

Abstract
The chemokine CXCL14/BRAK participates in immune surveillance by recruiting dendritic cells. CXCL14 gene expression is altered in a number of cancers, but protein expression levels have not been investigated. Here we report that CXCL14 protein can be expressed in primary epithelial cells, however in several immortalized and cancer cell lines this protein is targeted for polyubiquitylation and proteasomal degradation. We determined the NMR structure of CXCL14 to identify motifs controlling its expression. CXCL14 adopts the canonical chemokine tertiary fold but contains a unique five amino acid insertion (41VSRYR45) relative to other CXC chemokines. Deletion or substitution of key residues within this insertion prevented proteasomal degradation. Furthermore, we defined a 15 amino acid fragment of CXCL14 that is sufficient to induce proteasomal degradation. This study elucidates a post-translational mechanism for the loss of CXCL14 in cancer and a novel mode of chemokine regulation.
Keywords: NMR spectroscopy, CXCL14, chemokine, proteasome, cancer, ubiquitin
Introduction
Chemokines are a large family of chemotactic cytokines that play a pivotal role in a number of cancer-related processes including cell migration, metastasis and angiogenesis 1, 2, 3. Their participation in cancer metastasis is well documented and results from the altered expression patterns of G-protein coupled receptors (GPCR) common in metastatic cancers. For example, upregulation of the chemokine receptor CXCR4 in cancer cells promotes migration of metastatic breast, ovarian and malignant melanoma cells toward tissues, like lung, liver, lymph nodes and bone marrow, that constitutively express its cognate chemokine ligand CXCL12. Alternatively, tumors may avoid detection by the immune system and prevent an anti-tumor immune response by altering the local production of the chemokines themselves. CXCL14/BRAK is an orphan chemokine with tumor suppressive properties whose expression is altered in prostate cancer, head and neck squamous cell carcinomas, and some cervical squamous cell carcinomas 4, 5. Mechanistically, the tumor suppressive actions of CXCL14 are thought to occur through the inhibition of angiogenesis or the recruitment and activation of immature dendritic cells 6.
Previous studies probing CXCL14 expression patterns in several cancer types focused exclusively on mRNA levels without considering the regulation of CXCL14 at the protein level 7. Examining protein expression levels in this class of signaling molecules is important as cells can employ several regulatory methods to quickly modulate cytokines at the protein level. For example, TNFα is expressed as an inactive transmembrane protein that is activated when TNFα convertase (TACE) cleaves TNFα from its membrane anchor to yield a soluble protein 8. Similarly, TACE cleaves the membrane anchored chemokine CX3CL1/Fractalkine from its mucin stalk to yield soluble CX3CL1 9, 10. Determining the mechanisms used to regulate cytokines is critical to understanding their biological role.
Protein degradation is one post-translational mechanism that effectively deactivates regulatory molecules. The 26S proteasome is a large multi-subunit complex located in the nucleus and cytosol that accounts for the bulk of cellular protein turnover, and is mediated, in part, by ubiquitin modification of target proteins 11. Specific sequence determinants controlling ubiquitylation and turnover have been identified for a few substrate proteins. For example, conserved recognition sequences (DSGXXS) within the HIV VPU protein, ß-catenin and IκBα exhibit striking similarities 12, 13. Other sequence elements responsible for recognition by the Skp1-Cullin/Cdc53-F-box protein SCF E3 ubiquitin ligase complex, such as the p27Kip1 motif, found in cyclins D and E have also been well characterized 14, 15, 16. However, for the majority of proteins recognized by the ubiquitylation machinery the precise sequence and structural motifs have not been identified.
Thus far, chemokines have not been reported as targets for ubiquitin-mediated degradation. In this report, we show for the first time that a chemokine, CXCL14, is degraded by ubiquitin-mediated proteolysis in cancer and immortalized cells, but not in normal epithelial cells. We determined the structure of CXCL14 by NMR spectroscopy to identify structural elements that may play a role in regulating protein stability. Analysis of the CXCL14 structure and sequences from 15 CXC family members revealed an extended loop containing a five amino acid insertion unique to CXCL14. We used mutagenesis to characterize the potential role of this insertion in polyubiquitin mediated degradation. The structural data, coupled with a series of alanine substitution and truncation mutants, defined a 15 residue ‘destruction box’ within CXCL14 that was both necessary and sufficient for polyubiquitin mediated degradation, a finding novel for any member of the chemokine family. The upregulation/dysregulation of the proteolytic machinery in cancer provides further rationale for the therapeutic use of proteasome inhibitors.
Results
26S proteasome inhibition restores CXCL14 expression
To investigate the function of CXCL14 in prostate cancer progression we attempted to create cell lines overexpressing CXCL14. Initially, we were unable to identify overexpressing clones, even using an affinity purified rabbit anti-CXCL14 antibody capable of detecting sub-nanogram protein levels. We subsequently employed an adenoviral system to transduce normal, primary prostate epithelial cells (PrEC) and the LNCaP prostate cancer cell line. Conditioned media were assayed three days post-transduction for the presence of secreted CXCL14 protein by western blot analysis, and cells were harvested to determine the presence of CXCL14 mRNA. CXCL14 protein was readily detectable in the media from PrEC primary cells, but absent in the media collected from LNCaP cancer cell line (Figure 1A). In contrast, CXCL14 mRNA was present in both cell types (Figure 1B).
Figure 1.
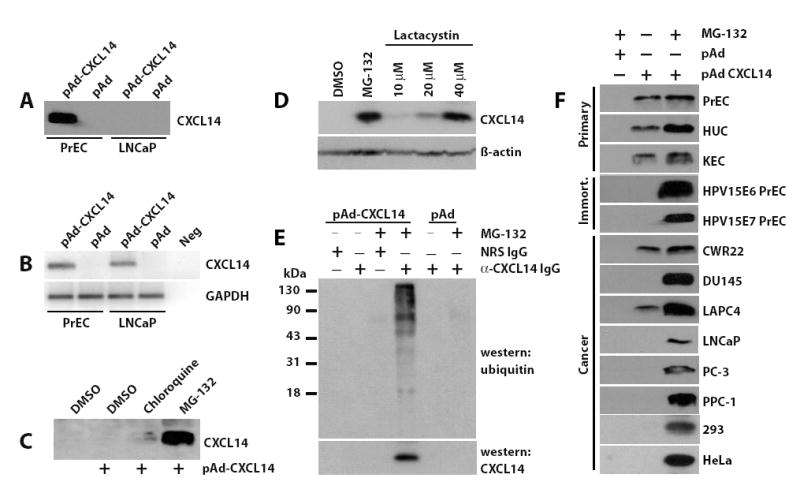
CXCL14 expression is regulated by the 26S proteasome and is a substrate for ubiquitylation. (a) Primary prostate epithelial cells (PrEC) and the prostate cancer cell line LNCaP were transduced for three days with CXCL14 expressing virus (pAd-CXCL14) or vector only virus (pAd). The conditioned media were analyzed by western blotting for CXCL14 expression. (b) Total RNA was isolated from the transduced cells, converted to cDNA and reverse-transcriptase PCR performed using primers designed to detect CXCL14 or GAPDH. PCR products were separated by agarose gel electrophoresis and detected by SYBR green staining. Neg, denotes the no DNA control. (c) LNCaP cells were transduced with CXCL14 expressing adenovirus (pAd-CXCL14) for 48 hours. Inhibitory doses of chloroquine (10 μM) and MG-132 (10 μM) were added for an additional 24 hours. The conditioned media were analyzed by western blotting for CXCL14 expression. Un-transduced LNCaP cells were included as a control treated with vehicle only (DMSO). (d) LNCaP cells expressing CXCL14 were treated for 24 hours with increasing concentrations of lactacystin. The conditioned media were analyzed by western blot analysis for CXCL14 expression. (e) LNCaP cells were transduced with CXCL14 expressing adenovirus (pAd-CXCL14) or vector alone. 48 hours post-transduction, cells were treated with 10 μM MG-132 or DMSO. Cells were harvested, lysed, and immunoprecipitated using the anti-CXCL14 antibody. Immune complexes were subjected to western blot analysis and probed with a mouse anti-ubiquitin or the rabbit anti-CXCL14 antibody. A high molecular weight complex was detected which is indicative of polyubiquitylated proteins. (f) Cells were transduced with vector (pAd) or CXCL14 expressing (pAd-CXCL14) adenovirus. After 48 hours post-transduction, cells were treated with MG-132 or DMSO. The media were analyzed for CXCL14 expression by western blot analysis. Without pAd-CXCL14 transduction the protein was undetectable in all cell lines. Primary cells utilized include: prostate epithelial cells, PrEC; human urothelial cells, HUC; and kidney epithelial cells, KEC. The immortalized PrEC lines include: HPV15E6, HPVE6 oncoprotein immortalized; and HPV15E7, HPVE7 oncoprotein immortalized. Cancer cell lines include: CWR22, DU145, LNCaP, LAPC4, PC-3, PPC-1, 293 and HeLa.
To test whether CXCL14 protein levels are regulated by post-translational mechanisms we used inhibitors of the two major protein degradation pathways. Chloroquine, an inhibitor of the lysosomal degradation pathway, and MG-132, a 26S proteasome inhibitor, were added to LNCaP cancer cells transduced with adenovirus capable of expressing CXCL14 (pAd-CXCL14) for 24 hours, and the medium from each culture was assayed for CXCL14 protein expression (Figure 1C). MG-132 treatment enabled robust CXCL14 expression, while chloroquine restored protein levels only minimally. To confirm that CXCL14 was in fact being targeted to the proteasome for degradation, we used a second more specific proteasome inhibitor, lactacystin, that does not inhibit thiol proteases and assayed cell lysates for expression 17. Lactacystin robustly conferred CXCL14 expression providing additional evidence of the involvement of the 26S proteasome and demonstrating that protein expression, not protein secretion, was impaired in the cancer cell lines (Figure 1D). Furthermore, polyubiquitylated CXCL14 was detected by western blot analysis using an anti-ubiquitin antibody in cell lysates from LNCaP cells treated with MG-132 and immunoprecipitated with an anti-CXCL14 antibody (Figure 1E), a characteristic feature of proteins degraded by the 26S proteasome. These data demonstrate that CXCL14 degradation in prostate cancer cells is mediated via polyubiquitylation and the 26S proteasome.
We next tested the ability of other epithelial cells to degrade CXCL14. Urogenital cell types were chosen, since we are focused on CXCL14 expression in prostate cancer. Three normal, primary epithelial cell types (prostate, urothelial and kidney), human papillomavirus (HPV) E6 and E7 oncogene immortalized PrEC lines and several other commonly used cancer cell lines were tested. All cell types were transduced with adenovirus capable of expressing CXCL14 and then treated with MG-132. The media collected from each cell line was analyzed for the presence of CXCL14 protein. All three primary cell lines readily expressed CXCL14 in the absence of proteasome inhibitors (Figure 1F). In contrast, MG-132 treatment was required to detect CXCL14 protein expression in the oncogene immortalized PrEC cells, 4 of 6 prostate cancer cell lines, and the 293 (kidney) and HeLa (cervical) cancer cell lines. The LAPC4 and CWR22 prostate cancer cell lines conferred CXCL14 protein expression in the absence of proteasome inhibition, a result similar to that of primary cell cultures. These observations indicate that normal cells have the capacity to express CXCL14, but that oncogene-mediated immortalization and transformation frequently initiates a process by which this chemokine is targeted to the ubiquitin-26S proteasome pathway.
CXCL14 structure determination
The ubiquitin mediated destruction of CXCL14 in cancer cells shown here is the first report of proteasomal targeting for any member of the chemokine family. In order to identify elements of CXCL14 that could enable polyubiquitylation, we constructed a sequence alignment of the 15 CXC chemokines (Figure 2). Comparison of CXCL14 with the other family members revealed a unique sequence, 41VSRYR45, that we speculated might participate in ubiquitin mediated degradation. This unusual sequence element was further examined by determining the three-dimensional structure of CXCL14 by NMR spectroscopy and probing the structural role of the VSRYR loop using a series of alanine replacement and truncation mutants.
Figure 2.

Amino acid sequence alignment of the human CXC chemokine family. CXC family members lacking the secretory signal pre-sequence were aligned using the ClustalW multiple sequence alignment tool. The conserved cysteine residues that define this family are shaded in gray. Disulfide linkages are indicated and residue numbering corresponds to CXCL14. CXC chemokines with an extended N- or C-terminus are denoted with an asterisk. CXCL16 was not included as it is a membrane protein.
Isotopically-labeled [U-15N/13C]-CXCL14 was bacterially expressed, oxidized and purified using a modified protocol previously described for the chemokines lymphotactin (XCL1) and stromal cell-derived factor 1-α (CXCL12) 18. Inspection of a 2D 15N-1H HSQC NMR spectrum of purified CXCL14 (Figure 3A) revealed a folded domain amenable to structural studies based on the number and uniformity of the resonances. The structure of CXCL14 is shown in Figure 3B and the structural statistics for the ensemble are presented in Table 1. CXCL14 adopts the canonical chemokine fold consisting of an N-terminal loop stabilized by two disulfide bonds, a three-stranded antiparallel β-sheet packed against a C-terminal α-helix and a single turn of 310 helix just prior to β-strand 1. The CXCL14 structure ensemble is characterized by backbone RMSD values of ~0.5 Å for most residues, excluding the N-loop (residues 1-13) and the extended 40s loop (residues 40-47) (Figure 3C). Low backbone RMSD values and the high heteronuclear 15N-1H NOE values measured for the majority of the residues are consistent with a well ordered protein (Figure 3C), though two regions of conformational disorder are present. The 15N-1H heteronuclear NOE values for N-loop residues 7-11 indicate significant flexibility on the pico- to nanosecond timescale, consistent with the disorder observed in the ensemble. In contrast, residues 41-48 appear disordered in the structural ensemble but display relatively high 15N-1H heteronuclear NOE values and broad resonances in the 15N-1H HSQC. These results are consistent with slow (μs–ms) conformational exchange in the 40s loop.
Figure 3.
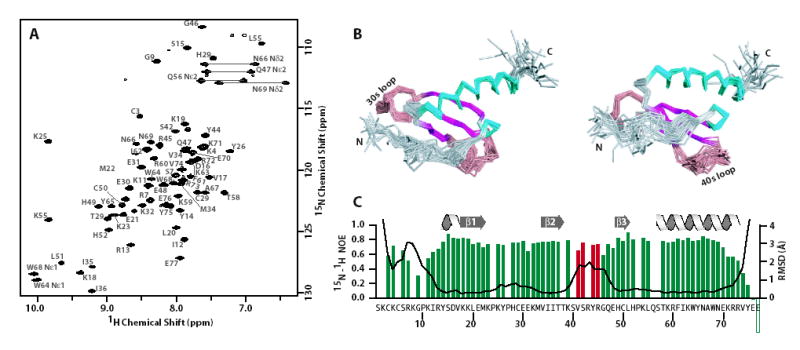
NMR data and structure of CXCL14. (a) Two-dimensional 1H-15N spectrum for [U-13C/15N]-CXCL14. (b) Ensemble of the 20 conformers shown as a Cα trace. The left and right panels are related by at 45° rotation about the horizontal axis. The loop, β-sheet and α-helical elements are shown in gray/salmon, pink and blue, respectively. (c) Backbone (N, Ca, C′) RMSD values (black line) and 1H-15N heteronuclear NOE values (green and red bars) plotted for each residue of CXCL14. Red bars denote the 41VSRYR45 insertion and highlights the discrepancy between the backbone RMSD and 1H-15N heteronuclear NOE values.
Table 1.
Statistics for the 20 CXCL14 conformers.
| Experimental constraints | ||
| Distance constraints | ||
| Long | 209 | |
| Medium [1<(i–j)≤5] | 133 | |
| Sequential [(i–j)=1] | 186 | |
| Intraresidue [i=j] | 366 | |
| Total | 894 | |
| Dihedral angle constraints (φ and ψ) | 97 | |
| Average atomic R.M.S.D. to the mean structure (Å) | ||
| Residues 13-40, 49-73 | ||
| Backbone (Cα, C′, N) | 0.49 ± 0.08 | |
| Heavy atoms | 1.00 ± 0.11 | |
| Deviations from idealized covalent geometry | ||
| Bond lengths | RMSD (Å) | 0.015 |
| Torsion angle violations | RMSD (°) | 1.4 |
| WHATCHECK quality indicators | ||
| Z-score | −2.83 ± 0.40 | |
| RMS Z-score | ||
| Bond lengths | 0.74 ± 0.02 | |
| Bond angles | 0.79 ± 0.03 | |
| Bumps | 0 ± 0 | |
| Lennard-Jones energya (kJ mol−1) | −1,690 ± 61 | |
| Constraint violations | ||
| NOE distance | Number > 0.5 Å | 0 ± 0 |
| NOE distance | RMSD (Å) | 0.024 ± 0.002 |
| Torsion angle violations | Number > 5 ° | 0.3 ± 0.66 |
| Torsion angle violations | RMSD (°) | 0.958 ± 0.192 |
| Ramachandran statistics (% of all residues) | ||
| Most favored | 84.3 ± 3.6 | |
| Additionally allowed | 10.1 ± 3.2 | |
| Generously allowed | 3.2 ± 1.7 | |
| Disallowed | 2.4 ± 2.1 | |
Nonbonded energy was calculated in XPLOR-NIH.
The CXCL14 sequence is clearly unusual in that it lacks N-terminal residues essential for biological activity of other CXC chemokines and contains a unique insertion (41VSRYR45) between strands β2 and β3 (the 40s loop) (Figure 2). Despite these differences, the CXCL14 structure resembles other chemokines like interleukin-8 (IL-8/CXCL8) (Figure 4A). On close inspection, however, we noticed significant differences in the positioning of the N-loop, a conserved structural element connecting the N-terminal cysteines to the first strand of the β-sheet. The N-loop is well defined in most chemokine structures owing to extensive hydrophobic interactions with residues of the C-terminal α-helix. In IL-8, for example, P16 packs against the aromatic side chain of W57 near the top of the C-terminal helix (Figure 4B) 19. These contacts are absent in CXCL14, and 15N-1H NOE values show that the corresponding residues (7RKGPK11) are mobile on ps-ns timescales (Figure 3C). As in all other chemokine structures, a 310 helix (residues 14YSD16) connects the N-loop to β-strand 1 and packs tightly against the long α-helix. This anchoring occurs through an aromatic clamp formed by F61 and W64 stacking on either side of the Y14 phenyl ring (Figure 4B). By compensating for the loss of other stabilizing contributions from the flexible N-loop residues in CXCL14, the aromatic clamp may help to maintain the chemokine fold.
Figure 4.
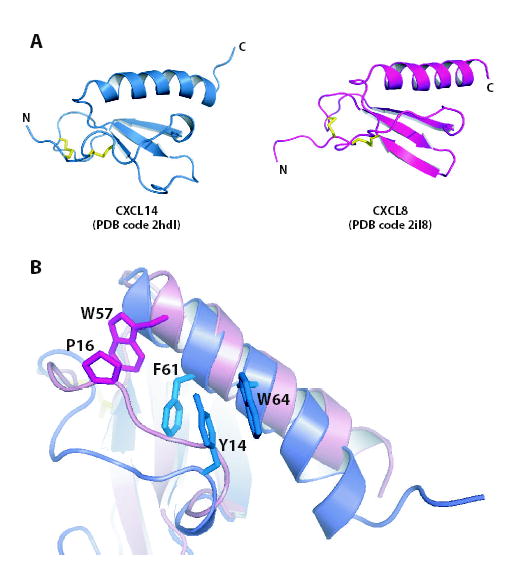
Comparison of CXCL14 and CXCL8. (a) Ribbon diagrams of CXCL14 (PDB entry 2hdl) and CXCL8 (PDB entry 1icw). Disulfide bonds are shown in yellow. (b) Overlay of CXCL14 and CXCL8 colored as in (a). Residues anchoring the N-loop to the C-terminal α-helix are shown as sticks.
Most CXC chemokines form dimers in solution with Kd values ranging from ~0.5–200 μM depending on the solution conditions 20, 21, 22. The oligomeric state of CXCL14 was probed using a series of 2D 15N-1H HSQC NMR spectra collected under varying conditions of 0 to 200 mM sodium chloride, protein concentrations ranging from 0.01 to 2 mM, and pH ranging from 5.0 to 7.4. Inspection of the resulting data suggested that CXCL14 exists only as a monomer under the solution conditions tested (data not shown).
Identification of the CXCL14 ‘Destruction Box’
To determine the sequence elements responsible for regulating CXCL14 destruction in cancer cells, green fluorescent protein (GFP) was N-terminally fused to various CXCL14 constructs as previously described 23. In this system, degradation of the fusion protein eliminates the GFP signal. LNCaP cells expressing GFP fused to mature CXCL14 (GFP-CXCL141-77) exhibited a significantly reduced fluorescence compared to GFP controls reflecting CXCL14-targeted degradation of the fusion protein (Figure 5A and B). Application of proteasome inhibitors restored the fluorescence intensity of the fusion protein (data not shown).
Figure 5.
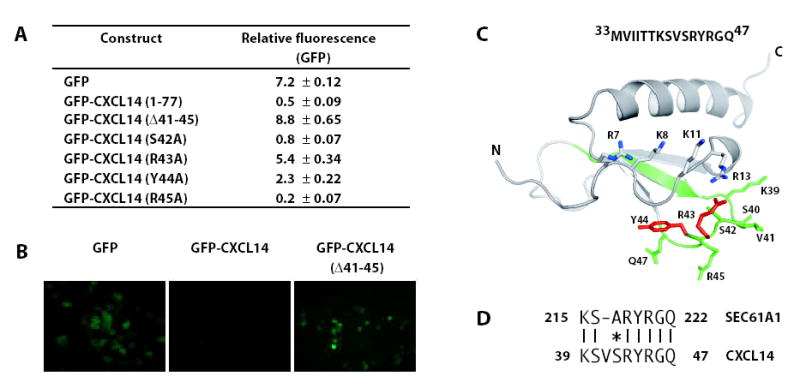
Functional analysis of the CXCL14 extended loop. (a) Modified CXCL14 constructs were generated and fused to GFP. The amino acid numbering reflects that of the mature chemokine. Flow cytometry was used to determine the average fluorescence intensity value in the transfected cells. The average of three experiments ± the standard deviation after standardizing for the transfection efficiency is shown. (b) Fluorescent microscopy images of cells expressing the indicated constructs. (c) The 15 amino acid sequence corresponding to the ‘destruction box’ is shown and highlighted in green on the three-dimensional structure of CXCL14. The 9 residues aligning with the SEC61A1 peptide, which includes the 5 residues insertion unique to CXCL14, are shown as sticks. Residues that restored the GFP signal when mutated to alanine are highlighted in Red. Basic residues in the N-loop are shown as sticks. (d) A region of the CXCL14 destruction domain aligns with highest identity to human SEC61A1, a target for proteolysis mediated through UBC6, a ubiquitin-conjugating enzyme involved in the degradation of proteins from the ER.
To test the functional role of the extended loop, the 41VSRYR45 sequence was deleted from the GFP-CXCL14 fusion. Removal of these five amino acids produced a marked increase in GFP fluorescence (Figure 5A). Next, site-directed mutagenesis was performed to identify the critical residues for this activity. Residues from S42 to R45 were individually mutated to alanine. Valine 41 was not changed since mouse CXCL14 contains a methionine at this position and is also efficiently degraded (data not shown). The S42A and R45A mutations failed to restore the GFP signal (Figure 5A). In contrast, the R43A substitution, and to a lesser extent Y44A, restored the GFP signal. These results identify specific residues within CXCL14 40s loop insertion that are necessary for proteolysis to occur.
Using the GFP fusion reporter assay we set out to determine the minimal amino acid sequence necessary and sufficient for promoting CXCL14 degradation. A series of N- and C-terminal CXCL14 deletion constructs were generated, transfected into cells and the GFP fluorescence intensity measured (Table 2). A 15 amino acid peptide corresponding to CXCL14 residues 33MVIITTKSVSRYRGQ47 fused to GFP was sufficient to mediate the loss of GFP signal. Consistent with this finding and the GFP-CXCL14Δ41-45 data, the GFP fusion construct lacking the 15 amino acids (GFP-CXCL14Δ33-47) was stable, identifying this region as a ‘destruction box’ containing the information necessary and sufficient to mediate proteasomal degradation.
Table 2.
Analysis of GFP-CXCL14 truncation/deletion mutants.
| Construct | Relative fluorescence (GFP) |
|---|---|
| GFP | 7.2 ± 0.12 |
| GFP-CXCL14 (1-77) | 0.5 ± 0.09 |
| GFP-CXCL14 (29-77) | 0.8 ± 0.04 |
| GFP-CXCL14 (39-77) | 4.0 ± 0.31 |
| GFP-CXCL14 (29-67) | 0.6 ± 0.09 |
| GFP-CXCL14 (29-57) | 0.6 ± 0.14 |
| GFP-CXCL14 (29-47) | 0.4 ± 0.06 |
| GFP-CXCL14 (29-42) | 9.2 ± 0.16 |
| GFP-CXCL14 (33-47) | 0.5 ± 0.04 |
| GFP-CXCL14 (41-45) | 2.9 ± 0.07 |
| GFP-CXCL14 (Δ33-47) | 7.6 ± 0.38 |
Discussion
These studies provide the first insight into the regulation of CXCL14 at the protein level. Proteasomal degradation of CXCL14 is the first report of ubiquitin-mediated regulation of any chemokine. The ability to recognize and destroy this chemokine is profound and restricted to transformed cells. Each of the three normal epithelial cell types we employed (prostate, urothelial and kidney) expressed CXCL14 protein consistently. In contrast, eight of ten epithelial cell lines that were either immortalized with an oncoprotein or were of cancer origin degraded CXCL14 below detectable levels in the absence of 26S proteasome inhibitors. These data suggest that CXCL14 ubiquitylation is commonly activated in cancer or transformed cells. Biologically, the tight regulation of CXCL14 protein levels would provide an effective means of controlling CXCL14 signaling and could help tumors avoid detection by the immune system.
CXCL14 inhibits angiogenesis 6 and when overexpressed suppresses tumor growth 24, 25. The absence of detectable CXCL14 protein in head and neck and prostate cancer suggests that the loss of CXCL14 expression may be an important step in cancer progression. Normal CXCL14 production would promote tumor infiltration by dendritic cells that can initiate an anti-tumor immune response 26. Alternatively, the observed proteasomal degradation of CXCL14 may reflect the induction of a specific degradation pathway in transformed cells that also targets a subset of proteins that remain to be identified. For example, the degradation of CXCL14 in cancer cells may represent an induction of endoplasmic reticulum (ER) stress pathways which are hallmarked by enhanced proteasomal degradation of proteins in the secretory pathway 27. Further investigation of the CXCL14 degradation pathway should provide insight into the mechanisms used by transformed cells.
The differential pattern of CXCL14 expression in transformed and non-transformed cells suggests that the degradation is regulated. Protein phosphorylation works jointly with ubiquitylation to regulate proteolysis. In preliminary studies, we have shown that the protein kinase inhibitor staurosporine confers CXCL14 expression (data not shown). Our ongoing studies indicate that CXCL14 is not a direct target of phosphorylation, suggesting that the requisite protein kinase activity targets proteins within the ubiquitin-dependent degradation machinery. The activation of a specific kinase may therefore be indirectly responsible for selective CXCL14 degradation.
The structure determined for CXCL14 revealed that this protein adopts the canonical chemokine fold. Comparisons with other CXC chemokines revealed several features unique to CXCL14 including a truncated N-terminus, an extended 40s loop, an aromatic rich α-helix and a disordered N-loop. Insertion of the 41VSRYR45 sequence in the 40s loop between strands β2 and β3 does not alter the canonical chemokine fold, but instead presents these residues in a flexible, solvent exposed form that could be readily accessible to an E3 ubiquitin ligase. This loop is essential for efficient proteasomal degradation of CXCL14, in particular residues R43 and Y44. While the VSRYR motif in isolation cannot induce proteasomal degradation, a larger CXLC14 fragment encompassing this loop (residues 33–47) was sufficient to act as a functional ‘destruction box’. Our truncation data suggests that one or more residues in the β2 strand (residues 34–38) are required for efficient targeting of CXCL14 to the proteasome. Side chains from β2 that are embedded within in the CXCL14 tertiary structure are unlikely to play a role, but I35 and T37 are solvent exposed and could participate in the recruitment of an E3 ubiquitin ligase (Figure 5C).
The flexibility of the N-loop in CXCL14 is unusual in comparison to other chemokine structures. While the N-loop and the extended 40s loop are distant in primary sequence, together they combine to form a surface with enhanced structural plasticity that may be important for recognition by the ubiquitylation machinery. One aspect of BRAK degradation that remains unresolved is the site of ubiquitylation. The destruction box (residues 33–47) contains only one lysine residue, K39, but alanine substitution at this position had no effect on proteasomal degradation (data not shown). Thus the 40s loop seems to serve as a recognition element rather than a direct substrate for ubiquitin ligation. In contrast, the N-loop contains four basic residues, including two lysines, K8 and K11 (Figure 5C), either of which could serve as targets for ubiquitin conjugation leading to the proteasomal degradation of CXCL14. While we have demonstrated the importance of residues from the 41VSRYR45 motif, the role of the nearby N-loop remains to be investigated.
The region of CXCL14, 33MVIITTKSVSRYRGQ47, that we identified as necessary and sufficient for proteasomal targeting (Figure 5C) was compared with the NCBI human protein database to identify similar sequences in other proteins. The best match was SEC61A1 (Figure 5D), which contains a sequence nearly identical to CXCL14 residues 39–47. The similarity may be meaningful, since SEC61 is a target for proteolysis mediated through UBC6, a ubiquitin-conjugating enzyme involved in the degradation of proteins from the ER 28. It is unknown whether the novel 41VSRYR45 insertion is necessary for chemotactic activity or serves exclusively as a regulatory motif, but its novelty suggests that as a substrate for ubiquitin-mediated proteasomal degradation CXCL14 may be unique in the chemokine family.
The current findings have implications for the therapeutic use of 26S proteasome inhibitors in the treatment of cancer. Differential regulation of the proteasomal machinery in cancer and normal cell types has been described previously, and our results demonstrate the aberrant ubiquitin-mediated degradation of CXCL14 in transformed cells. This difference is exploited by the chemotherapeutic agent Velcade (reviewed in 29), an inhibitor of the 26S proteasome, which markedly increases the susceptibility of cancer cells to apoptosis and inhibits angiogenesis 30. Our results suggest a mechanistic rationale for the use of proteasome inhibitors on tumors containing activated protein degradation machinery. Furthermore, elucidating the pathways involved in CXCL14 proteolysis will undoubtedly identify further differences in the 26S proteasome machinery between normal and cancer cells and offer new avenues for the development of chemotherapeutic agents.
Materials and Methods
Cloning
Full-length human CXCL14 (GenBank accession BC003513) was cloned into the pENTR/D-TOPO vector (Invitrogen) and recombined into the adenoviral vector pAd/CMV/V5-DEST (Invitrogen). The pEXPRESS GFP expression vector 31 was used to generate CXCL14-GFP fusion proteins. All CXCL14 constructs were cloned in frame to the GFP C-terminus. Site-directed mutations were created using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). For the production of recombinant CXCL14 protein, CXCL14 was cloned into a modified pQE30 vector (Qiagen, Valencia, CA) 18. To accommodate the TEV protease recognition sequence, a glycine was appended to the amino terminus of mature CXCL14. Isotopically-labeled proteins for NMR were produced using M9 minimal media containing 15N-ammonium chloride and/or 13C-glucose as the sole nitrogen and carbon sources. All constructs were verified by sequence analysis 32.
Protein expression, purification and refolding
The CXCL14 expression plasmid, pQE30T-CXCL14, was transformed into E. coli strain SG13009[pRPEP4] (Qiagen). Protein expression was then induced by the addition of isopropyl-β-D-thiogalactopyranoside for five hours at 37ºC. Recombinant CXCL14 was found exclusively in the insoluble fraction of the harvested cells. Protein was isolated, purified by metal affinity chromatography and separated from the octa-His affinity tag 18. Formation of the two CXCL14 disulfide bonds was accomplished by dropwise addition of the cleaved protein into 200 ml of oxidation buffer (50 mM Tris, pH 8.0, 200 mM NaCl), followed by dialysis of the oxidation reaction against 4 liters of oxidation buffer. Oxidized CXCL14 was concentrated and separated from aggregated protein on a HiTrapS column (Pharmacia, Piscataway, NJ) using a 0.5 – 1 M sodium chloride gradient in 100 mM sodium phosphate pH 6.5. Fractions containing CXCL14 were pooled and purified to homogeneity by reversed-phased high performance liquid chromatography. The purity and identity of all CXCL14 variants were confirmed by matrix-assisted laser desorption ionization mass spectrometry.
Antibody production and purification
The Polyclonal Antibody Service at the University of Wisconsin Medical School Laboratory Animal Resources generated a polyclonal antibody to mature CXCL14 using standard procedures. An IgG fraction was purified using ImmunoPure IgG Binding Kit (Pierce Biotechnology, Rockford, IL). The IgG fraction was then affinity-purified over a CXCL14 affinity column generated using the Aminolink Plus Immobilization Kit (Pierce Biotechnology) and desalted with a PD-10 gel filtration column (Amersham Bioscience, Piscataway, NJ).
Adenovirus generation and transduction
Virus was generated with the ViraPower adenoviral expression system (Invitrogen). Viral stocks were titered using plaque forming assays. All transductions in this study were carried out with a multiplicity of infection of 1.
Tissue acquisition and cell culture
All human tissue was acquired under Internal Review Board approval. Primary cells were cultured from human prostate, ureter and kidney explants as previously described 33, 34. Transformation and culture of the Human Papillomavirus E6 (HPVE6) and Human Papillomavirus E7 (HPVE7) oncoprotein immortalized prostate epithelial cell lines has been described 33. Prostate cancer cell lines were obtained from ATCC (Manassas, VA) and maintained in DMEM with 5% FCS. MG-132 was obtained from Calbiochem (San Diego, CA). Chloroquine and Lactacystin were purchased from Sigma (St. Louis, MO).
Western blot analysis
The conditioned media were precipitated with 10% trichloroacetic acid. The precipitate was solubilized with 1 × SDS buffer loading dye prior to western analysis. Intracellular protein was obtained as previously described with the exception that a ubiquitin protein extraction buffer (UPEB; 150 mM NaCl, 50 mM Tris-HCl (pH 7.5), 5 mM EDTA, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, and 10 nM MG-132) was used 35. Antibodies were obtained from the following sources: CXCL14, generated here; α-tubulin, Oncogene (AB-1); anti-Ubiquitin monoclonal, Santa Cruz Biotechnology (Santa Cruz, CA, P4D1).
Ubiquitin co-immunoprecipitation
LNCaP cells (3 × 106/plate) were lysed in UPEB and homogenized through a 23-gauge needle. The homogenate was cleared by centrifugation followed by the addition of protein A/G Sepharose (Pierce Biotechnology) and the pre-bleed rabbit IgG fraction. To the pre-cleared lysate, 0.5 μg/ml of rabbit anti-CXCL14 antibody and protein A/G Sepharose was added and the samples were mixed at 4°C for two hours. The immune complexes were washed twice in UPEB.
NMR spectroscopy
NMR samples were prepared in buffers containing 100 mM sodium phosphate, pH 6.5, 150 mM sodium chloride, 0.02% sodium azide and 10% 2H2O. All NMR data were acquired at 25 °C on a Bruker 600 MHz spectrometer equipped with a triple-resonance CryoProbeTM and processed with NMRPipe software 36. Backbone 1H, 15N and 13C resonance assignments were obtained in an automated manner using the program Garant 37, with peaklists from 3D HNCO, HNCACO, HNCA, HNCOCA, HNCACB, CCONH and 2D 1H/15N HSQC spectra generated manually with XEASY 38 or automatically with SPSCAN. Sidechain assignments were completed manually from 3D HCCONH, HCCH-TOCSY and 13C(aromatic)-edited NOESY-HSQC spectra.
Structural calculation and analysis
Distance constraints were obtained from 3D 15N-edited NOESY-HSQC, 13C-edited NOESY-HSQC, and 13C(aromatic)-edited NOESY-HSQC spectra (τmix = 80 ms). Backbone ψ and φ dihedral angle constraints were generated from secondary shifts of the 1Hα, 13Cα, 13Cβ, 13C′, and 15N nuclei using the program TALOS 39. Initial structures were generated in an automated manner using the NOEASSIGN module of the torsion angle dynamics program CYANA 40, followed by iterative manual refinement to eliminate constraint violations. The 20 conformers with the lowest target function were chosen for further refinement by X-PLOR, in which physical force field terms and explicit water solvent molecules were added to the experimental constraints 41. The coordinates for these structural models, along with the restraints employed, have been deposited in the Protein Data Bank (PDB entry 2hdl) and at the Biological Magnetic Resonance Data Bank (BMRB entry 7229).
GFP reporter assays
LNCaP cells were transfected with Effectene (Qiagen, Valencia, CA). Cells were harvested 24 hours post-transfection and analyzed by flow cytometry for GFP expression (FACScan, BD Biosciences, San Jose, CA). Using FlowJo software (BD Biosciences), the percentage of cells transfected was determined. To quantitate the ability of GFP-CXCL14 fusion proteins to be degraded, the average fluorescence intensity value in the transfected cells was first determined and then standardized for transfection efficiency. Each construct was assayed in triplicate and averaged.
Acknowledgments
We thank Dr. Rolf Jakobi for providing the pEXPRESS GFP plasmid. This work was supported in part by NIH grant AI058072 to BFV.
Footnotes
Coordinates and related data have been deposited at PDB (2HDL) and NMR data at BMRB (7229).
References
- 1.Rossi D, Zlotnik A. The biology of chemokines and their receptors. Annu Rev Immunol. 2000;18:217–42. doi: 10.1146/annurev.immunol.18.1.217. [DOI] [PubMed] [Google Scholar]
- 2.Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verastegui E, Zlotnik A. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–6. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 3.Belperio JA, Keane MP, Arenberg DA, Addison CL, Ehlert JE, Burdick MD, Strieter RM. CXC chemokines in angiogenesis. J Leukoc Biol. 2000;68:1–8. [PubMed] [Google Scholar]
- 4.Schwarze SR, Luo J, Isaacs WB, Jarrard DF. Modulation of CXCL14 (BRAK) expression in prostate cancer. Prostate. 2005;64:67–74. doi: 10.1002/pros.20215. [DOI] [PubMed] [Google Scholar]
- 5.Shurin GV, Ferris RL, Tourkova IL, Perez L, Lokshin A, Balkir L, Collins B, Chatta GS, Shurin MR. Loss of new chemokine CXCL14 in tumor tissue is associated with low infiltration by dendritic cells (DC), while restoration of human CXCL14 expression in tumor cells causes attraction of DC both in vitro and in vivo. J Immunol. 2005;174:5490–8. doi: 10.4049/jimmunol.174.9.5490. [DOI] [PubMed] [Google Scholar]
- 6.Shellenberger TD, Wang M, Gujrati M, Jayakumar A, Strieter RM, Burdick MD, Ioannides CG, Efferson CL, El-Naggar AK, Roberts D, Clayman GL, Frederick MJ. BRAK/CXCL14 is a potent inhibitor of angiogenesis and a chemotactic factor for immature dendritic cells. Cancer Res. 2004;64:8262–70. doi: 10.1158/0008-5472.CAN-04-2056. [DOI] [PubMed] [Google Scholar]
- 7.Frederick MJ, Henderson Y, Xu X, Deavers MT, Sahin AA, Wu H, Lewis DE, El-Naggar AK, Clayman GL. In vivo expression of the novel CXC chemokine BRAK in normal and cancerous human tissue. Am J Pathol. 2000;156:1937–50. doi: 10.1016/S0002-9440(10)65067-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–33. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 9.Tsou CL, Haskell CA, Charo IF. Tumor necrosis factor-alpha-converting enzyme mediates the inducible cleavage of fractalkine. J Biol Chem. 2001;276:44622–6. doi: 10.1074/jbc.M107327200. [DOI] [PubMed] [Google Scholar]
- 10.Garton KJ, Gough PJ, Blobel CP, Murphy G, Greaves DR, Dempsey PJ, Raines EW. Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1) J Biol Chem. 2001;276:37993–8001. doi: 10.1074/jbc.M106434200. [DOI] [PubMed] [Google Scholar]
- 11.Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 12.Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. beta-catenin is a target for the ubiquitin-proteasome pathway. Embo J. 1997;16:3797–804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DiDonato J, Mercurio F, Rosette C, Wu-Li J, Suyang H, Ghosh S, Karin M. Mapping of the inducible IkappaB phosphorylation sites that signal its ubiquitination and degradation. Mol Cell Biol. 1996;16:1295–304. doi: 10.1128/mcb.16.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsvetkov LM, Yeh KH, Lee SJ, Sun H, Zhang H. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr Biol. 1999;9:661–4. doi: 10.1016/s0960-9822(99)80290-5. [DOI] [PubMed] [Google Scholar]
- 15.Won KA, Reed SI. Activation of cyclin E/CDK2 is coupled to site-specific autophosphorylation and ubiquitin-dependent degradation of cyclin E. Embo J. 1996;15:4182–93. [PMC free article] [PubMed] [Google Scholar]
- 16.Diehl JA, Zindy F, Sherr CJ. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 1997;11:957–72. doi: 10.1101/gad.11.8.957. [DOI] [PubMed] [Google Scholar]
- 17.Kisselev AF, Goldberg AL. Proteasome inhibitors: from research tools to drug candidates. Chem Biol. 2001;8:739–58. doi: 10.1016/s1074-5521(01)00056-4. [DOI] [PubMed] [Google Scholar]
- 18.Peterson FC, Elgin ES, Nelson TJ, Zhang F, Hoeger TJ, Linhardt RJ, Volkman BF. Identification and characterization of a glycosaminoglycan recognition element of the C chemokine lymphotactin. J Biol Chem. 2004;279:12598–604. doi: 10.1074/jbc.M311633200. [DOI] [PubMed] [Google Scholar]
- 19.Clore GM, Appella E, Yamada M, Matsushima K, Gronenborn AM. Three-dimensional structure of interleukin 8 in solution. Biochemistry. 1990;29:1689–96. doi: 10.1021/bi00459a004. [DOI] [PubMed] [Google Scholar]
- 20.Lowman HB, Fairbrother WJ, Slagle PH, Kabakoff R, Liu J, Shire S, Hebert CA. Monomeric variants of IL-8: effects of side chain substitutions and solution conditions upon dimer formation. Protein Sci. 1997;6:598–608. doi: 10.1002/pro.5560060309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Veldkamp CT, Peterson FC, Pelzek AJ, Volkman BF. The monomer-dimer equilibrium of stromal cell-derived factor-1 (CXCL 12) is altered by pH, phosphate, sulfate, and heparin. Protein Sci. 2005;14:1071–81. doi: 10.1110/ps.041219505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holmes WD, Consler TG, Dallas WS, Rocque WJ, Willard DH. Solution studies of recombinant human stromal-cell-derived factor-1. Protein Expr Purif. 2001;21:367–77. doi: 10.1006/prep.2001.1402. [DOI] [PubMed] [Google Scholar]
- 23.Fiebiger E, Hirsch C, Vyas JM, Gordon E, Ploegh HL, Tortorella D. Dissection of the dislocation pathway for type I membrane proteins with a new small molecule inhibitor, eeyarestatin. Mol Biol Cell. 2004;15:1635–46. doi: 10.1091/mbc.E03-07-0506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwarze SR, Luo J, Isaacs WB, Jarrard DF. Modulation of CXCL14 (BRAK) expression in prostate cancer. Prostate. 2005 doi: 10.1002/pros.20215. [DOI] [PubMed] [Google Scholar]
- 25.Shurin GV, Ferris R, Tourkova IL, Perez L, Lokshin A, Balkir L, Collins B, Chatta GS, Shurin MR. Loss of new chemokine CXCL14 in tumor tissue is associated with low infiltration by dendritic cells (DC), while restoration of human CXCL14 expression in tumor cells causes attraction of DC both in vitro and in vivo. J Immunol. 2005;174:5490–8. doi: 10.4049/jimmunol.174.9.5490. [DOI] [PubMed] [Google Scholar]
- 26.Fushimi T, O’Connor TP, Crystal RG. Adenoviral gene transfer of stromal cell-derived factor-1 to murine tumors induces the accumulation of dendritic cells and suppresses tumor growth. Cancer Res. 2006;66:3513–22. doi: 10.1158/0008-5472.CAN-05-1493. [DOI] [PubMed] [Google Scholar]
- 27.Kadowaki H, Nishitoh H, Ichijo H. Survival and apoptosis signals in ER stress: the role of protein kinases. J Chem Neuroanat. 2004;28:93–100. doi: 10.1016/j.jchemneu.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 28.Biederer T, Volkwein C, Sommer T. Degradation of subunits of the Sec61p complex, an integral component of the ER membrane, by the ubiquitin-proteasome pathway. Embo J. 1996;15:2069–76. [PMC free article] [PubMed] [Google Scholar]
- 29.Adams J. The proteasome: a suitable antineoplastic target. Nat Rev Cancer. 2004;4:349–60. doi: 10.1038/nrc1361. [DOI] [PubMed] [Google Scholar]
- 30.Drexler HC. Activation of the cell death program by inhibition of proteasome function. Proc Natl Acad Sci U S A. 1997;94:855–60. doi: 10.1073/pnas.94.3.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jakobi R, McCarthy CC, Koeppel MA. Mammalian expression vectors for epitope tag fusion proteins that are toxic in E. coli. Biotechniques. 2002;33:1218–20. 1222. doi: 10.2144/02336bm07. [DOI] [PubMed] [Google Scholar]
- 32.Maniatis T, Fritsch EF, Sambrook J. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1986. [Google Scholar]
- 33.Jarrard DF, Sarkar S, Shi Y, Yeager TR, Magrane G, Kinoshita H, Nassif N, Meisner L, Newton MA, Waldman FM, Reznikoff CA. p16/pRb pathway alterations are required for bypassing senescence in human prostate epithelial cells. Cancer Res. 1999;59:2957–64. [PubMed] [Google Scholar]
- 34.Yeager TR, DeVries S, Jarrard DF, Kao C, Nakada SY, Moon TD, Bruskewitz R, Stadler WM, Meisner LF, Gilchrist KW, Newton MA, Waldman FM, Reznikoff CA. Overcoming cellular senescence in human cancer pathogenesis. Genes Dev. 1998;12:163–74. doi: 10.1101/gad.12.2.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwarze SR, Shi Y, Fu VX, Watson PA, Jarrard DF. Role of cyclin-dependent kinase inhibitors in the growth arrest at senescence in human prostate epithelial and uroepithelial cells. Oncogene. 2001;20:8184–92. doi: 10.1038/sj.onc.1205049. [DOI] [PubMed] [Google Scholar]
- 36.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 37.Bartels C, Billeter M, Güntert P, Wüthrich K. Automated Sequence-specific NMR Assignments of Homologous Proteins using the program GARANT. J Biomol NMR. 1996;7:207–213. doi: 10.1007/BF00202037. [DOI] [PubMed] [Google Scholar]
- 38.Bartels C, Xia TH, Billeter M, Güntert P, Wüthrich K. The Program XEASY for Computer-Supported NMR Spectral Analysis of Biological Macromolecules. J Biomol NMR. 1995;5:1–10. doi: 10.1007/BF00417486. [DOI] [PubMed] [Google Scholar]
- 39.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 40.Herrmann T, Guntert P, Wuthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol. 2002;319:209–27. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- 41.Linge JP, Williams MA, Spronk CA, Bonvin AM, Nilges M. Refinement of protein structures in explicit solvent. Proteins. 2003;50:496–506. doi: 10.1002/prot.10299. [DOI] [PubMed] [Google Scholar]