

Abstract
Diseases of the brain and spinal cord represent especially daunting challenges for cell-based strategies of repair, given the multiplicity of cell types within the adult central nervous system, and the precision with which they must interact in both space and time. Nonetheless, a number of diseases are especially appropriate for cell-based therapy, in particular those in which single phenotypes are lost, and in which the re-establishment of vectorially specific connections is not entirely requisite for therapeutic benefit. We review here a set of potential therapeutic indications that meet these criteria as potentially benefiting from the transplantation of neural stem and progenitor cells. These include: (i) transplantation of phenotypically restricted neuronal progenitor cells into diseases of a single neuronal phenotype, such as Parkinson's disease; (ii) implantation of mixed progenitor pools into diseases characterized by the loss of a limited number of discrete phenotypes, such as spinal cord injury and the motor neuronopathies; (iii) transplantation of glial and nominally oligodendrocytic progenitor cells as a means of treating disorders of myelin; and (iv) transplantation of neural stem cells as a means of treating lysosomal storage disorders and other diseases of enzymatic deficiency. Among the diseases potentially approachable by these strategies, the myelin disorders, including the paediatric leucodystrophies as well as adult traumatic and inflammatory demyelinations, may present the most compelling targets for cell-based neurological therapy.
Keywords: leucodystrophy, oligodendrocyte, glial progenitor cell, transplantation, neural stem cell
Studies of lower species have informed us of the role that newly added neurons may play in the natural histories of adult neural networks (Goldman 1998; Gage 2002), while the identification and mapping of persistent neural stem cells (NSCs) and progenitor cells in the human central nervous system (CNS) have served to highlight the potential for structural neuroplasticity in adulthood (Kirschenbaum et al. 1994; Pincus et al. 1998; Roy et al. 2000b; Sanai et al. 2004). At the same time, these studies have served to emphasize the inadequacy of the adult human brain's response to disease and injury, which is typified by a lack of clinically significant regeneration in all but the youngest brains. Against this backdrop, we have witnessed an explosion in our understanding of stem cell biology. Pluripotential embryonic stem (ES) cells, neural tissue-derived stem and phenotype-specified progenitor cells have all been investigated for their ability to generate neurons and glia, and the molecular mechanisms by which they do so. This review will focus on the potential utility of NSCs and progenitor cells as substrates for structural repair of the brain and spinal cord. It will emphasize only a few selected clinical targets for which stem and progenitor cell therapy might be the most appropriate and imminent. We and others have recently reviewed strategies for inducing endogenous progenitor cells to effect structural repair (Goldman 2004, 2005b; Lindvall et al. 2004; Palmer & Gage 2006), so that this emerging topic will not be covered here. Similarly, we will not consider cell therapy in disorders such as stroke, Huntington's disease and Alzheimer's disease, which have also been recently reviewed (Lindvall et al. 2004; Chmielnicki & Goldman 2005), and for which significant gaps in our understanding of their disease mechanisms may slow clinical translation (Goldman 2005a). Rather, we will focus here on cell transplant-based treatment of a small cadre of carefully chosen neurological disorders, for which our understanding of both the disease process and our intended cellular vector may be sufficient to anticipate therapeutic success once the two are appropriately paired. These potential targets of cell therapy include: Parkinson's disease (PD), a model neurodegeneration of a single neuronal phenotype; spinal cord disease, both the traumatic and degenerative manifestations of which are typically limited to a discrete number of cell phenotypes; and the leucodystrophies and acquired myelin disorders, in which oligodendrocyte loss is the principal endpoint of a wide variety of pathologies.
1. Acquisition and preparation of engraftable human neural progenitor cells
Cell transplantation strategies for CNS disease require the acquisition of human NSC and progenitor cells in both high purity and large number. Until phenotypically restricted neuronal and oligodendrocytic progenitors can be safely and adequately derived in purity from human embryonic stem (hES) cells, cell therapeutic strategies will remain dependent on the use of allografted human progenitors derived from both foetal and adult tissues. Multipotential NSCs are abundant in foetal tissues, and continue to line the forebrain ventricles in adults (Morshead et al. 1994; Weiss et al. 1996), while neuronal progenitor cells also remain within the ventricular wall, throughout its extensions to the olfactory bulb (Lois & Alvarez-Buylla 1993; Luskin 1993) and hippocampus. In addition, a larger pool of glial progenitors also pervade both the ventricular zone (VZ) and tissue parenchyma (Levine et al. 2001). To address the need for large quantities of engraftable cells, several groups have established lines of human NSCs, by exposing initially uncommitted cells continuously to mitogens in serum-deficient culture (Vescovi et al. 1993; Morshead et al. 1994; Palmer et al. 1995; Gritti et al. 1996; Flax et al. 1998; Keyoung et al. 2001). These lines give rise to functionally mature neurons both in vitro (Carpenter et al. 1999; Svendsen et al. 1999; Vescovi et al. 1999) and in vivo (Brustle et al. 1998; Fricker et al. 1999; Keyoung et al. 2001). However, these preparations have typically been generated by preferential expansion over prolonged periods in vitro, and are thus exposed from inception not only to mitogens, but also to neuronal and glial paracrine agents in the initially heterogeneous cultures. As a result, NSCs generated by such means may differ in their expansion characteristics, gene expression and differentiation competence from the parental cells from which they derive.
To address this weakness of selective expansion as a means of isolating stem and progenitor cells, surface antigen-based sorting has also been used for their selective extraction (Uchida et al. 2000; Lee et al. 2005). Although currently available antigenic markers for human NSCs are not sufficiently specific to purify these cells from human samples (Uchida et al. 2000), separation based upon a combination of size, lectin binding and antigenicity has been reported to do so in rodents (Rietze et al. 2001; Capela & Temple 2002). Specific stem and progenitor cell types may also be isolated by progenitor cell-selective reporter gene expression (Wang et al. 1998). Individual phenotypes may be identified by transducing either tissue dissociates or cultured cells with fluorescent reporters placed under the control of promoters for genes selectively activated in progenitor cells (Chalfie et al. 1994; figure 1). The fluorescent progenitor cells may then be isolated by fluorescence-activated cell sorting (FACS). By this strategy of promoter-based sorting, neuronal progenitors have been extracted from both the adult human VZ and hippocampus (Roy et al. 2000a,b), using Tα1-specified, green fluorescent protein (GFP)-based FACS (Wang et al. 1998). Similarly, NSCs have been isolated from the human VZ and hippocampus (Roy et al. 2000a,b; Wang et al. 2000; Kawaguchi et al. 2001; Keyoung et al. 2001; Mignone et al. 2004), based on their transcription of nestin and musashi1, two genes expressed by NSCs (Roy et al. 2000a; Keyoung et al. 2001). Oligodendrocyte progenitors were also isolated from human brain tissue, based on their activation of the early oligodendrocyte promoter CNP2 (Roy et al. 1999); these cells were found to express membrane gangliosides recognized by the A2B5 antibody, permitting their surface antigen-based extraction and purification. This latter approach has proven critical to isolating engraftable populations of glial progenitor cells sufficient for both preclinical and clinical assessment.
Figure 1.

Sources, isolation and use of defined progenitor phenotypes. This figure schematizes the methods of isolating prospectively defined neural progenitor phenotypes from a variety of human cell and tissue sources, and highlights several of the experimental purposes to which these cells may be allocated and devoted. EGFP, enhanced green fluorescent protein; MAGS, magnetic activated cell sorting; PSA-NCAM; polysialylated neural cell adhesion molecule; LTR, long terminal repeat. Adapted from Goldman (2005b).
Progenitors for the major categories of neuronal and glial phenotypes of the adult brain may thus be directly extracted from adult tissue. However, these adult cells are not necessarily competent to generate all of the transmitter-restricted neuronal phenotypes of the human brain. Rather, progenitors fated to give rise to discrete neuronal phenotypes may be transient in nature, yielding phenotypically restricted derivatives during a narrow window of embryonic development, and neither before nor after. As a result, obtaining engraftable populations of neuronal progenitor cells of pre-selected and defined phenotype has proven challenging. Other approaches have been developed to address this problem. First, phenotypically restricted neuronal progenitor cells may thus be derived from the developing brain and spinal cord, and immortalized as such. Second, they may be generated and expanded from NSCs removed at the time point and from the site at which the desired phenotypes would otherwise have been generated in vivo; in at least some instances, these isolated NSCs may continue to generate their typical position- and transmitter-defined progeny in vitro, after extraction and propagation. As a third approach, selected neuronal phenotypes may now be generated from hES cells, though only a small number of neuronal phenotypes have thus far been specifically generated through this strategy, and their purity has been insufficient to permit clinical engraftment. Nonetheless, this approach may hold great promise as a source of engraftable cells for selected neurological diseases. One prominent example of a phenotypically restricted neurological disease that has been the target of each of these approaches is PD, which is characterized by the specific and selective loss of nigrostriatal dopaminergic projection neurons; the uniform pathology and single phenotype involvement of PD lends itself to treatment strategies based on the replacement of a single neuronal cell type.
2. Cell-based strategies for treating Parkinson's disease
Parkinson's disease was arguably the first preclinical and clinical target of cell-based therapy among neurological and neurodegenerative diseases. PD is characterized by the progressive deterioration of dopaminergic neurons in the substantia nigra of the midbrain (Jenner & Olanow 1998). Since these neurons project to the caudate and putamen of the neostriatum, there is a significant decrease in neostriatal dopamine in PD. Cell-based therapies have thus far focused on implanting foetal midbrain cells, which include those destined to become dopaminergic neurons, into the neostriatum (Olanow et al. 1996; Lindvall 1999). These donor dopaminergic neurons are able to ameliorate Parkinsonian symptoms only when a sufficient number of them survive, avoid immune rejection and make appropriate postsynaptic connections with striatal neurons (Bjorklund & Lindvall 2000). Despite promising studies in experimental models (Kordower et al. 1998; Hauser et al. 1999), foetal cell grafts to adult PD patients have thus far yielded generally poor results, characterized by limited efficacy and significant morbidity, the latter manifested as medication-independent, refractory dyskinesias (Freed et al. 2001; Hagell et al. 2002). The potential causes of these weak clinical results are legion; they include the relative scarcity of dopaminergic cells in the donor tissues, the heterogeneity of cell types in each donor sample and variability among those samples, as well as the often non-uniform tissue distributions of the engrafted cells, and their lack of appropriate afferent control when placed into the striatum, among others (Piccini et al. 2005). Yet, the relative simplicity of the major pathology of PD, with its loss of a unifocal and phenotypically homogeneous neuronal population, along with PD's manifest epidemiological importance, has continued to spur stem cell-based strategies for its cure. As a result, these initial disappointments in using foetal nigral grafts to treat PD have spurred research into generating more uniform and accessible populations of human dopaminergic neurons, using defined dopaminergic progenitors rather than foetal tissue as source.
3. Embryonic stem cells as sources of dopaminergic neurons
To address these issues, a number of groups explored the possibility of generating highly enriched populations of dopaminergic neurons from mesencephalic progenitors (Studer et al. 1998; Sawamoto et al. 2001). Tissue-derived progenitors proved capable of giving rise to functional dopaminergic neurons, in sufficient numbers and uniformity so as to permit the functional recovery of rats rendered Parkinsonian by 6-hydroxydopamine (6-OHDA; Studer et al. 1998; Sawamoto et al. 2001). Yet preparing engraftable quantities of dopaminergic progenitors from foetal tissue has proven difficult, because of the small numbers of NSCs that may be purified from the first trimester midbrain, and the impracticality and controversial nature of sampling large numbers of abortuses for this purpose. As a result, a number of groups have begun to assess hES cells as a potential source of dopaminergic neurons. This possibility was opened by an elegant series of developmental studies, in which FGF8 and sonic hedgehog (SHH) were implicated as tandem initiators of dopaminergic neurogenesis in vivo (Ye et al. 1998). By replicating these conditions in vitro, McKay and colleagues first generated dopaminergic neurons from ES cells (Lee et al. 2000), and later achieved greater degrees of dopaminergic enrichment by mimicking the oxygen tension of the developing midbrain (Studer et al. 2000). Further improvements at replicating an in vivo environment by stromal cell co-culture were attended by even greater degrees of dopaminergic enrichment (Kawasaki et al. 2000; Kim et al. 2006). When implanted into 6-OHDA lesioned rats, in which nigrostriatal dopaminergic afferents are largely lost, such enriched populations of ES-derived dopaminergic cells proved able to restore functional normalcy to the dopamine-depleted animals (Kim et al. 2002). Similar results were obtained using monkey ES cell-derived dopaminergic neurons allografted into MPTP-lesioned adult cynomolgus monkeys, in which treatment-associated behavioural improvement was noted by 10 weeks after transplantation (Takagi et al. 2005).
These promising studies notwithstanding, the use of ES cell-derived dopaminergic neurons remains limited though by some of the same caveats that limit tissue-derived progenitor implantation. First, even highly enriched ES-derived dopaminergic preparations are by no means pure, and may be contaminated with serotonergic and GABAergic neurons, as well as by glia (Goridis & Rohrer 2002). The co-introduction of these other transmitter phenotypes, without further dopaminergic selection, might yield as yet unpredictable interactions among the engrafted cells, both synaptic and presynaptic. Furthermore, the long-term survival and phenotypic stability of engrafted primate and human ES-derived neurons have been problematic in both rodent (Park et al. 2004) and primate (Takagi et al. 2005) models of experimental Parkinson's disease. In addition, any ES cells that might have escaped in vitro differentiation have the potential for undifferentiated expansion, potentially yielding teratomas or their derivative tumours after implantation (Bjorklund et al. 2002). Thus, even though tumours have not yet been reported in primate or human ES transplants to the CNS (Park et al. 2004; Nistor et al. 2005; Takagi et al. 2005), one must be sceptical. No long-term survival studies have yet been reported that would allow one to adequately assess the neoplastic potential of human ES cell grafts over time. Fundamentally, the risk of uncontrolled expansion by remnant ES cells is sufficiently high that complete abolition of undifferentiated ES cells from donor grafts would certainly lend more confidence to the safe clinical use of ES-derived dopaminergic neurons. Promoter-specified isolation strategies, based on both GFP-based sorting (Roy et al. 2005) and antibiotic selection (Li et al. 1998), have allowed the enrichment of increasingly defined phenotypes from hES cells. Yet whether these approaches can be scaled up to achieve the abolition of undesired phenotypes from the large numbers of cells required for clinical transplants remains to be seen. To be sure, once dopaminergic progenitors do indeed become available in purity and abundance, a number of still unresolved issues need to be addressed before the clinical utility of these cells can become likely. These include the lack of afferent control of cells implanted into the caudate-putamen, their non-uniform and unpredictable dispersal, and the lack of nigrostriatal reconstruction upon alternative implantation into the substantia nigra; each of these issues was identified as problematic in foetal mesencephalic transplants, and each remains an impediment to early clinical translation.
4. Cell-based strategies for treating spinal cord disease
The spinal cord has been an attractive target for cell-based therapy, in part because of the dearth of available treatment options for spinal cord injury (SCI), but also because of the rapid pace of advance in our understanding of how to effect axonal regeneration in the injured cord (Gao et al. 2004; Hu & Strittmatter 2004; Nikulina et al. 2004), without which cellular replacement alone would be of limited benefit.
Spinal cord injury is associated with the loss of both neurons and glia. Flexion–extension injuries, contusion and anterior spinal artery ischaemic events, as may occur in trauma, spinal cord stroke and iatrogenic occlusion, can all result in segmental loss of both the superficial white matter tracts, and the various neuronal populations of the central grey. Thus, cell-based strategies for reconstituting the injured spinal cord must accommodate the need to replace multiple cell types. NSC implants into the injured spinal cord have thus been used under the premise that regionally appropriate phenotypes might be generated from undifferentiated cells in response to local signals competent to induce cell type-specification. Yet the extent to which such context-dependent phenotypic differentiation of undifferentiated NSCs can be relied upon to appropriately regenerate regions of local damage is unclear. Donor cell differentiation can vary with both the differentiated state of the donor cells and the developmental stage of the recipient, as well as with the structural integrity and normalcy of the intended target region. Similarly, the ability of neurons derived from non-homotypic NSCs to appropriately follow local position-dependent cues in axonal pathfinding remains unclear. Nonetheless, Okano et al. noted that NSCs implanted into sites of SCI yielded behavioural improvement compared to untransplanted controls (Ogawa et al. 2002). Similarly, Olson et al. found that neurogenin2-transduced NSCs, thereby primed to neuronal differentiation, yielded functional benefit after contusion injury (Hofstetter et al. 2005). In both cases, improvement was associated with local neurogenesis and synaptogenesis that might have served to establish alternative pathways for neurotransmission in the injured segments of the cord. Nonetheless, the benefits of NSC transplantation might also have resulted from donor–host humoral interactions, by which the implanted donor cells may have supported the survival and regeneration of host axons and their segmental connections, without requiring the functional or synaptic maturation of the donor-derived neurons, or their integration within the host neural network (Ourednik et al. 2002).
It is important to note here that the lack of control that can be exercised on the differentiated fate of implanted NSCs, or on the connections achieved by their neuronal daughters, is a real source of concern, since inappropriate phenotypic differentiation can prove ineffective or worse. For instance, rats transplanted with NSCs after thoracic weight-drop injury developed forelimb allodynia and heightened pain sensitivity that were associated with aberrant axonal sprouting following ectopic astrocytic differentiation (Hofstetter et al. 2005). In the complex disease environment of acute traumatic injury, the very environmental malleability of NSCs may prove undesirable. This in turn suggests that more phenotypically restricted progenitor cells might behave more predictably and reliably upon host engraftment.
Besides the loss of neurons and astrocytes seen in spinal cord injuries and infarcts, many cord injuries are demyelinative; the sensory tracts of the posterior columns and the descending motor tracts of the corticospinal pathways are both frequent victims of flexion–extension and contusion injuries of the cord. These surface pathways are also especially predisposed to ischaemic damage following cord oedema, with the draining veins on the posterolateral surfaces of the spinal cord being especially predisposed to congestion and hypoperfusion when oedematous. As a result, the transplantation of myelinogenic glial progenitors has been an especially appealing strategy for treating SCI. Indeed, this approach has been considered as a potential treatment for other segmental demyelinations, such as those that occur in transverse myelitis. Rao and colleagues demonstrated that foetal tissue-derived glial-restricted progenitors implanted into the contused rat spinal cord dispersed widely, with both astrocytic and oligodendrocytic maturation (Han et al. 2004). Although neither myelination nor the net efficiency of oligodendrogliogenesis were reported in this study, it nonetheless suggested the potential utility of glial progenitor grafts in carefully selected spinal cord injuries, especially those with limited involvement of the posterior columns and lateral funiculi.
Oligodendrocytes derived from human ES cells have also been reported to myelinate demyelinated foci in spinal cord contusions (Nistor et al. 2005). This latter observation paralleled earlier studies that reported myelination in the injured spinal cord by implanted murine ES cells (Brustle et al. 1999). However, neither of these studies isolated glial progenitors or oligodendrocytes prior to transplantation, and neither followed animals for the long periods of time required to ensure the long-term survival and phenotypic stability of the engrafted cells. In particular, these ES-based approaches are currently limited by the potential of any persistent undifferentiated ES cells in the donor pool to yield teratomas or germinomas after implantation. Thus, donor cell populations must be completely depleted of any persistent undifferentiated ES cells, before hES cell-based therapy of SCI—or of any other CNS disease target—may be safely contemplated. As a result, the implantation of tissue-derived glial progenitor cells may prove the more clinically feasible option, at least in the near future. Even then, any effective cell-based treatment of SCI will need to be accompanied by treatments designed to maximize extant cell survival (Wang et al. 2004), axonal extension and synaptogenesis (Pearse et al. 2004), as well as engraftment and differentiation. As with so many other disease targets, cell therapy of SCI will be unlikely to effect clinically significant restoration of function, without concurrent modulation of the disease environment in such a way as to favour cell integration and axonal regeneration, while suppressing ongoing disease pathology.
5. Cell-based therapy of motor neuron diseases
In contrast to the many cell types lost in SCI, the major degenerative diseases of the spinal cord, including both amyotrophic lateral sclerosis (ALS) and the spinal muscular atrophies of children, as well as several major viral infections of the spinal cord, including poliomyelitis and West Nile virus, are characterized by a selective loss of spinal motor neurons. Motor neuron disease was an early target of stem cell-based therapy, but while initial attempts at using ES cell implants to treat experimental motor neuron disease did yield some functional benefit, this was not accompanied by appreciable motor neuronal differentiation (Kerr et al. 2003). Indeed, this valuable initial effort suggested the need for engrafting phenotypically restricted cells in such disease models, rather than being held hostage to the stochastic differentiation of multipotential stem cells. Fortunately, motor neurons have proven especially amenable to generation from human ES cells (Li et al. 2005), largely because of the well-studied development of motor neurons in normal ontogeny and its recapitulation in murine ES cells (Wichterle et al. 2002), in which SHH and retinoic acid proved sufficient to induce motor neurons from human embryoid bodies.
As an alternative means of generating phenotypically restricted spinal neurons, Roy et al. transduced human foetal spinal neuroepithelium with retroviruses overexpressing human telomerase reverse transcriptase (hTERT), the rate-limiting component of human telomerase. The resultant telomerase-immortalized cells included lines biased to generate neurons; one in particular preferentially generated spinal interneurons, but also generated a minor cohort of motor neurons, that could be isolated by FACS based on Hb9 promoter-driven GFP expression (Roy et al. 2004). Like their human ES-derived counterparts, the motor neurons generated from these hTERT-immortalized progenitors were post-mitotic, both antigenically and physiologically appropriate, and functionally appropriate after engraftment (Roy et al. 2004). However, just as the use of ES cells as a source of donor neurons poses the potential for adventitiously introducing as-yet undifferentiated cells that may prove tumorigenic, the use of neurons derived from a telomerase-immortalized precursor line may similarly present the potential for undifferentiated expansion from inadvertently admitted precursor cells. Again, the issue of pre-implantation purification will need to be addressed before either of these otherwise highly promising sources of phenotypically restricted donor cells may be used as clinical vectors.
It is also important to note that despite the availability of highly enriched human motor neurons from both human ES cells and telomerase-immortalized progenitors, and the identification of genetic tags that allow their isolation and targeting (Arber et al. 1999; Roy et al. 2004; Nakano et al. 2005), the specific replacement of motor neurons presents a formidable challenge. Spinal cord neurons are the frequent victims of multilevel disease, with classical ALS involving most rostrocaudal segments of the CNS. Motor neurons generated from ES cells or hTERT-immortalized native spinal progenitors are essentially non-migratory, and would need to be delivered to sites of need; such multifocal targeting would be problematic at best. Indeed, the replacement of lost motor neurons in vivo requires not only multisegmental delivery or induction, but also the re-establishment of appropriate afferent innervation, and the long-distance extension of their axons, through often degenerating nerve roots, to specific loci in the distant musculature. Our poor understanding of much of the biology underlying these processes suggests that despite much recent progress in generating motor neurons from both ES cells and their tissue-derived homologues, cell-based treatment of the motor neuronopathies remains a difficult goal.
6. Glial progenitor cell-based therapy of demyelinating disease
The demyelinating diseases of the brain are especially attractive targets for cell-based therapeutic strategies, since they are caused by the loss of a single cell type, the oligodendrocyte. Oligodendrocytes are the sole source of myelin in the adult CNS, and their loss or dysfunction is at the heart of a wide variety of diseases of both children and adults. Indeed, from the standpoint of the recipient, given their relative homogeneity of the affected phenotype, the diseases of myelin are especially attractive targets for cell-based therapy of brain and spinal cord disease. Fortuitously, from the standpoint of donor acquisition, glial progenitor cells very likely constitute the most abundant progenitor cell in both the adult and mid-gestation foetal brain (Scolding et al. 1998; Nunes et al. 2003). These cells may be isolated and harvested in bulk from human brain tissue, and are readily biased to oligodendrocyte phenotype (figure 1). As a result, they have been assessed in a variety of models of dysmyelinating diseases.
In adults, oligodendrocytic loss is causal in disease as diverse as the vascular leucoencephalopathies and multiple sclerosis. When transplanted into lysolecithin-lesioned adult rat brain, adult human oligodendrocyte progenitor cells (OPCs) were able to quickly mature as oligodendrocytes and myelinate residual denuded host axons, but with relatively low efficiency compared to the robust myelination seen in congenitally hypomyelinated brain. Similarly, systemic administration of NSCs into mice subjected to experimental allergic encephalomyelitis resulted in some degree of local engraftment, oligodendrocytic maturation and myelination (Pluchino et al. 2003), although the robustness and stability of donor cell-mediated remyelination remains unclear (Pluchino et al. 2004). These observations highlight the importance of the disease environment in permitting appropriate oligodendrocytic differentiation and myelination (Franklin 2002). Thus, while human OPCs would seem effective agents by which to remyelinate acutely demyelinated brain tissue, the complexity of the adult disease environment may make such targets less imminently approachable than their paediatric counterparts. At the very least, cell-based therapeutic strategies for adult demyelination, especially those intended to remyelinate the inflammatory lesions of multiple sclerosis, will require aggressive disease modification and immunosuppression as adjuncts to cell delivery.
7. The childhood disorders of myelin as therapeutic targets
The early dysmyelinations of the paediatric leucodystrophies comprise especially appropriate targets for progenitor cell-based therapy. Children suffer from a variety of hereditary diseases of myelin failure or loss, that include: (i) the hypomyelinating diseases, such as Pelizaeus–Merzbacher disease and hereditary spastic paraplegia, which represent primary disorders of myelin formation; (ii) the metabolic demyelinations and lysosomal storage disorders, such as metachromatic leucodystrophy, Tay-Sachs, Sandhoff's and Krabbe's diseases, as well as adrenoleucodystrophy and the mucopolysaccharidoses; and (iii) gross disorders of tissue loss, such as vanishing white matter disease and Canavan's disease, in which oligodendrocytes are early targets (reviewed in Kaye 2001; Powers 2004). In addition, a variety of hereditary metabolic disorders that are manifested by early neuronal loss, such as the organic acidurias and neuronal ceroid lipofuscinoses (NCLs), are accompanied by early oligodendrocyte loss. Moreover, even more common paediatric afflictions, such as cerebral palsy, are due in part to a perinatal loss of oligodendrocytes and their precursors (Back & Rivkees 2004; Follett et al. 2004; Robinson et al. 2005).
Their mechanistic heterogeneity notwithstanding, all of these conditions include as a prominent feature the loss of oligodendrocytes and central myelin. To assess the potential of cell-based treatment for congenital dysmyelination, Windrem et al. transplanted sorted human OPCs of both foetal and adult origin into newborn shiverer mice, a dysmyelinated mouse deficient in myelin basic protein (MBP; Windrem et al. 2002, 2003, 2004). The donor OPCs dispersed widely throughout the shiverer forebrain white matter, such that single neonatal injections of OPCs into the lateral ventricles and adjacent callosum yielded abundant donor cell infiltration of the entire corpus callosum, fimbria and internal and external capsules, as well as the deep subcapsular white matter to the level of the cerebral peduncles (figure 2a–h). Although the dorsal brainstem and cerebellum were not infiltrated by cells introduced through a forebrain route, addition of a single intracerebellar injection at birth proved sufficient to substantially infiltrate the cerebellar white matter and peduncles, allowing donor engraftment contiguous with that of the forebrain and ventral brainstem (figure 3a–c). Overall, these cells migrated as widely as did immortalized murine NSCs, which were reported by Snyder and colleagues to be similarly capable of context-dependent differentiation and myelination (Yandava et al. 1999).
Figure 2.
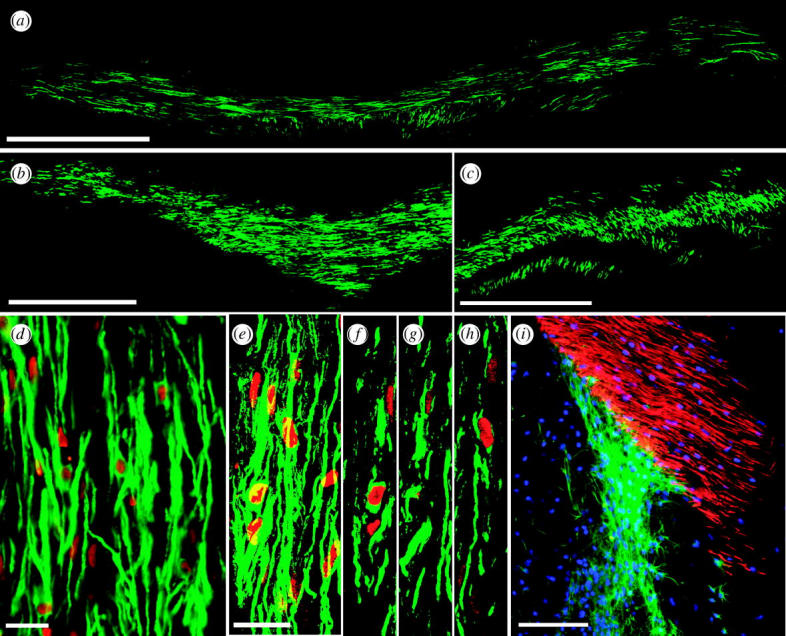
Myelination by engrafted OPCs. (a–c) Implanted human foetal OPCs myelinated extensive regions of shiverer mouse forebrain. (a, b) MBP expression by sorted human OPCs, implanted into neonatal shiverer mice, indicates that large regions of the corpus callosum ((a) and (b), different mice) have myelinated by 12 weeks (MBP, green). (c) Donor-derived myelin extended throughout the internal capsules. (d–h) New myelin was exclusively derived from human donor cells. (d) MBP (green), in a shiverer callosum three months after neonatal graft, is associated with human donor cells, identified by human nuclear antigen (hNA, red). (e–h) Confocal images of implanted shiverer callosum, with human cells (hNA, red) surrounded by associated MBP (green). ((e) Merged images of (f–h), 1 μm apart.) (i) OPCs were recruited as oligodendrocytes or astrocytes in a context-dependent manner, such that they matured as MBP+ oligodendrocytes in the white matter, but as glial fibrillary acidic protein (GFAP+) astrocytes in both white and grey matter. Panel (i) shows the striatocallosal border of a shiverer brain, three months after human foetal OPC transplant (hNA, blue). Donor-derived MBP (red) fills the callosum, while donor-derived GFAP+ (green) astrocytes predominate in the striatum and ventricular wall. Scale bar, 1 mm (a–c), 100 μm (d), 20 μm (e–h), 200 μm (i). Adapted from Goldman (2005b) and Windrem et al. (2004).
Figure 3.

Dual site cell transplantation. Widespread infiltration of human donor OPCs into the shiverer brain and brainstem follows neonatal injection into the cisterna magnum and corpus callosum. (a) This animal was injected on P0 with 1×105 cells each into the cisterna magnum(*), and corpus callosum (CC), then sacrificed at day 30 and stained for human nuclear antigen (red, arrow) to identify donor cells. SC, superior colliculus. (b) This animal was treated identically as that in (a), except that it was given an additional injection into the cerebellar central white matter. It was sacrificed two months later, by which time it exhibited not only widespread forebrain and brainstem cell dispersal, but also early MBP production throughout the cerebellar white matter (green). (c) An assessment of five different measures of engraftment success, derived from another set of shiverers sacrificed at 12 weeks, and assessed by selectively scoring areas of robust myelination, so as to estimate maximal efficacy of engraftment. Scored metrics include: (c-1) proportion of human nuclear antigen (hN+) donor cells expressing MBP; (c-2) density of myelinating donor cells per mm3; (c-3) number of axons ensheathed per donor cell and per MBP+ donor cell; (c-4) donor cell density at 12 weeks, compared between fimbria, callosum and cerebellar white matter (WM).
Neonatally implanted human donor OPCs developed as astrocytes and myelinating oligodendrocytes in a context-dependent fashion, such that those donor cells that engrafted presumptive white matter developed as oligodendrocytes, while those invading cortical and subcortical grey developed largely as astrocytes (figure 2i; Windrem et al. 2004). The majority of donor cells engrafted the white matter, such that after neonatal intraventricular and intracallosal injection, the corpus callosum typically expressed MBP throughout its mediolateral extent, along its entire length in the sagittal plane, and throughout the dorsoventral extent of the internal capsules to the cerebral peduncles (Windrem et al. 2004; figure 2a–c). Donor-derived myelin effectively ensheathed host shiverer axons, as validated by both confocal imaging (figure 4a,b), and by the ultrastructural observation of donor-derived myelin with major dense lines (figure 4c,d), indicating effective myelin compaction, of which native shiverer oligodendrocytes are incapable. In addition, confocal analysis revealed the presence of nodes of Ranvier between donor-derived myelinated segments, and the paranodal expression of contactin-associated protein (Caspr) suggesting functionally appropriate nodal architecture (figure 4e). Given the widespread dispersal of donor cells, their high-density engraftment and myelination, and their architecturally appropriate and quantitatively significant ensheathment of host axons (figure 3c), these results strongly suggested the feasibility of neonatal progenitor cell implantation as a potential therapeutic strategy in the congenital disorders of myelin.
Figure 4.

Axonal ensheathment and myelin compaction by engrafted human progenitor cells. (a) A confocal micrograph showing a triple immunostain for MBP (red), human nuclear antigen (blue) and neurofilament protein (green). In this image, all MBP immunostaining is derived from the sorted human OPCs, whereas the NF+ axons are those of the mouse host. Arrows identify murine axons ensheathed by human MBP. (b) A 2 μm deep composite of optical sections, taken through the corpus callosum of a shiverer recipient sacrificed 12 weeks after foetal OPC implantation. Shiverer axons were scored as ensheathed when the yellow index lines intersected an NF+ axon abutted on each side by MBP. The asterisk indicates the field enlarged in the inset. (c, d) Representative electron micrographs of 16-week-old shiverer homozygotes, implanted with human OPCs shortly after birth. The images show shiverer axons ensheathed by densely compacted myelin. The asterisk indicates the field enlarged in the inset. Inset: major dense lines are noted between lamellae, providing electron microscopic confirmation of myelination. (e) High power confocal images of MBP+ donor-derived myelin sheaths (green) at internodal junctions, characterized by expression of Caspr protein (red) at the paranodal borders. Left, a single 0.4 μm optical section; right, a z-stack composite. Caspr staining confirmed nodes of Ranvier between adjacent donor-derived myelinated segments (Einheber et al. 1997); these results suggest physiologically appropriate conduction support by donor-derived myelin. Scale bar, 20 μm (a), 40 μm (b), 1 μm (c, d). Adapted from Goldman (2005b) and Windrem et al. (2004).
8. Cell-based strategies for treating lysosomal storage disorders and metabolic leucodystrophies
In many of the metabolic disorders of myelin, such as Krabbe's and Canavan's diseases, oligodendrocytes are essentially bystanders, killed by toxic metabolites emanating from cells deficient in one or more critical enzymes. Since the engraftment of glial progenitor cells is associated with astrocytic as well as oligodendrocytic production, and since both the subcortical and cortical grey are infiltrated with donor-derived astrocytes after early implantation, glial progenitors would seem an especially promising vehicle for the distribution of enzyme-producing cells throughout otherwise deficient brain parenchyma. On that basis, several groups have begun to assess the ability of enzymatically competent, effectively wild-type glial progenitor cells to delay or ameliorate the signs and symptoms of the lysosomal storage disorders and other metabolic leucodystrophies. In addition, undifferentiated NSCs are being assessed with much the same logic, in that they can migrate widely throughout the developing nervous system, and can to some extent differentiate and integrate in a regionally appropriate fashion. As such, they can be used as delivery vehicles for both normally expressed and genetically overexpressed enzymes deficient in their hosts. Snyder and colleagues first established this principle of cell-based rescue of enzymatically deficient host cells by wild-type donor NSC implantation in a mouse model of MPS-VII, Sly's disease, in which myc-transduced NSCs were implanted neonatally and observed to migrate widely and restore lost enzymatic function broadly in the recipient forebrain (Snyder et al. 1995). The same group subsequently reported expression of β-hexosaminidase upon engraftment of transduced NSCs into recipient mice (Lacorazza 1996), though functional benefits accruing to engraftment-associated enzyme expression have not yet been reported. Similarly, Pellegatta et al. (2006) recently engrafted twitcher mice, a murine model of Krabbe's globoid cell leucodystrophy, with cultured NSCs transduced to overexpress galactocerebrosidase, the enzyme deficient in Krabbe's disease. Although the engrafted cells did not survive well in the highly inflammatory twitcher brain, they migrated appropriately to active sites of demyelination, in a manner akin to that noted in adults by Martino et al. (Pluchino et al. 2003, 2004). One might hope that in recipients immunosuppressed to reduce donor cell rejection, future trials may be better able to assess the capacity of engrafted NSCs to restore lost structure and function. A recently approved investigational new drug application to assess the use of human NSC allografts in treating Batten's disease (NCL2) speaks to the efforts that may be anticipated in this field over the next several years.
9. Defining optimal cellular vectors for the leucodystrophies
Both foetal and adult OPCs have been proposed as potential cellular vectors for cell therapy of the congenitally dysmyelinated CNS. Yet foetal and adult-derived OPCs have been observed to behave quite differently after neonatal xenograft (Windrem et al. 2004). Isolates of human OPCs derived from adult white matter myelinated recipient brain much more rapidly than did foetal OPCs; adult-derived progenitors achieved widespread myelination by just four weeks after graft, while cells derived from late second trimester foetuses took over three months to do so. The adult OPCs also generated oligodendrocytes more efficiently than foetal glial progenitors, and ensheathed more axons per donor cell than did foetal cells. In contrast, foetal glial progenitors emigrated more widely and engrafted more efficiently, differentiating as astrocytes in grey matter regions and oligodendrocytes in white matter (figure 5).
Figure 5.

Foetal and adult OPCs differed substantially in their speed and efficiency of myelinogenesis. (a) Adult-derived human OPCs (hNA, red) achieved dense MBP (green) by four weeks after graft. (b) In contrast, foetal OPCs expressed no MBP at four weeks, and none until 12 weeks. (c, d) Low- and high-power images of the callosal–fimbrial junction of a shiverer brain; dense myelination by 12 weeks after perinatal delivery of adult OPCs. (e, f) Adult OPCs developed mature myelin ultrastructure and major dense lines within five weeks of perinatal injection. (e, f) Myelin in a shiverer homozygote five weeks after perinatal injection of adult OPCs. Mice injected with foetal OPCs exhibited no evidence of myelination at this time point. (g) The distribution of hNA+ adult OPCs (red), four weeks after implant. (h) A higher proportion of adult OPCs developed MBP expression than did foetal OPCs, when assessed 12 weeks after transplant. (i) Foetal OPCs nonetheless engrafted more efficiently and in higher numbers than did adult OPCs. *p<0.05; **p<0.005, Student's t-test. (j) A plot comparing the number of ensheathed axons per donor cell achieved by foetal and adult-derived OPCs. Ensheathment was defined by confocal-imaged MBP+ enwrapping NF+ axons, and was measured as a function of total donor cell number (left), and of MBP+ donor-derived oligodendrocytes (right). The difference between foetal and adult donor ensheathment efficiencies was significant by Mann–Whitney (p<0.02). Scale bar, 100 μm (a, b), 1 mm (c), 30 μm (d), 1 μm (e, f). Adapted from Windrem et al. (2004).
The divergent behaviours of foetal and adult-derived glial progenitors suggest their respective use for different disease targets. Adult OPCs, by virtue of their oligodendrocytic bias and rapid myelination, may be most appropriate for diseases of acute oligodendrocytic loss, such as subcortical infarcts and post-inflammatory demyelinated lesions. In contrast, foetal progenitors may prove more effective for treating the congenital leucodystrophies, since their extensive emigration better assures uniform and widespread dispersal, while their astrocytic differentiation and invasion of grey matter may offer the correction of enzymatic deficits in deficient cortex. Indeed, perinatal grafts of foetal progenitor cells might prove a means of simultaneously myelinating and correcting enzymatic deficiencies in the paediatric leucodystrophies (Snyder et al. 1995). The lysosomal storage disorders present especially attractive targets in this regard, since wild-type lysosomal enzymes may be released by integrated donor cells, and taken up by deficient host cells through the mannose-6-phosphate receptor pathway (Urayama et al. 2004). As a result, a relatively small number of donor glia may provide sufficient enzymatic activity to correct the underlying catalytic deficit and storage disorder of a much larger number of host cells (Jeyakumar et al. 2005). That being said, little data currently exists with regards to the number or proportion of wild-type cells required to achieve local correction of enzymatic activity and substrate clearance in any storage disorder, and these values will probably need to be obtained for each disease target. Similarly, effective cell doses, delivery sites and time frames will need to be established in models of congenital hypomyelination before clinical trials of progenitor-based therapy can be contemplated. Moreover, the efficiency of myelination required for significant benefit remains unclear, as functional improvement may require remyelination over much if not the entire linear extent of each recipient axon. These caveats notwithstanding, there is reason for optimism that cell-based therapy of the paediatric myelin disorders, in particular for the primary dysmyelinations, such as Pelizaeus–Merzbacher disease (Hudson et al. 2004), vanishing white matter disease (van der Knaap et al. 1997) and the spastic diplegic forms of cerebral palsy (Back & Rivkees 2004), may not be far off.
10. Overview
Clinically meaningful cell replacement in the diseased CNS will require a rigorous identification of those diseases most amenable to rational cell-based therapy. We have specified three such targets, broadly defined as PD, SCI and demyelinating disease, the latter encompassing both the paediatric leucodystrophies and storage disorders, as well as conditions of adult demyelination. These seemingly diverse disorders are all characterized by relatively simple neuropathology and known pathophysiology, and each involves the loss of a limited number of cell phenotypes. In this regard, the glial diseases stand out as the especially promising initial targets for cell transplant-based therapy of CNS disease. Using a common strategy of glial progenitor cell implantation, paediatric diseases as diverse as the leucodystrophies, lysosomal storage diseases and cerebral palsy, and such adult acquired demyelinations as transverse myelitis, multiple sclerosis and subcortical stroke, may all be approachable as therapeutic targets. Moreover, the same technologies used to enrich progenitor cells for transplantation yield isolates amenable to both immortalization and gene expression analyses as well. This has led to the generation of lines of specific types of neuronal progenitors that may prove useful not only as cellular vectors for transplantation, but also as tools for understanding the signalling pathways and growth control of native progenitors. Using such information, endogenous progenitor cells can be targeted for directed mobilization and phenotypic induction, whether by neurotrophic cytokines, or by their small molecule mimetics. Indeed, by mobilizing endogenous progenitors in vivo, we can hope to mitigate the need for transplantation in many disease settings. In others however, such as diseases involving the widespread loss of cells across the neuraxis, and disorders in which endogenous progenitor cells themselves are lost or deficient, therapeutic strategies based on cell transplantation will be as necessary as they now appear promising. Together, these distinct approaches to cell-based therapy should serve to establish effective tissue repair as a therapeutic endpoint in clinical neurology.
Acknowledgements
I thank Drs Neeta Roy and Fraser Sim for their contributions to the studies discussed, and Dr Sim for drawing figure 1. I apologize to the many authors whose relevant work we have not been able to cite in this short review. S.A.G. is supported by NINDS, the National Multiple Sclerosis Society, the A-T Children's Project and the CNS Foundation, Berlex Bioscience, and Q Therapeutics.
Footnotes
One contribution of 13 to a Theme Issue ‘The regenerating brain’.
References
- Arber S, Han B, Mendelsohn M, Smith M, Jessell T.M, Sockanathan S. Requirement for the homeobox gene Hb9 in the consolidation of motor neuron identity. Neuron. 1999;23:659–674. doi: 10.1016/s0896-6273(01)80026-x. 10.1016/S0896-6273(01)80026-X [DOI] [PubMed] [Google Scholar]
- Back S, Rivkees S. Emerging concepts in periventricular white matter injury. Semin. Perinatol. 2004;6:405–414. doi: 10.1053/j.semperi.2004.10.010. 10.1053/j.semperi.2004.10.010 [DOI] [PubMed] [Google Scholar]
- Bjorklund A, Lindvall O. Cell replacement therapies for central nervous system disorders. Nat. Neurosci. 2000;3:537–544. doi: 10.1038/75705. 10.1038/75705 [DOI] [PubMed] [Google Scholar]
- Bjorklund L, Sanchez-Pernaute R, Chung S, Jenkins B, Wahlestedt C, Kim K.M, Isacson O. Embryonic stem cells develop into functional dopaminergic neurons after transplantation in a Parkinson rat model. Proc. Natl Acad. Sci. USA. 2002;99:2344–2349. doi: 10.1073/pnas.022438099. 10.1073/pnas.022438099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustle O, Choudhary K, Karram K, Huttner A, Murray K, Dubois-Dalcq M, McKay R. Chimeric brains generated by intraventricular transplantation of fetal human brain cells into embryonic rats. Nat. Biotechnol. 1998;16:1040–1044. doi: 10.1038/3481. [DOI] [PubMed] [Google Scholar]
- Brustle O, Jones K.N, Learish R.D, Karram K, Choudhary K, Wiestler O.D, Duncan I.D, McKay R.D. Embryonic stem cell-derived glial precursors: a source of myelinating transplants. Science. 1999;285:754–756. doi: 10.1126/science.285.5428.754. 10.1126/science.285.5428.754 [DOI] [PubMed] [Google Scholar]
- Capela A, Temple S. LeX/ssea-1 is expressed by adult mouse CNS stem cells, identifying them as nonependymal. Neuron. 2002;35:865–875. doi: 10.1016/s0896-6273(02)00835-8. 10.1016/S0896-6273(02)00835-8 [DOI] [PubMed] [Google Scholar]
- Carpenter M, Cui X, Hu Z, Jackson J, Sherman S, Seiger A, Wahlberg L. In vitro expansion of a multipotent population of human neural progenitor cells. Exp. Neurol. 1999;158:265–278. doi: 10.1006/exnr.1999.7098. 10.1006/exnr.1999.7098 [DOI] [PubMed] [Google Scholar]
- Chalfie M, Tu Y, Euskirchen G, Ward W, Prasher D. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- Chmielnicki E, Goldman S. Stem cells and cell-based therapy in neurodegenerative disease. In: Beal M.F, Lang A, Ludolph A, editors. Neurodegenerative diseases. Cambridge University Press; Cambridge, UK: 2005. pp. 347–362. [Google Scholar]
- Einheber S, Zannazi G, Ching W, Scherer S, Milner T, Peles E, Salzer J. The axonal membrane protein Caspr, a homologue of neurexin IV, is a component of the septate-like paranodal junctions that assemble during myelination. J. Cell Biol. 1997;139:1495–1506. doi: 10.1083/jcb.139.6.1495. 10.1083/jcb.139.6.1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flax J, Aurora S, Yang C, Simonin C, Willis A, Sidman R, Wolfe J, Kim S, Snyder E. Engraftable human neural stem cells respond to developmental cues, replace neurons, and express foreign genes. Nat. Biotechnol. 1998;16:1033–1039. doi: 10.1038/3473. 10.1038/3473 [DOI] [PubMed] [Google Scholar]
- Follett P, Deng W, Dai W, Talos D, Massilon L, Rosenberg P, Volpe J, Jensen F. Glutamate receptor-mediated oligodendrocyte toxicity in periventricular leukomalacia: a protective role for topiramate. J. Neurosci. Res. 2004;24:4412–4420. doi: 10.1523/JNEUROSCI.0477-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin R.J.M. Why does remyelination fail in multiple sclerosis? Nat. Rev. Neurosci. 2002;3:705–714. doi: 10.1038/nrn917. 10.1038/nrn917 [DOI] [PubMed] [Google Scholar]
- Freed C, et al. Transplantation of embryonic dopamine neurons for severe Parkinson's disease. N. Engl. J. Med. 2001;344:710–719. doi: 10.1056/NEJM200103083441002. 10.1056/NEJM200103083441002 [DOI] [PubMed] [Google Scholar]
- Fricker R, Carpenter M, Winkler C, Greco C, Gates M, Bjorklund A. Site-specific migration and neuronal differentiation of human neural progenitor cells after transplantation in the adult rat brain. J. Neurosci. 1999;19:5990–6005. doi: 10.1523/JNEUROSCI.19-14-05990.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage F.H. Neurogenesis in the adult brain. J. Neurosci. 2002;22:612–613. doi: 10.1523/JNEUROSCI.22-03-00612.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, et al. Activated CREB is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron. 2004;44:609–621. doi: 10.1016/j.neuron.2004.10.030. 10.1016/j.neuron.2004.10.030 [DOI] [PubMed] [Google Scholar]
- Goldman S. Adult neurogenesis: from canaries to the clinic. J. Neurobiol. 1998;36:267–286. 10.1002/(SICI)1097-4695(199808)36:2%3C267::AID-NEU12%3E3.0.CO;2-B [PubMed] [Google Scholar]
- Goldman S.A. Directed mobilization of endogenous neural progenitor cells: the intersection of stem cell biology and gene therapy. Curr. Opin. Mol. Ther. 2004;6:466–472. [PubMed] [Google Scholar]
- Goldman S.A. Neurology and the stem cell debate. Neurology. 2005a;64:1675–1676. doi: 10.1212/01.WNL.0000165312.12463.BE. [DOI] [PubMed] [Google Scholar]
- Goldman S.A. Stem and progenitor cell-based therapy of the human central nervous sytem. Nat. Biotechnol. 2005b;23:862–871. doi: 10.1038/nbt1119. 10.1038/nbt1119 [DOI] [PubMed] [Google Scholar]
- Goridis C, Rohrer H. Specification of catecholaminergic and serotonergic neurons. Nat. Neurosci. 2002;3:531–541. doi: 10.1038/nrn871. [DOI] [PubMed] [Google Scholar]
- Gritti A, et al. Multipotential stem cells from the adult mouse brain proliferate and self-renew in response to basic fibroblast growth factor. J. Neurosci. 1996;16:1091–1100. doi: 10.1523/JNEUROSCI.16-03-01091.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagell P, et al. Dyskinesias following neural transplantation in Parkinson's disease. Nat. Neurosci. 2002;5:627–628. doi: 10.1038/nn863. [DOI] [PubMed] [Google Scholar]
- Han S.W, Liu Y, Tyler-Polsz C, Rao M.S, Fischer I. Transplantation of glial-restricted precursor cells into the adult spinal cord. Glia. 2004;45:1–16. doi: 10.1002/glia.10282. 10.1002/glia.10282 [DOI] [PubMed] [Google Scholar]
- Hauser R, Freeman T, Snow B, Nauert M, Gauger L, Kordower J, Olanow C. Long-term evaluation of bilateral fetal nigral transplantation in Parkinson disease. Arch. Neurol. 1999;56:179–187. doi: 10.1001/archneur.56.2.179. 10.1001/archneur.56.2.179 [DOI] [PubMed] [Google Scholar]
- Hofstetter C, Holmstrom N, Lilja J, Schweinhardt P, Hao J, Spenger C, Kurpad S, Frisen J, Olson L. Allodynia limits the usefulness of intraspinal neural stem cell grafts; directed differentiation improves outcome. Nat. Neurosci. 2005;8:346–353. doi: 10.1038/nn1405. 10.1038/nn1405 [DOI] [PubMed] [Google Scholar]
- Hu F, Strittmatter S.M. Regulating axon growth within the postnatal central nervous system. Semin. Perinatol. 2004;28:371–378. doi: 10.1053/j.semperi.2004.10.001. 10.1053/j.semperi.2004.10.001 [DOI] [PubMed] [Google Scholar]
- Hudson L, Garbern J, Kamholz J. Pelizaeus–Merzbacher disease. In: Lazzarini R, editor. Myelin biology and disorders. vol. 2. Elsevier Academic Press; San Diego, CA: 2004. pp. 867–886. [Google Scholar]
- Jenner P, Olanow C.W. Understanding cell death in Parkinson's disease. Ann. Neurol. 1998;44:S72–S84. doi: 10.1002/ana.410440712. [DOI] [PubMed] [Google Scholar]
- Jeyakumar M, Dwek R, Butters T, Platt F. Storage solutions: treating lysosomal disorders of the brain. Nat. Rev. Neurosci. 2005;6:1–12. doi: 10.1038/nrn1725. [DOI] [PubMed] [Google Scholar]
- Kawaguchi A, et al. Nestin-GFP transgenic mice: visualization of the self-renewal and multipotency of CNS stem cells. Mol. Cell. Neurosci. 2001;17:259–273. doi: 10.1006/mcne.2000.0925. 10.1006/mcne.2000.0925 [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Mizuseki K, Nishikawa S, Kaneko S, Kuwana Y, Nakanishi S, Nishikawa S.I, Sasai Y. Induction of midbrain dopaminergic neurons from ES cells by stromal cell-derived inducing activity. Neuron. 2000;28:31–40. doi: 10.1016/s0896-6273(00)00083-0. 10.1016/S0896-6273(00)00083-0 [DOI] [PubMed] [Google Scholar]
- Kaye E. Update on genetic disorders affecting white matter. Pediatr. Neurol. 2001;24:11–24. doi: 10.1016/s0887-8994(00)00232-0. 10.1016/S0887-8994(00)00232-0 [DOI] [PubMed] [Google Scholar]
- Kerr D.A, et al. Human embryonic germ cell derivatives facilitate motor recovery of rats with diffuse motor neuron injury. J. Neurosci. 2003;23:5131–5140. doi: 10.1523/JNEUROSCI.23-12-05131.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyoung H.M, Roy N.S, Benraiss A, Louissaint A, Jr, Suzuki A, Hashimoto M, Rashbaum W.K, Okano H, Goldman S.A. High-yield selection and extraction of two promoter-defined phenotypes of neural stem cells from the fetal human brain. Nat. Biotechnol. 2001;19:843–850. doi: 10.1038/nbt0901-843. 10.1038/nbt0901-843 [DOI] [PubMed] [Google Scholar]
- Kim J, et al. Dopamine neurons derived from embryonic stem cells function in an animal model of Parkinson's disease. Nature. 2002;418:50–56. doi: 10.1038/nature00900. 10.1038/nature00900 [DOI] [PubMed] [Google Scholar]
- Kim D, et al. Stromal cell-derived inducing activity, Nurr1 and signaling molecules synergistically induce dopaminergic neurons from mouse ES cells. Stem Cells. 2006;24:557–567. doi: 10.1634/stemcells.2005-0233. 10.1634/stemcells.2005-0233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschenbaum B, Nedergaard M, Preuss A, Barami K, Fraser R.A, Goldman S.A. In vitro neuronal production and differentiation by precursor cells derived from the adult human forebrain. Cereb. Cortex. 1994;4:576–589. doi: 10.1093/cercor/4.6.576. [DOI] [PubMed] [Google Scholar]
- Kordower J, Freeman T, Chen E, Mufson E, Sanberg P, Hauser R, Snow B, Olanow C. Fetal nigral grafts survive and mediate clinical benefit in a patient with Parkinson's disease. Mov. Disord. 1998;13:383–393. doi: 10.1002/mds.870130303. 10.1002/mds.870130303 [DOI] [PubMed] [Google Scholar]
- Lacorazza H.D. Expression of human β-hexosaminidase α-subunit gene (the gene defect of Tay-Sachs disease) in mouse brains upon engraftment of transduced progenitor cells. Nat. Med. 1996;2:424–429. doi: 10.1038/nm0496-424. 10.1038/nm0496-424 [DOI] [PubMed] [Google Scholar]
- Lee S.-H, Lumelsky N, Studer L, Auerbach J, McKay R. Efficient generation of midbrain and hindbrain neurons from mouse embryonic stem cells. Nat. Biotechnol. 2000;18:675–679. doi: 10.1038/76536. 10.1038/76536 [DOI] [PubMed] [Google Scholar]
- Lee A, Kessler J, Read T, Kaiser C, Corbeil D, Huttner W, Johnson J, Wechsler-Reya R. Isolation of neural stem cells from the postnatal cerebellum. Nat. Neurosci. 2005;8:723–729. doi: 10.1038/nn1473. 10.1038/nn1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine J.M, Reynolds R, Fawcett J.W. The oligodendrocyte precursor cell in health and disease. Trends Neurosci. 2001;24:39–47. doi: 10.1016/s0166-2236(00)01691-x. 10.1016/S0166-2236(00)01691-X [DOI] [PubMed] [Google Scholar]
- Li M, Pevny L, Lovell-Badge R, Smith A. Generation of purified neural precursors from embryonic stem cells by lineage selection. Curr. Biol. 1998;8:971–974. doi: 10.1016/s0960-9822(98)70399-9. 10.1016/S0960-9822(98)70399-9 [DOI] [PubMed] [Google Scholar]
- Li X, Du Z, Zarnowska E, Pankratz M, Hansen L, Pearce R, Zhang S.-C. Specification of motoneurons from human embryonic stem cells. Nat. Biotechnol. 2005;23:215–221. doi: 10.1038/nbt1063. 10.1038/nbt1063 [DOI] [PubMed] [Google Scholar]
- Lindvall O. Cerebral implantation in movement disorders: state of the art. Mov. Disord. 1999;14:201–205. doi: 10.1002/1531-8257(199903)14:2<201::aid-mds1001>3.0.co;2-q. 10.1002/1531-8257(199903)14:2%3C201::AID-MDS1001%3E3.0.CO;2-Q [DOI] [PubMed] [Google Scholar]
- Lindvall O, Kokaia Z, Martinez-Serrano A. Stem cell therapy for human neurodegenerative disorders—how to make it work. Nat. Med. 2004;10(Suppl.):S42–S50. doi: 10.1038/nm1064. 10.1038/nm1064 [DOI] [PubMed] [Google Scholar]
- Lois C, Alvarez-Buylla A. Proliferating subventricular zone cells in the adult mammalian forebrain can differentiate into neurons and glia. Proc. Natl Acad. Sci. USA. 1993;90:2074–2077. doi: 10.1073/pnas.90.5.2074. 10.1073/pnas.90.5.2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luskin M.B. Restricted proliferation and migration of postnatally generated neurons derived from the forebrain subventricular zone. Neuron. 1993;11:173–189. doi: 10.1016/0896-6273(93)90281-u. 10.1016/0896-6273(93)90281-U [DOI] [PubMed] [Google Scholar]
- Mignone J, Kukekov V, Chiang A, Steindler D, Enikolopov G. Neural stem and progenitor cells in nestin-GFP transgenic mice. J. Comp. Neurol. 2004;469:311–324. doi: 10.1002/cne.10964. 10.1002/cne.10964 [DOI] [PubMed] [Google Scholar]
- Morshead C.M, Reynolds B.A, Craig C.G, McBurney M.W, Staines W.A, Morassutti D, Weiss S, van der K.D. Neural stem cells in the adult mammalian forebrain: a relatively quiescent subpopulation of subependymal cells. Neuron. 1994;13:1071–1082. doi: 10.1016/0896-6273(94)90046-9. 10.1016/0896-6273(94)90046-9 [DOI] [PubMed] [Google Scholar]
- Nakano T, Windrem M, Zappavigna V, Goldman S.A. Identification of a conserved 125 base-pair Hb9 enhancer that specifies gene expression to spinal motor neurons. Dev. Biol. 2005;283:474–485. doi: 10.1016/j.ydbio.2005.04.017. 10.1016/j.ydbio.2005.04.017 [DOI] [PubMed] [Google Scholar]
- Nikulina E, Tidwell J.L, Dai H.N, Bregman B.S, Filbin M.T. The phosphodiesterase inhibitor rolipram delivered after a spinal cord lesion promotes axonal regeneration and functional recovery. Proc. Natl Acad. Sci. USA. 2004;101:8786–8790. doi: 10.1073/pnas.0402595101. 10.1073/pnas.0402595101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nistor G, Totoiu M, Haque N.S, Carpenter M, Kieirstead H. Human embryonic stem cells differentiate into oligodendrocytes in high purity and myelinate after spinal cord contusion. Glia. 2005;49:385–396. doi: 10.1002/glia.20127. 10.1002/glia.20127 [DOI] [PubMed] [Google Scholar]
- Nunes M.C, Roy N.S, Keyoung H.M, Goodman R.R, McKhann G, Jiang L, Kang J, Nedergaard M, Goldman S.A. Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat. Med. 2003;9:439–447. doi: 10.1038/nm837. 10.1038/nm837 [DOI] [PubMed] [Google Scholar]
- Ogawa Y, et al. Transplantation of in vitro-expanded fetal neural progenitor cells results in neurogenesis and functional recovery after spinal cord contusion injury in adult rats. J. Neurosci. Res. 2002;69:925–933. doi: 10.1002/jnr.10341. 10.1002/jnr.10341 [DOI] [PubMed] [Google Scholar]
- Olanow C, Kordower J, Freeman T. Fetal nigral transplantation as a therapy for Parkinson's disease. Trends Neurosci. 1996;19:102–109. doi: 10.1016/s0166-2236(96)80038-5. 10.1016/S0166-2236(96)80038-5 [DOI] [PubMed] [Google Scholar]
- Ourednik J, Ourednik V, Lynch W.P, Schachner M, Snyder E. Neural stem cells display an inherent mechanism for rescuing dysfunctional neurons. Nat. Biotechnol. 2002;20:1103–1110. doi: 10.1038/nbt750. 10.1038/nbt750 [DOI] [PubMed] [Google Scholar]
- Palmer T, Gage F. Mobilizing neural precursor cells in the adult nervous system. In: Rao M, editor. Neural development and stem cells. Humana Press; Totowa, NJ: 2006. pp. 343–370. [Google Scholar]
- Palmer T.D, Ray J, Gage F.H. FGF-2-responsive neuronal progenitors reside in proliferative and quiescent regions of the adult rodent brain. Mol. Cell. Neurosci. 1995;6:474–486. doi: 10.1006/mcne.1995.1035. 10.1006/mcne.1995.1035 [DOI] [PubMed] [Google Scholar]
- Park S, et al. Generation of dopaminergic neurons in vitro from human embryonic stem cells treated with neurotrophic factors. Neurosci. Lett. 2004;359:99–103. doi: 10.1016/j.neulet.2004.01.073. 10.1016/j.neulet.2004.01.073 [DOI] [PubMed] [Google Scholar]
- Pearse D, Pereira F, Marcillo A, Bates M.L, Berrocal Y, Filbin M.T, Bunge M.B. cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat. Med. 2004;10:610–616. doi: 10.1038/nm1056. 10.1038/nm1056 [DOI] [PubMed] [Google Scholar]
- Pellegatta S, Tunici P, Poliani P, Dolcetta D, Cajola L, Colombelli C, Ciusani E, DiDonato S, Finocchiaro G. The therapeutic potential of neural stem/progenitor cells in murine globoid cell leukodystrophy is conditioned by macrophage/microglial activation. Neurobiol. Dis. 2006;21:314–323. doi: 10.1016/j.nbd.2005.07.016. [DOI] [PubMed] [Google Scholar]
- Piccini P, Pavese N, Hagell P, Reimer J, Bjorklund A, Oertel W, Quinn N, Brooks D, Lindvall O. Factors affecting the clinical outcome after neural transplantation in Parkinson's disease. Brain. 2005;128:2977–2986. doi: 10.1093/brain/awh649. 10.1093/brain/awh649 [DOI] [PubMed] [Google Scholar]
- Pincus D.W, et al. Fibroblast growth factor-2/brain-derived neurotrophic factor-associated maturation of new neurons generated from adult human subependymal cells. Ann. Neurol. 1998;43:576–585. doi: 10.1002/ana.410430505. 10.1002/ana.410430505 [DOI] [PubMed] [Google Scholar]
- Pluchino S, et al. Injection of adult neurospheres induces recovery in a chronic model of multiple sclerosis. Nature. 2003;422:688–694. doi: 10.1038/nature01552. 10.1038/nature01552 [DOI] [PubMed] [Google Scholar]
- Pluchino S, Furlan R, Martino G. Cell-based remyelinating therapies in multiple sclerosis: evidence from experimental studies. Curr. Opin. Neurol. 2004;17:247–255. doi: 10.1097/00019052-200406000-00003. 10.1097/00019052-200406000-00003 [DOI] [PubMed] [Google Scholar]
- Powers J. The leukodystrophies: overview and classification. In: Lazzarini R.A, editor. Myelin biology and disorders. vol. 2. Elsevier Academic Press; San Diego, CA: 2004. pp. 663–690. [Google Scholar]
- Rietze R, Valcanis G, Brooker G, Thomas T, Voss A, Bartlett P. Purification of a pluripotent neural stem cell from the adult mouse brain. Nature. 2001;412:736–739. doi: 10.1038/35089085. 10.1038/35089085 [DOI] [PubMed] [Google Scholar]
- Robinson S, Petelenz K, Li Q, Cohen M, Dechant A, Tabrizi N, Bucek M, Lust D, Miller R. Developmental changes induced by graded prenatal systemic hypoxic-ischemic insults in rats. Neurobiol. Dis. 2005;18:568–581. doi: 10.1016/j.nbd.2004.10.024. 10.1016/j.nbd.2004.10.024 [DOI] [PubMed] [Google Scholar]
- Roy N.S, Wang S, Harrison-Restelli C, Benraiss A, Fraser R.A, Gravel M, Braun P.E, Goldman S.A. Identification, isolation, and promoter-defined separation of mitotic oligodendrocyte progenitor cells from the adult human subcortical white matter. J. Neurosci. 1999;19:9986–9995. doi: 10.1523/JNEUROSCI.19-22-09986.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy N.S, et al. Promoter-targeted selection and isolation of neural progenitor cells from the adult human ventricular zone. J. Neurosci. Res. 2000a;59:321–331. doi: 10.1002/(sici)1097-4547(20000201)59:3<321::aid-jnr5>3.0.co;2-9. 10.1002/(SICI)1097-4547(20000201)59:3%3C321::AID-JNR5%3E3.0.CO;2-9 [DOI] [PubMed] [Google Scholar]
- Roy N.S, et al. In vitro neurogenesis by progenitor cells isolated from the adult human hippocampus. Nat. Med. 2000b;6:271–277. doi: 10.1038/73119. 10.1038/73119 [DOI] [PubMed] [Google Scholar]
- Roy N, et al. Telomerase-immortalization of neuronally restricted progenitor cells derived from the human fetal spinal cord. Nat. Biotechnol. 2004;22:297–305. doi: 10.1038/nbt944. 10.1038/nbt944 [DOI] [PubMed] [Google Scholar]
- Roy N, Nakano T, Xuing L, Kang J, Nedergaard M, Goldman S.A. Enhancer-specified GFP-based FACS purification of human spinal motor neurons from embryonic stem cells. Exp. Neurol. 2005;196:224–234. doi: 10.1016/j.expneurol.2005.06.021. 10.1016/j.expneurol.2005.06.021 [DOI] [PubMed] [Google Scholar]
- Sanai N, et al. Unique astrocyte ribbon in adult human brain contains neural stem cells but lacks chain migration. Nature. 2004;427:740–744. doi: 10.1038/nature02301. 10.1038/nature02301 [DOI] [PubMed] [Google Scholar]
- Sawamoto K, et al. Generation of dopaminergic neurons in the adult brain from mesencephalic precursor cells labeled with a nestin-GFP transgene. J. Neurosci. 2001;21:3895–3903. doi: 10.1523/JNEUROSCI.21-11-03895.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scolding N, Franklin R, Stevens S, Heldin C.H, Compston A, Newcombe J. Oligodendrocyte progenitors are present in the normal adult human CNS and in the lesions of multiple sclerosis [see comments] Brain. 1998;121:2221–2228. doi: 10.1093/brain/121.12.2221. 10.1093/brain/121.12.2221 [DOI] [PubMed] [Google Scholar]
- Snyder E.Y, Taylor R.M, Wolfe J.H. Neural progenitor cell engraftment corrects lysosomal storage throughout the MPS VII mouse brain. Nature. 1995;374:367–370. doi: 10.1038/374367a0. 10.1038/374367a0 [DOI] [PubMed] [Google Scholar]
- Studer L, Tabar V, McKay R. Transplantation of expanded mesencephalic precursors leads to recovery in Parkinsonian rats. Nat. Neurosci. 1998;1:290–295. doi: 10.1038/1105. 10.1038/2774 [DOI] [PubMed] [Google Scholar]
- Studer L, Csete M, Lee S, Kabbani N, Walikonis J, Wold B, McKay R. Enhanced proliferation, survival and dopaminergic differentiation of CNS precursors in lowered oxygen. J. Neurosci. 2000;20:7377–7738. doi: 10.1523/JNEUROSCI.20-19-07377.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svendsen C, Caldwell M, Ostenfeld T. Human neural stem cells: isolation, expansion and transplantation. Brain Pathol. 1999;9:499–513. doi: 10.1111/j.1750-3639.1999.tb00538.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi Y, et al. Dopaminergic neurons generated from monkey embryonic stem cells function in a Parkinson primate model. J. Clin. Invest. 2005;115:102–108. doi: 10.1172/JCI21137. 10.1172/JCI200521137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida N, Buck D.W, He D, Reitsma M.J, Masek M, Phan T.V, Tsukamoto A.S, Gage F.H, Weissman I.L. Direct isolation of human central nervous system stem cells. Proc. Natl Acad. Sci. USA. 2000;97:14 720–14 725. doi: 10.1073/pnas.97.26.14720. 10.1073/pnas.97.26.14720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urayama A, Grubb J, Sly W, Banks W. Developmentally regulated mannose 6-phophate receptor-mediated transport of a lysosomal enzyme across the blood–brain barrier. Proc. Natl Acad. Sci. USA. 2004;101:12 658–12 663. doi: 10.1073/pnas.0405042101. 10.1073/pnas.0405042101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Knaap M, Barth P, Gabreels F, Franzoni E, Begeer J, Stroink H, Rotteveel J, Valk J. A new leukoencephalopathy with vanishing white matter. Neurology. 1997;48:845–855. doi: 10.1212/wnl.48.4.845. [DOI] [PubMed] [Google Scholar]
- Vescovi A.L, Reynolds B.A, Fraser D.D, Weiss S. bFGF regulates the proliferative fate of unipotent (neuronal) and bipotent (neuronal/astroglial) EGF-generated CNS progenitor cells. Neuron. 1993;11:951–966. doi: 10.1016/0896-6273(93)90124-a. 10.1016/0896-6273(93)90124-A [DOI] [PubMed] [Google Scholar]
- Vescovi A.L, et al. Isolation and cloning of multipotential stem cells from the embryonic human CNS and establishment of transplantable human stem cells lines by epigenetic stimulation. Exp. Neurol. 1999;156:71–83. doi: 10.1006/exnr.1998.6998. 10.1006/exnr.1998.6998 [DOI] [PubMed] [Google Scholar]
- Wang S, Wu H, Jiang J, Delohery T.M, Isdell F, Goldman S.A. Isolation of neuronal precursors by sorting embryonic forebrain transfected with GFP regulated by the T alpha 1 tubulin promoter. Nat. Biotechnol. 1998;16:196–201. doi: 10.1038/nbt0298-196. 10.1038/nbt0298-196 [DOI] [PubMed] [Google Scholar]
- Wang S, Roy N.S, Benraiss A, Goldman S.A. Promoter-based isolation and fluorescence-activated sorting of mitotic neuronal progenitor cells from the adult mammalian ependymal/subependymal zone. Dev. Neurosci. 2000;22:167–176. doi: 10.1159/000017437. 10.1159/000017437 [DOI] [PubMed] [Google Scholar]
- Wang X, et al. P2X7 receptor inhibition improves recovery after spinal cord injury. Nat. Med. 2004;10:821–827. doi: 10.1038/nm1082. 10.1038/nm1082 [DOI] [PubMed] [Google Scholar]
- Weiss S, Dunne C, Hewson J, Wohl C, Wheatley M, Peterson A.C, Reynolds B.A. Multipotent CNS stem cells are present in the adult mammalian spinal cord and ventricular neuroaxis. J. Neurosci. 1996;16:7599–7609. doi: 10.1523/JNEUROSCI.16-23-07599.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichterle H, Lieberam I, Porter J.A, Jessell T.M. Directed differentiation of embryonic stem cells into motor neurons. Cell. 2002;110:385–397. doi: 10.1016/s0092-8674(02)00835-8. 10.1016/S0092-8674(02)00835-8 [DOI] [PubMed] [Google Scholar]
- Windrem M.S, Roy N.S, Wang J, Nunes M, Benraiss A, Goodman R, McKhann G.M, Goldman S.A. Progenitor cells derived from the adult human subcortical white matter disperse and differentiate as oligodendrocytes within demyelinated lesions of the rat brain. J. Neurosci. Res. 2002;69:966–975. doi: 10.1002/jnr.10397. 10.1002/jnr.10397 [DOI] [PubMed] [Google Scholar]
- Windrem M.S, Roy N.S, Nunes M, Goldman S.A. Identification, selection and use of adult human oligodendrocyte progenitor cells. In: Zigova T, Snyder E, editors. Neural stem cells for brain repair. Humana Press; New York, NY: 2003. pp. 69–88. [Google Scholar]
- Windrem M.S, Nunes M.C, Rashbaum W.K, Schwartz T.H, Goodman R.A, McKhann G, Roy N.S, Goldman S.A. Fetal and adult human oligodendrocyte progenitor cell isolates myelinate the congenitally dysmyelinated brain. Nat. Med. 2004;10:93–97. doi: 10.1038/nm974. 10.1038/nm974 [DOI] [PubMed] [Google Scholar]
- Yandava B.D, Billinghurst L.L, Snyder E.Y. “Global” cell replacement is feasible via neural stem cell transplantation: evidence from the dysmyelinated shiverer mouse brain. Proc. Natl Acad. Sci. USA. 1999;96:7029–7034. doi: 10.1073/pnas.96.12.7029. 10.1073/pnas.96.12.7029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye W, Shimamura K, Rubenstein J, Hynes M, Rosenthal A. FGF and Shh signals control dopaminergic and serotinergic cell fate in the anterior neural plate. Cell. 1998;93:755–766. doi: 10.1016/s0092-8674(00)81437-3. 10.1016/S0092-8674(00)81437-3 [DOI] [PubMed] [Google Scholar]