

Abstract
In Escherichia coli photolyase, excitation of the FAD cofactor in its semireduced radical state (FADH•) induces an electron transfer over ≈15 Å from tryptophan W306 to the flavin. It has been suggested that two additional tryptophans are involved in an electron transfer chain FADH• ← W382 ← W359 ← W306. To test this hypothesis, we have mutated W382 into redox inert phenylalanine. Ultrafast transient absorption studies showed that, in WT photolyase, excited FADH• decayed with a time constant τ ≈ 26 ps to fully reduced flavin and a tryptophan cation radical. In W382F mutant photolyase, the excited flavin was much longer lived (τ ≈ 80 ps), and no significant amount of product was detected. We conclude that, in WT photolyase, excited FADH• is quenched by electron transfer from W382. On a millisecond scale, a product state with extremely low yield (≈0.5% of WT) was detected in W382F mutant photolyase. Its spectral and kinetic features were similar to the fully reduced flavin/neutral tryptophan radical state in WT photolyase. We suggest that, in W382F mutant photolyase, excited FADH• is reduced by W359 at a rate that competes only poorly with the intrinsic decay of excited FADH• (τ ≈ 80 ps), explaining the low product yield. Subsequently, the W359 cation radical is reduced by W306. The rate constants of electron transfer from W382 to excited FADH• in WT and from W359 to excited FADH• in W382F mutant photolyase were estimated and related to the donor–acceptor distances.
Electron transfer is ubiquitous in enzymatic catalysis and crucial to life (1, 2). However, most of these reactions escape from direct observation in macroscopic samples because they proceed faster than any synchronization process. An exception to this situation is found in proteins using light-induced electron transfer, where a short (laser) flash can be used to trigger the reaction. Photolyase is one of the few proteins that, on one hand, provide this possibility and, on the other hand, have been structurally resolved. Thus, it permits us to study intraprotein electron transfer processes directly and individually and to correlate them with structural data. In contrast to the extensively studied photosynthetic reaction centers (3), where electron transfer proceeds mostly via cofactors, in photolyase the electron transfer chain has been proposed to comprise several intrinsic amino acids [presumably three tryptophan residues in Escherichia coli photolyase (4, 5) and an additional tyrosine in Anacystis nidulans photolyase (6)]. This feature makes the system suitable for mutation studies, providing an excellent tool to relate the structure with the intraprotein electron transfer function in a native system (see ref. 7 for comparable artificially photosensitized proteins). We note that a similar chain of three tryptophans is also conserved in cryptochromes (8–10), a related class of flavoproteins that function as blue-light photoreceptors in a wide range of organisms.
DNA photolyase is found in a variety of organisms, ranging from bacteria to multicellular eukaryotes (11). It catalyzes the repair of major UV-induced DNA lesions, cyclobutane pyrimidine dimers, or (6-4) photoproducts, using the energy of near-UV or blue light (12, 13). Photolyase contains a FAD molecule as the essential catalytic cofactor that is functional only when it is in its fully reduced state (FADH–). Three-dimensional structures have been obtained for the photolyases from E. coli (14), from the cyanobacterium A. nidulans (15) and from the thermophilic bacterium Thermus thermophilus (16). Photolyase binds specifically to damaged DNA. After complex formation, the repair reaction is presumably initiated by electron transfer from photoexcited FADH– to the pyrimidine dimer (17).
In vitro, the fully reduced state FADH– can be reached by a separate photoreaction, called photoactivation, as it brings the enzyme into its catalytically active form. Excitation of the dark stable semireduced radical form FADH• leads to its complete reduction by means of intraprotein electron transfer to the excited flavin from a tryptophan residue. External electron donors can reduce the oxidized tryptophan and thus stabilize the fully reduced FADH–. In the absence of external donors, electron back transfer from reduced FADH– to the oxidized tryptophan reverses the light reaction (charge recombination). Therefore, isolated photolyase is typically found with its cofactor in the semireduced form FADH•. This result sets it apart from most other flavoproteins, whose flavin cofactor is normally oxidized or fully reduced and whose catalytic mechanism does not require light. “Artificial” electron transfer starting from excited oxidized or fully reduced flavins has been studied in several flavoproteins (18–20), whereas redox photochemistry of flavin radicals is so far only known for photolyase.
According to the crystal structures, a chain of three conserved tryptophans connects the FAD with the aqueous phase at the protein surface. In E. coli photolyase, this putative electron transfer chain comprises the solvent exposed W306, the intermediate W359, and W382, which is proximal to the flavin. The function of W306 as electron donor has been established by the observation that its mutation prevents photoactivation (21). W382 and W359 were suggested as intermediates because the reduction kinetics of excited FADH• (≈30 ps, ref. 5) seemed too fast for direct electron transfer over the large distance between W306 and the flavin (shortest “edge-to-edge distance” between the aromatic/conjugated systems, R = 14.8 Å). The following reaction sequence was proposed (5): (i) electron transfer from W382 to excited FADH• (R = 4.2 Å) in ≈30 ps; (ii) electron transfer from W359 to the W382 cation radical (R = 5.2 Å) in <10 ns (not resolved); (iii) electron transfer from W306 to the W359 cation radical (R = 3.9 Å) in <10 ns (not resolved); and (iv) proton release from the W306 cation radical to the aqueous phase in ≈300 ns, yielding the energetically relaxed neutral form of the tryptophanyl radical (readily reduced by external reductants). Electrostatic free energy calculations indicated that this reaction sequence is energetically feasible (22).
According to the crystal structure of E. coli photolyase (14), the W382 cation radical should be unable to deprotonate as there is no potential proton acceptor within 5 Å of the ring nitrogen that would release the proton. For W359, there is a buried water molecule close to the ring nitrogen, but its function as proton acceptor is questionable (discussed in ref. 5). Potential competitive internal electron donors (E. coli photolyase contains 12 further tryptophan residues and 14 tyrosine residues) are positioned such that their distances to the members of the suggested electron transfer chain significantly exceed, with one exception, the relevant electron transfer distances within the chain. The exception is W316, which is slightly closer to W382 (R = 3.7 Å) than W359 (R = 4.2 Å). Whether electron transfer from W316 to the W382 cation radical is energetically feasible, is not known.
Thus, structural data are largely consistent with the suggested electron transfer chain FADH• ← W382 ← W359 ← W306. Nevertheless, no direct proof for the involvement of W359 and W382 existed so far.
Here, we present studies on a mutant (W382F) where the tryptophan proximal to FAD has been replaced by phenylalanine, which has a similar geometry but is virtually unable to donate/accept electrons, interrupting the proposed electron transfer chain. Thus, the W382 mutant can provide direct evidence for or against the involvement of W382 in the native process. Our spectroscopic characterization on picosecond, millisecond and second time scales showed a dramatic loss of electron transfer efficiency for the mutant, along with a 3-fold increase in the lifetime of excited FADH•. A very small absorption change remaining on a millisecond time scale nevertheless allowed us to detect and to characterize low yield electron transfer in the mutant. Our findings suggest that this residual electron transfer proceeds via the two remaining tryptophans, W359 and W306, just as in the WT, but that the first electron transfer step is slowed according to the larger distance between FAD and W359 compared with the FAD-W382 distance in WT E. coli photolyase.
Materials and Methods
Mutagenesis. The photolyase W382F mutant was obtained from pKE (23), which is pKK223-3 harboring the E. coli WT photolyase gene, by the QuikChange mutagenesis method (Stratagene) by using GGCAGCCAATAACGGTGGCTTTCAGTGGGCCGCTTCAACCGG as primer. The mutated gene was overexpressed in E. coli KY29 (24), a photoreactivation-deficient strain in which the endogenous photolyase gene has been inactivated by replacement with a chloramphenicol resistance marker (25).
Proteins. After growth at 37°C in ampicillin and chloramphenicol-containing medium, mutant photolyase was induced with isopropyl-β-d-thiogalactopyranoside at 27°C. Cells were destructed by sonication in the presence of 10 mM 2-mercaptoethanol (ME). After centrifugation (twice) at 43,000 × g for 1 h, photolyase was purified by chromatography (twice) on heparin-Sepharose fast flow resin (Amersham Pharmacia) by elution with a 0.1–1.1 M NaCl gradient in 0.01 M potassium phosphate (pH 7.0), 10 mM ME, followed by chromatography on SP-Sepharose fast flow resin (Amersham Pharmacia) eluted with a 0.04–0.3 M NaCl gradient. The final preparation was estimated to be >98% pure. E. coli WT photolyase was overexpressed and purified as described (26).
Sample Preparation and Characterization. Samples were stored at –80°C in 10 mM phosphate buffer (pH 7.0), containing also 0.25 M NaCl, 18% (wt/wt) glycerol, and 5 mM ME. Immediately before starting measurements, polyacrylamide chromatography microcolumns were used to exchange the storing buffer for the standard measuring buffer containing 20 mM Tris·HCl (pH 7.4) and 0.2 M NaCl, or other buffers if indicated. Steady state absorption spectrometry using a Uvikon 922 UV/Vis spectrometer (Kontron, Milan) confirmed that mutant E. coli photolyase had very similar absorption characteristics as the WT, i.e., the flavin cofactor was present in the neutral radical state FADH•.
Time-Resolved Absorption Spectroscopy. For measurements on the picosecond time scale, the cell with an optical path of 1 mm was held at 10°C. These measurements were performed with a 30-Hz repetition rate pump-probe setup as described (27). The pump beam was centered at 620 nm and adapted to provide pulses of ≈200 fs full-width at half-maximum (FWHM) that excited ≈10% of the FADH• present. The pump beam was polarized at magic angle (54.7°) with respect to the white light continuum probe pulse. Transient spectra were recorded with ≈1 nm resolution by dispersing the probe beam in a polychromator, and detection was with a charge-coupled device camera.
For measurements on millisecond and seconds time scales, the cuvette with an optical path of 10 mm for the measuring beam and 4 mm for the excitation beam was held at 12°C.
The laboratory-built spectrometer for the millisecond scale (6) had a time resolution of 0.5 ms. Measuring light was selected by 10-nm bandpass filters from a 100-W tungsten halogen lamp. An Nd:YAG laser-pumped optical parametric oscillator (Rainbow, Quantel, Les Ulis, France) provided 630-nm pulses of typically 15 mJ/cm2 energy and 5-ns duration to initiate the photoreaction. WT photolyase signals were obtained from single flashes; mutant photolyase signals are averages over 128 flashes at a repetition rate of 1.983 Hz. The signal was detected by an FND 100Q Si photodiode (EG & G, Vaudreuil, QC, Canada), preamplified by a Tektronix AM502 (40 dB; DC-3 kHz, 1 MΩ) and registered by a Tektronix DSA 602A digital oscilloscope.
For photoaccumulation measurements on a seconds time scale, continuous actinic light was provided by an 800-W tungsten lamp. The collimated beam was filtered by a 30-mm water cell and a long pass OG570 filter and then focused into the sample cell. As external reducing agent, 30 mM ME was added.
Data Processing. For picosecond data, flash-induced absorbance changes have been averaged over wavelength intervals of 10 nm width. Flash-induced absorbance changes ΔAλ at different wavelengths λ have then been described by fitting to them globally the following model function:
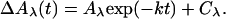 |
[1] |
Here, k is the λ-independent global decay rate constant, Aλ + Cλ is the amplitude extrapolated to time 0 (“initial amplitude”) at wavelength λ. The amplitude at the end of the fitting window, Cλ (“final amplitude”), as function of wavelength λ, describes the “resting spectrum.”
Results and Discussion
Ultrafast Absorption Spectroscopy Reveals W382 as the Primary Electron Donor to Excited FADH•. Absorbance changes accompanying formation of the excited state of FADH• and subsequent electron transfer in E. coli photolyase were monitored on a picosecond time scale. Fig. 1a shows kinetic traces for both WT and mutant E. coli photolyase typical for two different spectral regions. The visible spectral region (400–600 nm) is characterized by the presence of ground state absorption of FADH• and the potential photoproducts: FADH– and oxidized tryptophan. In the near IR spectral region above 700 nm, on the contrary, neither FADH• nor FADH– nor oxidized tryptophan absorbs in the ground state, so that only absorbance changes due to absorption of the excited state of FADH• (FADH•*) are expected. Accordingly, absorbance changes in this spectral window (see Fig. 1a Lower) decayed both in WT and mutant photolyases almost completely to zero within the detection time range of 250 ps. Exponential fits yielded decay time constants of 24 ps for WT photolyase and 80 ps for the mutant. As shown in the Inset, the initial absorbance increase in the 650- to 850-nm region was virtually identical for WT and mutant photolyase (the smaller amplitude of the mutant is due to its lower concentration). Above 700 nm, the transient absorption spectrum did not change until the signal had completely decayed (not shown).
Fig. 1.

Absorbance changes in WT and mutant E. coli photolyase on a picosecond time scale. (a) Time traces of the absorbance changes at 500 nm (Upper) and 700 nm (Lower). Incidence of the pump pulse marks time 0. Solid lines are simulated exponential decays with τ = 27 and 81 ps for WT and mutant at 500 nm and τ = 24 and 80 ps for WT and mutant at 700 nm. The signals were normalized such that the fit curves coincide at time 0. (Inset) The spectra of the absorbance changes in the 650- to 850-nm region. Throughout all figures, open symbols mark the WT, and filled symbols mark the W382F mutant. The optical densities at 620 nm (OD620) were 0.14 for WT, and, for W382F samples, 0.11 for detection in the near IR and 0.065 in the visible. (b) Spectra of absorbance changes in the 400- to 600-nm region for WT (Upper) and mutant (Lower) E. coli photolyase. Circles, initial spectra; triangles, resting spectra.
Time traces in the visible spectral region (e.g., 500 nm; Fig. 1a Upper) revealed decay time constants that were essentially the same as in the red/near IR, i.e., τ = 27 ps for the WT and τ = 81 ps for the mutant. A significant difference was observed in the degree of the decay: whereas in WT ≈66% of the initial absorbance change at 500 nm decayed in the 250-ps time window, >90% decayed in mutant photolyase. For the mutant, the accessible time window was not sufficient to monitor the decay completely. The resting spectra, along with those of the initial absorbance changes in the 400- to 600-nm spectral region are shown in Fig. 1b. The initial spectra for both WT and mutant show a major bleaching band around 500 nm and a second, minor band around 580 nm. We attribute these two bands to the initial bleaching of the FADH• absorption. The lack of bleaching at 550 nm where there is also ground state absorption of FADH• indicates that the excited state of FADH• has some absorption at 550 nm, besides the strong excited state absorption bands around 420 (see Fig. 1b) and 700 nm (Fig. 1a Inset). We have previously shown that the WT resting spectrum (Fig. 1b Upper, open triangles) in the 450- to 600-nm region is well described by the difference [(FADH– + tryptophan cation radical) – (FADH•+ neutral tryptophan)], i.e., the difference of product and reactant spectra for an electron transfer from tryptophan to FADH• (5, 28). For the mutant, there is hardly any resting spectrum detectable. The virtual lack of a resting state strongly indicates an inhibition or drastic reduction of the electron transfer as compared with the WT. This finding constitutes direct evidence that W382 is in fact the primary donor of electrons to the excited FADH• in WT photolyase. Replacement of W382 by phenylalanine blocks electron transfer from W382 to FADH•* (k2 = 0 in Fig. 2a) and thus increases the lifetime of FADH•* from ≈26 ps to ≈80 ps. Because electron transfer from W359 to FADH•* can be neglected here (as estimated below k3 ≈ 4.2 × 107 s–1), the 80-ps lifetime in the mutant should equal the intrinsic lifetime for relaxation of FADH•* to the ground state, i.e., k1 ≈ (80 ps)–1 = 1.25 × 1010 s–1. In WT, the observed decay rate kobs ≈ (26 ps)–1 = 3.85 × 1010 s–1 is due to competition between the intrinsic decay of FADH•* (k1) and electron transfer from W382 (k2). Assuming similar decay of FADH•* in WT and mutant, the electron transfer rate constant k2 can be estimated from kobs = k1 + k2, yielding k2 = 2.6 × 1010 s–1.
Fig. 2.

Components of the electron transfer chain in E. coli photolyase. (a) Reaction scheme of light-induced forward electron transfer in E. coli photolyase. Solid arrows indicate reactions in the WT. The broken arrow indicates the suggested first electron transfer step in the mutant. (b) Spatial arrangement of the components of the electron transfer chain in E. coli photolyase (from 1dnp.pdb; ref. 14): FAD, W382 (in the mutant replaced by a phenylalanine), W359, and the surface-exposed tryptophan W306. Distances are measured between closest contacts of the ring systems.
In a previous picosecond study on WT E. coli photolyase (29), transient absorption spectra and kinetics similar to ours were observed in the near IR; in the visible region, however, the difference spectra taken between 40 ps and 1 ns after excitation were completely different from ours. In particular in ref. 29, no bleaching was observed in the main ground state absorption region of FADH• around 500–600 nm. A possible origin of the disagreement is the use of 355-nm excitation flashes that could excite species other than FADH• (for example fully oxidized FAD; see ref. 30). Under excitation at 620 nm, as in our study, FADH• is the only species being excited.
Residual Millisecond Signal in W382F Photolyase Indicates a Low-Yield FADH• Photoreduction. If electron transfer to the excited FADH• is inhibited in W382F mutant photolyase, formation of fully reduced FADH– by photoaccumulation should be inhibited as well. This prediction was first checked by monitoring the absorption at 500 nm under continuous illumination of WT and mutant photolyases in the presence of an external electron donor (Fig. 3a Inset; for reasons of visibility, the mutant trace has been multiplied by ten). Interestingly, we could detect photoaccumulation of FADH– in the W382F mutant, although with a very low yield (≈0.5% of the amplitude in WT photolyase). Similarly, in the absence of external electron donors (Fig. 3a; the mutant trace has again been multiplied by ten), we could detect a very small (≈0.5% of the yield in WT photolyase) flash-induced bleaching at 580 nm that decayed on a millisecond time scale in a similar way as the well characterized (5) charge recombination between FADH– and the oxidized terminal tryptophan W306 in WT E. coli photolyase. These observations may indicate that the intermediate tryptophan W359 can transfer an electron to FADH•* just bypassing the replaced W382. Because of the larger distance to FAD, electron transfer from W359 (k3 in Fig. 2a) should be slower than from the proximal tryptophan W382 (k2), explaining a low electron transfer yield in competition with the intrinsic decay (k1) of the excited flavin. Alternatively, the excited flavin could be reduced by another tryptophan or a tyrosine residue (see introduction).
Fig. 3.

Absorbance changes in WT and mutant E. coli photolyase on a millisecond time scale. (a) Time traces for absorbance changes at 580 nm after an excitation flash at time 0, OD620 was 0.09 for the WT and 0.1 for the mutant photolyase. (Inset) Accumulation of absorbance changes at 500 nm under continuous illumination in the presence of 30 mM ME. OD620 = 0.16 for mutant and WT. Note that, for visibility, the mutant signals have been multiplied by 10. (b) Time course of absorbance changes of W382F photolyase at 410, 510, and 580 nm (OD620 = 0.1). (Inset) The initial amplitudes of these three curves (squares) are overlaid over a suitably scaled spectrum of the absorbance changes in the WT (solid line, taken from ref. 5). (c) Time course of normalized absorbance changes at 580 nm of W382F photolyase at various pH values as indicated (higher pH values correspond to longer signal lifetimes). The excitation flash marks time 0 (OD620 ≈ 0.1). (Inset) The decadic logarithm of the decay rate constants in s–1 is plotted vs. pH for both WT (open squares) and W382F mutant (filled squares) E. coli photolyase. Buffer was 20 mM Mes below pH 7.0 and 20 mM Tris above pH 7.0.
To decide this issue, we performed a series of experiments with the aim of characterizing the small millisecond signal in the mutant and comparing it with the behavior of WT photolyase (Fig. 3 b and c). Because of the very small amplitudes, we restricted the spectral analysis to three characteristic wavelengths 410, 510, and 580 nm (Fig. 3b). The relative amplitudes at these wavelengths (Inset) fit well with the spectrum for the FADH–/neutral (deprotonated) tryptophanyl radical state that had been observed (5) in the WT. This spectral behavior argues for tryptophan and against tyrosine as electron donor (the tyrosine radical is known to absorb at 410 nm; refs. 6 and 31). In Fig. 3c, we compare the WT and mutant pH dependence of flash-induced absorbance changes at 580 nm that in the WT reflect charge recombination between FADH• and the neutral W306 radical. For the WT (time traces not shown), lowering the pH over two units led to an increase in recombination rate of more than one order of magnitude, as reported (5) and explained by the need to reprotonate the neutral W306 radical before electron back transfer. In the mutant, kinetics and pH-dependence were similar to those in the WT (Fig. 3c qInset), which suggests that W306 also is the partner of FADH– in a recombination reaction in W382F mutant photolyase.
Possible Pathways of Forward Electron Transfer and Recombination. Assuming W306 to be the terminal electron donor to FADH• in the mutant too, and that the mutation does not induce major structural changes of the protein matrix, W359 would be best positioned to serve as an intermediate in electron transfer from W306 to excited FADH•.
According to the larger distance from FAD to W359 as compared with W382, electron transfer would be slower and its yield respectively lower. In fact, based on the model in Fig. 2a, and the observed ratio of ≈0.005 between electron transfer yields in mutant and WT photolyases, we can obtain k3 from the relation for the yields in mutant and WT: k3/(k1 + k3) = 0.005 × k2/(k1 + k2). With the values for k1 and k2 found above, i.e., k1 ≈ 1.25 × 1010 s–1 and k2 ≈ 2.6 × 1010 s–1, we arrive at k3 ≈ 4.2 × 107 s–1 as an estimate of the primary electron transfer rate in the W382F mutant. To test whether this rate is consistent with the distance between the flavin and W359 as the primary electron donor in the mutant, we use the simple “Moser-Dutton ruler” (32, 33). This semiempirical rule of thumb relates electron transfer rates k (in s–1) in proteins with edge-to-edge distances between partners R (in Å) and the relevant energetics (reaction free energy ΔG and reorganization energy λ, both in eV):
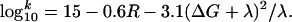 |
[2] |
Using for the distance between W359 and FAD the value R = 9.9 Å from the crystal structure of WT photolyase (ref. 14; see Fig. 2b), the “optimal” rate (for the special case ΔG = –λ) is found as k ≈ 109 s–1. Under less favorable energetic conditions, a slower rate would result, i.e., the rate calculated from the observed decrease in yield (k3 ≈ 4.2 × 107 s–1) is compatible with the distance between FAD and W359. For a putative direct electron transfer from W306 to FADH•* (R = 14.8 Å), the same approach predicts an optimal rate of ≈106 s–1, which seems too slow to account for the observed product yield. These estimations, along with the spectral and pH-dependent kinetic data presented in Fig. 3, lead us to the suggestion that the low yield photoreduction of flavin observed in the W382F mutant on a millisecond time scale occurs through the electron transfer pathway FADH• ← W359 ← W306.
Besides W359, five other residues could, due to their short distance to the flavin, reduce excited FADH• with non-negligible yield in the W382F mutant, i.e., W384 (R = 6.3 Å), Y281 (R = 6.9 Å), W338 (R = 7.4 Å), W277 (R = 9.1 Å), and W316 (R = 9.2 Å). However, all these residues are >12 Å apart from W306, so that their oxidized form is less likely to be re-reduced by “forward” electron transfer from W306 than by electron back transfer from FADH–. The latter reaction is likely to occur on a submillisecond time scale, so that it would not have been detected in our millisecond experiments, but might have contributed to the small resting absorbance changes in our picosecond experiments on the W382F mutant.
With respect to the recombination between FADH– and the W306 radical, the similar kinetics in WT photolyase and in the W382F mutant (Fig. 3c Inset) could indicate that W382 is not involved in this reaction in the WT. The first step of a putative electron back transfer through the triple tryptophan chain, from W359 to the W306 cation radical, was calculated to be uphill by 0.2 eV (22). This high energy barrier may favor direct electron transfer from FADH– to the W306 cation radical, circumventing the other tryptophan(s), both in WT photolyase and in the W382F mutant. Alternatively, the overall recombination rate could be mainly determined by the reprotonation of the neutral W306 radical preceding the electron back transfer, as suggested recently (22).
Concluding Remarks. Using a mutagenesis approach, we provided spectroscopic evidence that, in E. coli photolyase, a chain of three conserved tryptophans is involved in light-induced electron transfer from solutes in the aqueous phase to the buried cofactor FADH•. In the photolyase model system, we have exploited the possibility of using ultrashort light pulses to build up sizeable populations of the electron transfer precursor and intermediates. This result allowed us to assess the unusual situation of three chemically identical, intraprotein, redox partners. We emphasize that similar chains may be functional in other electron-transfer proteins, where the synchronization by light pulses is impossible.
Intriguing questions concerning the kinetics, energetics, and mechanisms of the individual electron transfer steps during photoactivation and recombination in this ingenious electron transfer system must be addressed by design of new mutants, in combination with the development of spectroscopic strategies to distinguish between the different tryptophans.
Acknowledgments
We thank Jessica Gladdines for preparing the mutant photolyase gene, Drs. A. Yasui and K. Yamamoto for providing us with the pKE plasmid and E. coli KY29 strain, respectively, and Dr. J. H. J. Hoeijmakers for his continuous interest in this project.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: FADH•, FAD in its semireduced state; FADH•*, semireduced FAD in its excited state; FADH–, FAD in its fully reduced state; ME, 2-mercaptoethanol.
References
- 1.Marcus, R. A. & Sutin, N. (1985) Biochim. Biophys. Acta 811, 265–322. [Google Scholar]
- 2.Stubbe, J. & van der Donk, W. A. (1998) Chem. Rev. 98, 705–762. [DOI] [PubMed] [Google Scholar]
- 3.Hoff, A. J. & Deisenhofer, J. (1997) Phys. Rep. 287, 2–247. [Google Scholar]
- 4.Cheung, M. S., Daizadeh, I., Stuchebrukhov, A. A. & Heelis, P. F. (1999) Biophys. J. 76, 1241–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aubert, C., Vos, M. H., Mathis, P., Eker, A. P. M. & Brettel, K. (2000) Nature 405, 586–590. [DOI] [PubMed] [Google Scholar]
- 6.Aubert, C., Mathis, P., Eker, A. P. M. & Brettel, K. (1999) Proc. Natl. Acad. Sci. USA 96, 5423–5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Bilio, A. J., Crane, B. R., Wehbi, W. A., Kiser, C. N., Abu-Omar, M. M., Carlos, R. M., Richards, J. H., Winkler, J. R. & Gray, H. B. (2001) J. Am. Chem. Soc. 123, 3181–3182. [DOI] [PubMed] [Google Scholar]
- 8.Kanai, S., Kikuno, R., Toh, H., Ryo, H. & Todo, T. (1997) J. Mol. Evol. 45, 535–548. [DOI] [PubMed] [Google Scholar]
- 9.Brudler, R., Hitomi, K., Daiyasu, H., Toh, H., Kucho, K., Ishiura, M., Kanehisa, M., Roberts, V. A., Todo, T., Tainer, J. A. & Getzoff, E. D. (2003) Mol. Cell 11, 59–67. [DOI] [PubMed] [Google Scholar]
- 10.Giovani, B., Byrdin, M., Ahmad, M. & Brettel, K. (2003) Nat. Struct. Biol. 10, 489–490. [DOI] [PubMed] [Google Scholar]
- 11.Yasui, A. & Eker, A. P. M. (1998) in DNA Damage and Repair, DNA Repair in Higher Eukaryotes, eds. Nickoloff, J. A. & Hoekstra, M. F. (Humana, Totowa, NJ), Vol. 2, pp. 9–32. [Google Scholar]
- 12.Sancar, A. (1994) Biochemistry 33, 2–9. [DOI] [PubMed] [Google Scholar]
- 13.Carell, T., Burgdorf, L. T., Kundu, L. M. & Cichon, M. (2001) Curr. Opin. Chem. Biol. 5, 491–498. [DOI] [PubMed] [Google Scholar]
- 14.Park, H. W., Kim, S. T., Sancar, A. & Deisenhofer, J. (1995) Science 268, 1866–1872. [DOI] [PubMed] [Google Scholar]
- 15.Tamada, T., Kitadokoro, K., Higuchi, Y., Inaka, K., Yasui, A., de Ruiter, P., Eker, A. P. M. & Miki, K. (1997) Nat. Struct. Biol. 4, 887–891. [DOI] [PubMed] [Google Scholar]
- 16.Komori, H., Masui, R., Kuramitsu, S., Yokoyama, S., Shibata, T., Inoue, Y. & Miki, K. (2001) Proc. Natl. Acad. Sci. USA 98, 13560–13565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim, S.-T., Heelis, P. F., Okamura, T., Hirata, Y., Mataga, N. & Sancar, A. (1991) Biochemistry 30, 11262–11270. [DOI] [PubMed] [Google Scholar]
- 18.Mataga, N., Chosrowjan, H., Taniguchi, S., Tanaka, F., Kido, N. & Kitamura, M. (2002) J. Phys. Chem. B 106, 8917–8920. [Google Scholar]
- 19.Enescu, M., Lindqvist, L. & Soep, B. (1998) Photochem. Photobiol. 68, 150–156. [DOI] [PubMed] [Google Scholar]
- 20.Zhong, D. & Zewail, A. H. (2001) Proc. Natl. Acad. Sci. USA 98, 11867–11872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li, Y. F., Heelis, P. F. & Sancar, A. (1991) Biochemistry 30, 6322–6329. [DOI] [PubMed] [Google Scholar]
- 22.Popović, D. M., Zmirić, A., Zarić, S. D. & Knapp, E. W. (2002) J. Am. Chem. Soc. 124, 3775–3782. [DOI] [PubMed] [Google Scholar]
- 23.Takao, M., Oikawa, A., Eker, A. P. M. & Yasui, A. (1989) Photochem. Photobiol. 50, 633–637. [DOI] [PubMed] [Google Scholar]
- 24.Nakajima, S., Sugiyama, M., Iwai, S., Hitomi, K., Otoshi, E., Kim, S. T., Jiang, C. Z., Todo, T., Britt, A. B. & Yamamoto, K. (1998) Nucleic Acids Res. 26, 638–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akasaka, S. & Yamamoto, K. (1991) Mutat. Res. 254, 27–35. [DOI] [PubMed] [Google Scholar]
- 26.Eker, A. P. M., Yajima, H. & Yasui, A. (1989) Photochem. Photobiol. 60, 125–133. [DOI] [PubMed] [Google Scholar]
- 27.Martin, J.-L. & Vos, M. H. (1994) Methods Enzymol. 232, 416–430. [DOI] [PubMed] [Google Scholar]
- 28.Byrdin, M., Brettel, K., Aubert, C., Eker, A. P. M. & Vos, M. H. (2002) in Flavins and Flavoproteins 2002, eds. Chapman, S., Perham, R. & Scrutton, N. (Rudolph Weber, Agency for Scientific Publications, Berlin), pp. 701–706.
- 29.Okamura, T., Sancar, A., Heelis, P. F., Hirata, Y. & Mataga, N. (1989) J. Am. Chem. Soc. 111, 5967–5969. [Google Scholar]
- 30.Gindt, Y. M., Vollenbroek, E., Westphal, K., Sackett, H., Sancar, A. & Babcock, G. T. (1999) Biochemistry 38, 3857–3866. [DOI] [PubMed] [Google Scholar]
- 31.Bent, D. V. & Hayon, E. (1975) J. Am. Chem. Soc. 97, 2599–2606. [DOI] [PubMed] [Google Scholar]
- 32.Moser, C. C. & Dutton, P. L. (1992) Biochim. Biophys. Acta 1101, 171–176. [PubMed] [Google Scholar]
- 33.Page, C. C., Moser, C. C., Chen, X. & Dutton, P. L. (1999) Nature 402, 47–52. [DOI] [PubMed] [Google Scholar]