and Kv3 sequences from GenBank were used to assemble a T1 MSA template in clustalw (5) with the following default parameters: gap opening = 10.00, gap-extension penalty = 0.05, delay-divergent sequences = 40%, protein-weight matrix = block substitution-matrices series, residue-specific penalties enabled, hydrophilic penalties enabled, hydrophilic residues = G, P, S, N, D, Q, E, K, and R, gap separation distance = 8, and end-gap separation disabled.
On this MSA template, FamVal scores were calculated at all MSA positions x along the T1 sequence (Fig. 2). To refer to MSA positions, we will use Kv4 residue numbers for the convenience of discussion, unless noted otherwise. In our analysis of Kv3 and Kv4 T1, we increased the stringency of the FamVal cutoff to 2σ. FamVal scores greater than 2σ above the mean for both Kv4 and Kv3 occurred at only two MSA positions: 66 and 93. MSA position 66 is Leu in Kv4 and Arg in Kv3 (Fig. 2, purple bars), whereas MSA position 93 is Arg and Ala for Kv4 and Kv3, respectively. The crystal structure of a Kv3 T1 domain (6) revealed that MSA position 93 (63 in Kv3) is deep in the subunit interface, whereas MSA position 66 is located on an outer surface of the T1 tetramer (Fig. 3). Because MSA position 66 is located at the outer surface of the tetramer, this site could be involved in additional subfamily-specific functions other than tetramer assembly between T1 subunits (interaction with cytoplasmic proteins, for example). In contrast, MSA position 93 is located at the subunit interface and, thus, appeared well positioned to contribute directly to differentiating binding affinity between Kv4 and Kv3.
Fig. 2.

Actual FamVal scores of Kv4 and Kv3 T1 domains. Red and blue bars represent FamVal scores for Kv3 and Kv4 subfamilies, in units of σ above the mean. Each horizontal line represents 1 σ. Actual amino acid residues are shown below. Those positions highlighted in purple bars (MSA positions 66 and 93) are the only positions that scored above 2σ for both Kv4 and Kv3. The raw sequence alignment can be found at http://sbl.salk.edu/~mnanao/kvalign.txt.
Fig. 3.

Superposition of Kv3 (purple) and Kv4 T1 (green) structures. Only two neighboring subunits of four subunits are shown for clarity. The four-fold symmetry axis is vertical in this view. (a) Two positions (Arg-93 and Ala-63, or Leu-66 and Arg-35) that scored above 2σ for both Kv4 and Kv3, respectively, are labeled with their side chains. Interacting amino acids for Arg-93 are also shown only for Kv4: Asp-88 and Glu-110. Zinc atoms known to be essential for the assembly interface are shown as spheres: Kv3 (red) and Kv4 (yellow). Side chains of three cysteines and one histidine coordinating the zinc are shown only for Kv4 T1. Note that the coordination of zinc is identical by the same set of amino acids: three Cys and one His. (b) Atomic interactions in the region of the MSA position 93 (Arg-93 of Kv4 or Ala-63 of Kv3) are shown. Residue numbers are given for their true amino acid positions. They are Asp-88 and Glu-110 of Kv4 backbone (green) and His-58 and Val-80 of Kv3 backbone (purple).
Altered Binding Specificity of T1 Mutants at MSA Position 93. WT and point mutants of T1 of Kv3 and Kv4 subfamilies were derived from Aplysia (aKv3.1 T1) and rat Kv4.2 T1 (rKv4.2 T1), respectively, but we will use Kv3 and Kv4 as a simple notation for aKv3.1 T1 and rKv4.2 T1, respectively, with their mutation sites in parentheses. Kv4(R93A) exhibited greatly reduced expression relative to Kv4(WT). Kv4(L66R) did not produce any soluble protein. It is not clear why the surface-exposed Leu-66 does not readily accept a mutation to Arg. To test the assembly affinity, purified His-tagged Kv3(WT) (Fig. 4, lanes 2 and 5), Kv3(A63R) (Fig. 4, lanes 1 and 4), or Kv4(WT) (Fig. 4, lane 7) was used as bait and were incubated with either Kv4(WT) (Fig. 4, lanes 1–3) or Kv4(R93A) (Fig. 4, lanes 4–6). The protein mixture was subjected first to EDTA dissociation, followed by reassociation in ZnSO4 to facilitate the mixing of the subunits (22). This procedure allows monomerization and reassociation of the Kv3 and Kv4 subunits because the subunit interface of both Kv4 and Kv3 tetramers is stabilized primarily by zinc atoms coordinated by the conserved set of four Zn-coordinating amino acids (6, 22). The mixture was then isolated by Ni·NTA affinity and the identity of the subunits constituting the various tetramers was determined by SDS/PAGE (Fig. 4).
Fig. 4.

In vitro pull-down assay of purified Kv3 and Kv4 T1 mutants. Below the gels, bait proteins Kv3(WT), Kv3(A63R), and Kv4(WTL) that are tagged by N-terminal 6-histidine tags are shown (Upper). Molecular weights of these bands range from ≈12 to ≈15 kDa. To test self-association of Kv4, a Kv4 WT with a 16-aa C-terminal extension [Kv4(WTL)] was used. The difference in size allowed us to distinguish them by SDS/PAGE. (Lower) The test proteins [Kv4(WT) and Kv4(R93A)] that are not histidine-tagged. Black bars represent the presence of those bait and test proteins in the reaction mixtures. The presence of the corresponding bands in the gels indicates the assembly affinity between them.
Based on this pull-down assay, Kv4(WT) has extremely weak binding to Kv3(WT) (Fig. 4, lane 2), whereas Kv4(WT) is clearly pulled down by a longer, His-tagged form of Kv4(WT) as a positive control (Fig. 4, lane 7) through homotetrameric assembly with Kv4(L). However, when point mutations were introduced, Kv4(R93A) assembles with Kv3(WT) with increased affinity (Fig. 4, lane 5), as compared with virtually no detectable affinity between Kv4(WT) and Kv3(WT) (Fig. 4, lane 2). Similarly, Kv3(A63R) binds to Kv4(WT) (Fig. 4, lane 1), indicating that MSA position 93 provides favorable affinity in a reciprocal manner. When Kv3(A63R) was mixed with Kv4(R93A), the assembly between them was pronounced (Fig. 4, lane 4), reaching a level of affinity comparable to that of intra-subfamily assembly (Fig. 4, lane 7). Additionally, Kv4(WT) and Kv4(R93A) have no endogenous binding to the Ni·NTA resin, as seen in Fig. 4, lanes 3 and 6. The results are consistent with the idea that the position is indeed a strong molecular determinant for subfamily-specific assembly specificity.
Structural Basis for Kv4/Kv3 Discrimination at Position 93. To characterize the structural basis of assembly specificity between Kv3 and Kv4 T1 domains, we have compared the crystal structures of Kv3 and Kv4 tetramers. The crystal structure of the rKv4.2 tetramer was determined to 2.1-Å resolution by molecular-replacement methods (Table 1) and compared with the published structure of aKv3 T1 (6). The overall scaffold of rKv4.2 T1 is the same as the aKv3.1 tetramer (Fig. 3). Before this structure determination, we predicted that the Zn atom would be coordinated by the same conserved set of amino acids in all non-Kv1 tetramers as it is in the structure of aKv3.1 T1 (6, 22). This prediction has been verified structurally for the Kv4 subfamily in this study. Furthermore, we observe that there is an interesting structural difference between aKv3.1 T1 and rKv4.2 T1. The membrane-facing side (layer 4) of aKv3.1 T1 consists of two helical segments (referred to as α5 and α6 in aKv3.1 T1) with a structural kink in the middle (6), in contrast to the straight unbroken helix (α5) observed previously in Aplysia Shaker T1 (aKv1.1 T1). An unanswered question was whether these different conformations were due to a single residue insertion in non-Kv3 subfamily members or to the Zn-coordinating conformation. Specifically, all non-Kv1 subfamily members contain two critical Zn-coordinating Cys residues that are absent in Kv1 subfamily members. These Zn-coordinating residues are located in a loop that forms a kink between α5 and α6 in aKv3.1 T1. In our structure, rKv4.2 T1 displays a single unbroken helix (α5) like aKv1.1 T1, but Zn-coordinating amino acids maintain the same Zn-coordination geometry as aKv3.1 T1 (Fig. 3). This result indicates that the two-helix conformation present in aKv3.1 T1 is not required for zinc coordination. This result also indicates that such conformational variation is probably due to the one-residue deletion, unique to the Kv3 subfamily.
Detailed inspection of the structure around MSA position 93 indicates that subfamily discrimination is due to a combination of polar and steric effects. In rKv4.2 T1, Arg-93 makes contacts with two positions on the neighboring subunit: Glu-110 (position 80 of aKv3.1 T1) and Asp-88 (position 58 of aKv3.1 T1) that together form a relatively acidic pocket (Fig. 3b). By contrast, aKv3.1 T1 has a Val at position 80 (MSA position 93), and His at position 58 (MSA position 88). These two concurrent changes remove the acidic pocket of Kv4 and create a bulky, hydrophobic (or even basic because of a potentially protonated His) pocket. Interestingly, both His-58 and Val-80 also show high FamVal scores 4.62 and 2.16σ, respectively, for Kv3. Our results strongly support the hypothesis that Kv4 and Kv3 use favorable interactions between Arg/Ala at MSA position 93 on one side of the interface and Asp/His at MSA position 88 and Glu/Val at MSA position 110 on the other side to differentiate themselves from each other.
Discussion
We have developed an algorithm (FamVal) to identify positions that encode specificity in the Kv4 and Kv3 subfamily T1 domains. Kv4 and Kv3 T1 domains normally show no coassembly. In our analysis, we identified residues that could potentially switch specificity by looking for positions that gave high scores for both Kv4 and Kv3. FamVal identified positions 66 and 93 in Kv4 and the equivalent positions 35 and 63 in Kv3 as positions encoding subfamily specificity. Among these, the importance of MSA position 93 for subfamily specificity was experimentally evaluated by exchanging the residue at this position between Kv4 and Kv3. Interestingly, the Shaker equivalent of this MSA position 93, Asp-119, was identified in a yeast two-hybrid experiment as a critical position for Shaker T1 domain assembly (23). In our experiments, the point mutants at this position alone showed measurable, although weak, cross-assembly with the T1 domain of the opposite subfamily. This weak assembly is expected because only a single interface of the T1 domain was altered. The point mutant, thus, can only poorly compete for assembly versus WT subunits that have both interfaces compatible for assembly. Indeed, when the Kv4 mutant and the Kv3 mutant were mixed together, the efficiency of coassembly increased to a level reasonably comparable to that of WT sequences. Structural studies indicate that MSA position 93 (position 93 of rKv4.2) is present on the T1 interface, which is consistent with its proposed role in oligomerization specificity. Furthermore, specificity appears to be encoded by a combination of steric and electrostatic effects.
The FamVal algorithm is designed to identify amino acid positions that encode subfamily-specific properties in a common structural-fold family. Although we have used FamVal to study assembly specificity, it is not the only possible subfamily-specific property that can be studied. Other biochemical functions such as protein binding or substrate specificity in enzyme catalysis can also be identified. FamVal also offers the possibility of selecting different chemical properties or different biochemically validated test cases. Future versions of FamVal will support a statistical method for parameterization (selection of chemical-property sets). Although FamVal can identify specificity-encoding positions, the structural means by which the protein achieves such properties is, however, not directly reflected in the numerical FamVal scores. For example, in specific protein–protein interaction, no information can be gleaned about whether specificity is achieved by providing residues that enhance binding to family members, or by providing residues that block binding to non-family members. In addition, FamVal scores do not provide any information about functional coupling of MSA positions because they are calculated independently. Nevertheless, FamVal provides a powerful method that can be used to identify functionally important regions in a functionally grouped sequence family without any prior structural information. With the rapidly growing amount of sequence, structural, and functional data, the need emerges for methods to combine and unite these data (24, 25). FamVal offers one such tool for the integration of these data.
Acknowledgments
We thank Witek Kwiatkowski for help with the multidimensional formulation of FamVal. W.Z. was supported by the American Heart Association. S.C. was supported by National Institutes of Health Grant GM56653. P.J.P. was supported by National Institutes of Health Grants NS31583, NS37444, and HD24064.
Abbreviations: FamVal, Family Values; Kv, voltage-gated potassium; MSA, multiple sequence alignment; NTA, nitrilotriacetic acid; PDGF, platelet-derived growth factor; T1, tetramerization assembly.
Data deposition: The atomic coordinates for the T1 domain of rat voltage-gated K channel 4.2 (rKv4.2T1) have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1NN7).
References
- 1.Hannenhalli, S. S. & Russell, R. B. (2000) J. Mol. Biol. 303, 61–76. [DOI] [PubMed] [Google Scholar]
- 2.Livingstone, C. D. & Barton, G. J. (1993) Comput. Appl. Biosci. 9, 745–756. [DOI] [PubMed] [Google Scholar]
- 3.Lichtarge, O., Bourne, H. R. & Cohen, F. E. (1996) J. Mol. Biol. 257, 342–358. [DOI] [PubMed] [Google Scholar]
- 4.Casari, G., Sander, C. & Valencia, A. (1995) Nat. Struct. Biol. 2, 171–178. [DOI] [PubMed] [Google Scholar]
- 5.Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994) Nucleic Acids Res. 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bixby, K. A., Nanao, M. H., Shen, N. V., Kreusch, A., Bellamy, H., Pfaffinger, P. J. & Choe, S. (1999) Nat. Struct. Biol. 6, 38–43. [DOI] [PubMed] [Google Scholar]
- 7.Chen, Y. G., Hata, A., Lo, R. S., Wotton, D., Shi, Y., Pavletich, N. & Massagué, J. (1998) Genes Dev. 12, 2144–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jaumann, M., Tatje, D. & Hoppe, J. (1992) FEBS Lett. 302, 265–268. [DOI] [PubMed] [Google Scholar]
- 9.Lecuit, M., Dramsi, S., Gottardi, C., Fedor-Chaiken, M., Gumbiner, B. & Cossart, P. (1999) EMBO J. 18, 3956–3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seddon, A. P., Aviezer, D., Li, L. Y., Bohlen, P. & Yayon, A. (1995) Biochemistry 34, 731–736. [DOI] [PubMed] [Google Scholar]
- 11.Bairoch, A. & Apweiler, R. (2000) Nucleic Acids Res. 28, 45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dayhoff, M. O., ed. (1978) Atlas of Protein Sequence and Structure (Natl. Biomed. Res. Found., Washington, DC), 5th Ed., pp. 345–358.
- 13.Grantham, R. (1974) Science 185, 862–864. [DOI] [PubMed] [Google Scholar]
- 14.Janin, J. (1979) Nature 277, 491–492. [DOI] [PubMed] [Google Scholar]
- 15.Lehninger, A. L., Nelson, D. L. & Cox, M. M. (1993) Principles of Biochemistry (Worth, New York), p. 113.
- 16.Rose, G. D., Geselowitz, A. R., Lesser, G. J., Lee, R. H. & Zehfus, M. H. (1985) Science 229, 834–838. [DOI] [PubMed] [Google Scholar]
- 17.Roseman, M. A. (1988) J. Mol. Biol. 200, 513–522. [DOI] [PubMed] [Google Scholar]
- 18.Zimmerman, J. M., Eliezer, N. & Simha, R. (1968) J. Theor. Biol. 21, 170–201. [DOI] [PubMed] [Google Scholar]
- 19.Li, M., Jan, Y. N. & Jan, L. Y. (1992) Science 257, 1225–1230. [DOI] [PubMed] [Google Scholar]
- 20.Shen, N. V., Chen, X., Boyer, M. M. & Pfaffinger, P. J. (1993) Neuron 11, 67–76. [DOI] [PubMed] [Google Scholar]
- 21.Shen, N. V. & Pfaffinger, P. J. (1995) Neuron 14, 625–633. [DOI] [PubMed] [Google Scholar]
- 22.Jahng, A. W., Strang, C., Kaiser, D., Pollard, T., Pfaffinger, P. & Choe, S. (2002) J. Biol. Chem., 277, 47885–47890. [DOI] [PubMed] [Google Scholar]
- 23.Strang, C., Cushman, S. J., DeRubeis, D., Peterson, D. & Pfaffinger, P. J. (2001) J. Biol. Chem. 276, 28493–28502. [DOI] [PubMed] [Google Scholar]
- 24.Xenarios, I., Salwinski, L., Duan, X. J., Higney, P., Kim, S. M. & Eisenberg, D. (2002) Nucleic Acids Res. 30, 303–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eisenberg, D., Marcotte, E. M., Xenarios, I. & Yeates, T. O. (2000) Nature 405, 823–826. [DOI] [PubMed] [Google Scholar]