

Abstract
Integrative physiology emphasizes the importance of understanding multiple pathways with overlapping, complementary, or opposing effects and their interactions in the context of intact organisms. The DNA microarray technology, the most commonly used method for high-throughput gene expression profiling, has been touted as an integrative tool that provides insights into regulatory pathways. However, the physiology community has been slow in acceptance of these techniques because of early failure in generating useful data and the lack of a cohesive theoretical framework in which experiments can be analysed. With recent advances in both technology and analysis, we propose a concept of multidimensional integration of physiology that incorporates data generated by DNA microarray and other functional, genomic, and proteomic approaches to achieve a truly integrative understanding of physiology. Analysis of several studies performed in simpler organisms or in mammalian model animals supports the feasibility of such multidimensional integration and demonstrates the power of DNA microarray as an indispensable molecular tool for such integration. Evaluation of DNA microarray techniques indicates that these techniques, despite limitations, have advanced to a point where the question-driven profiling research has become a feasible complement to the conventional, hypothesis-driven research. With a keen sense of homeostasis, global regulation, and quantitative analysis, integrative physiologists are uniquely positioned to apply these techniques to enhance the understanding of complex physiological functions.
High throughput gene expression profiling has emerged over the last decade as one of the most important and powerful approaches in biomedical research. The rapid progress in this field is in large part driven by the development and application of DNA microarray techniques (Schena et al. 1995; Lockhart et al. 1996). With DNA microarray techniques, the mRNA expression level of thousands of genes or potentially the entire genome can be assessed simultaneously, while traditional methods only allow one or a few genes to be examined at a time. The power of microarray techniques has been clearly demonstrated in numerous studies (Chipping Forecast, 1999, 2002), particularly in genetics and cancer research. The rapidly increasing popularity of these techniques is evidenced by the number of publications involving microarrays, which exceeded 1000 in the year 2002 alone.
Changes in gene expression are often results of interactions between genetic materials and environmental stimuli, which underlie the regulation of various physiological responses, especially those with long-term implications. While the microarray technology has been touted as an integrative tool that provides insights into regulatory pathways, the physiology community has been slow in acceptance of these techniques because of early failure in generating useful data and the lack of a cohesive theoretical framework in which experiments can be analysed. With recent advance in both technology and analysis, it is appropriate to re-examine the use of microarray technology in the physiology laboratory.
In this review, we will address the question of how integrative physiologists can take advantage of these high-throughput gene expression profiling techniques in order to better understand organ function. The authors will present reasoning and evidence that these techniques, although appearing very ‘molecular’, are in fact particularly suitable and powerful for studying integrative physiology.
An overview of the DNA microarray technology
DNA microarray technology involves hybridizing target molecules derived from mRNA samples to probe molecules that have been immobilized as thousands of microspots on a solid support. The fluorescence or radioactivity incorporated in the target molecules can then be quantified in each microspot to reflect the number of target molecules corresponding to the genes represented by the probes in that spot. Figure 1 depicts the basic process of typical cDNA microarray techniques that have been utilized in our laboratories (Kita et al. 2002; Liang et al. 2002, 2003b; Yuan et al. 2003), which are derived from the prototype described by Brown and colleagues (Schena et al. 1995; Brown Laboratory homepage). Basically, thousands of cDNA clones are individually amplified by PCR and printed on glass slides using a robotic arrayer to produce microarrays of DNA probes. mRNAs from two samples (e.g. control and treated) are reverse-transcribed to cDNA and labelled with Cy3 and Cy5 fluorescent dyes, respectively. The labelled cDNAs are pooled and hybridized to a microarray. After unbound labelled cDNA is washed off the slide, fluorescence intensities of Cy3 and Cy5 are quantified in each spot using a laser confocal scanner. Fluorescence intensity data are then analysed to yield log-transformed ratios between Cy3 and Cy5 in each spot, which, with spotted DNA probes in excess, reflect the relative mRNA expression levels of the corresponding gene in the control and treated samples.
Figure 1. Basic process of a typical cDNA microarray experiment.

RT, reverse transcription.
The potentially enormous power of DNA microarrays has stimulated numerous technological innovations that improve or diversify microarray techniques. An important variant is oligonucleotide microarray in which specifically designed oligonucleotide probes are synthesized in situ (Lockhart et al. 1996) or printed in place of cDNA. Other significant technical modifications include non-contact printing (Okamoto et al. 2000), indirect labelling (Randolph & Waggoner, 1997; Hughes et al. 2001), and target (Van Gelder et al. 1990; Luo et al. 1999) or signal (Stears et al. 2000) amplification. Non-contact printing often uses ink jet-based techniques rather than the standard pin contact-based techniques to print DNA probes, and has the potential to increase spot density and be cost effective. Indirect labelling allows reverse transcription to be carried out prior to differential labelling and thus reduces the confounding effect of different labelling molecules on reverse transcription. Amplification of target mRNAs or fluorescence signals enables investigators to profile gene expression when only small amounts of RNA samples are available, although the quantitative fidelity of the amplification, particularly in the case of target amplification, remains a subject of hot debate. The use of DNA microarrays has also expanded from mRNA expression measurements to include for example genomic sequence analysis using oligonucleotide probes with specific sequence variations (Hacia, 1999). Furthermore, the microarray format has been adapted to study a wide variety of subjects such as gene functions in cells (Ziauddin & Sabatini, 2001), small molecule interactions (Kuruvilla et al. 2002), tissue profiling (Kononen et al. 1998), and protein activities (Zhu et al. 2001).
The DNA microarray as a molecular tool for studying integrative physiology
Concept. Integrative physiology is traditionally characterized by its focus on the study of whole organisms, especially whole animals. It emphasizes the interaction between multiple functional pathways and regulatory mechanisms with overlapping, complementary, or opposing effects in the context of intact organisms. Its ultimate goal is to understand quantitatively how an organism works as a whole. However, because of the formidable complexity of an intact organism, especially in mammalian model animals commonly used by physiologists, integrative physiology can become a victim of the so-called ‘black box’ phenomenon: the inability to interpret the mechanistic link between the input (such as an experimental manipulation) and the output (such as the functional alterations observed). In addition, the lack of appropriate tools may force integrative physiologists to abandon their emphasis on the global network of regulation or, worse yet, to yield themselves to the temptation of explaining the entire network based on the small parts that they are able to study.
Revolutionary progress in genomics along with an array of new tools, such as those for high throughput gene expression profiling, have brought new prospects and opportunities to integrative physiology. As schematized in Fig. 2, these new tools are beginning to make a multidimensional integration of physiology possible. At the top of this multidimensional integration is the traditional integrative physiology that categorizes, measures, and integrates the functions of an organism. An excellent example is the cardiovascular model established by Guyton and associates more than three decades ago (Guyton et al. 1972). This first dimension of integrative physiology is termed ‘physiome’ by some (Physiome Project website). At the bottom is the genome sequence, which is or soon will be available for many species including human, mouse and rat (Rat Genome Project website; Rat Genome Database website). High throughput assays can then be used to categorize and measure on a global scale genome sequence variations, mRNA expression, and protein expression, modification, and interaction, as well as small regulatory molecules. The results of these measurements will constitute what Vidal described as ‘a biological atlas’ (Vidal, 2001). These will then become the substance for a second dimension of integration that links the genome to transcriptome, proteome and function. The integration could and should also be carried out in yet another dimension that is across different environmental conditions that influence the organism as well as across various developmental stages.
Figure 2. Multi-dimensional integrative physiology.

The availability of genome sequences and a variety of high-throughput techniques (several examples shown in italic font) has made it possible to integrate physiology across functional pathways (1st dimension), regulatory levels (2nd dimension), and various conditions (3rd dimension). MS, mass spectrometry; SAGE, serial analysis of gene expression; SNP, single nucleotide polymorphism.
Such a multidimensional map of an organism is precisely what one would need in order to truly understand how an organism works as a whole. With a keen sense of homeostasis, global regulation, and quantitative analysis, integrative physiologists are uniquely prepared for creating and refining such multidimensional maps. The potential value of this new, multidimensional integrative physiology has been recognized by a number of investigators. Several terms, such as functional genomics (Science Functional Genomics website), physiological genomics (Cowley, 1997), systems biology (Kitano, 2002), and computational biology (Nature Focus on Computational Biology website), have been used to describe this concept of integrative physiology or to emphasize some aspects of it.
Approach. Can this actually be done in the foreseeable future? Obviously, an enormous amount of detail needs to be worked out, and new technologies and tools need to be developed if this is to be done with any level of efficiency. Already, a number of attempts, primarily involving the use of DNA microarray, have been made in this direction with various degrees of success. These studies were mostly performed in simpler model organisms and typically investigated only one dimension of the ‘biological atlas’. However, they provided examples of how high throughput expression profiling techniques such as DNA microarray can be used to assist the integration of physiology. The following section of this article will focus on analysing in some details the approaches used in several of these studies and how the results might be expanded.
Hughes and colleagues (Hughes et al. 2000) used DNA microarrays to produce a compendium of genome-wide expression profiles in yeast corresponding to 300 mutations and chemical treatments. They argued that these reference expression profiles could serve as ‘fingerprints’ to identify functions of unknown open reading frames (ORFs), which are segments of DNA potentially encoding proteins. If the mutation of an unknown ORF resulted in an expression profile that matches reference expression profiles associated with mutants of certain known genes, the unknown ORF would likely belong to the same functional pathway as those known genes. Matching of expression profiles was carried out through agglomerative hierarchical clustering with an error-weighted correlation coefficient as the similarity measure. The validity of this approach was first demonstrated by the grouping of known genes that was apparently consistent with their known affiliation with specific functional pathways. Several uncharacterized ORFs or drug treatments were then assigned or linked to certain functional pathways according to their clustering with known genes or treatments. For example, YER044c, an unknown ORF, was assigned to the yeast ergosterol biosynthesis pathways because its mutations resulted in expression profiles that clustered with a group of genes involved in this pathway. This was verified by showing that YER044c mutant yeasts accumulated more sterols and less ergosterol compared to the wild-type. The authors noted that identifying the role of YER044c would have been difficult using traditional approaches since the mutations of this ORF only had a modest effect on ergosterol biosynthesis. Thus, through the analysis of microarray-generated gene expression profiles in combination with gene targeting or pharmacological intervention, the understanding of the functional network of the yeast was being refined and improved at a pace exceeding the capacity of conventional methods. As acknowledged by the authors, this approach was based on the assumption that perturbations of most, if not all, functional pathways would be associated with unique expression profiles. Moreover, disruption of any steps of a pathway had to be sufficient to elicit responses characteristic of that pathway. The validity of these assumptions would be questionable especially in more complex mammalian systems where the boundaries between many pathways are ambiguous as a gene may be involved in several distinct pathways and different pathways may have overlapping functions.
Several other studies attempted to assemble global transcriptional regulatory networks in the yeast based on microarray data. In a study by Lee et al. (2002), chromatin immunoprecipitation and a promoter microarray were first used to identify approximately 4000 interactions between 106 transcriptional regulators and promoter regions based on an arbitrary P-value threshold that was believed to be stringent. The regulatory motifs revealed by this location analysis were then refined for common expression by reiterative matching of expression profiles obtained from a large number of microarray experiments. In this case, gene expression profiles were used to partially overcome the limitation of the location analysis. As an example, the approach was used to assemble a cell cycle transcriptional regulatory network. The regulatory network assembled was found to be largely consistent with previous findings and to suggest roles for several less well-characterized transcriptional regulators. Tavazoie et al. (1999) used a k-means algorithm with Euclidean distance as the similarity measure to cluster expression profiles obtained from a cell cycle microarray study (Cho et al. 1998). Some, but not all, gene clusters appeared to be enriched for genes with similar functions or contain common upstream cis-regulatory motifs. This work was extended to emphasize the combinatorial nature of transcriptional regulation through analysis of Euclidean distance-based expression coherence score utilizing several microarray datasets (Pilpel et al. 2001).
These studies demonstrated the possibility and the power of integrating genome, transcriptome, and physiome (see Fig. 2). It is also possible to integrate proteomic data into this paradigm. Ideker et al. (2001) carried out a proof-of-principle study for integrating genomic and proteomic analyses to refine a model of a metabolic pathway. Specifically, the galactose utilization pathway in yeast was systematically perturbed by a series of genetic deletions and environmental stresses. The resulting global changes of mRNA expression were measured by microarrays. Changes in protein expression were measured by tandem mass spectrometry using isotope-coded affinity tags to distinguish control and treated samples. The results of microarray and proteomic analysis were used to refine the initial model of this pathway. The integration could be extended to include other levels of regulation. For example, by comparing microarray-based mRNA expression profiles and protein–protein interaction profiles generated by yeast two-hybrid analyses, Ge et al. (2001) found that genes with similar mRNA expression profiles were more likely to encode interacting proteins.
The utilization of microarray data in the integration of regulatory networks has also been demonstrated in other species such as Drosophila (Furlong et al. 2001; Stathopoulos et al. 2002; Halfon & Michelson, 2002) and C. elegans (Kim et al. 2001).
Studies in simpler organisms often provide useful models that can be applied to the study of more complex mammalian organisms such as rats and mice that physiologists commonly use. Although the complexity of these mammalian organisms remains a major obstacle, high throughput expression profiling techniques in combination with other approaches are beginning to generate evidence supporting the feasibility of the multidimensional integration of mammalian physiology depicted in Fig. 2.
Utilizing a chromosomal substitution approach, Jacob and colleagues (Cowley et al. 2001; PhysGen website) have been generating a panel of consomic rats in which chromosomes of one strain are being replaced one at a time by those from another strain. Parental and consomic rats are functionally characterized by measuring several hundred phenotypes related to cardiovascular, renal and other functions. The analysis of the F2 population derived from two parental strains, Dahl salt-sensitive (SS) and Brown Norway (BN) rats, generated an extensive map that linked hundreds of phenotypes with specific regions of the genome (Stoll et al. 2001). To extend this link between the genome and the physiome, initial DNA microarray analyses have been carried out in SS and consomic SS-13BN/Mcw rats (Liang et al. 2002, 2003b). In SS-13BN/Mcw, chromosome 13 of SS has been replaced by that of BN. As a result, the blood pressure salt-sensitivity and salt-induced renal injury were substantially reduced compared to SS (Cowley et al. 2001). Using a custom-made microarray representing the majority of known rat genes, 50 genes were identified to exhibit distinct temporal expression profiles in the renal medulla between SS and SS-13BN/Mcw following dietary high-salt exposure for overnight, three days, or two weeks (Liang et al. 2003b). A Euclidean distance-based index was used as the similarity measure for expression profiles. Interestingly, 30 of these 50 genes could be assembled into two networks related to the regulation of blood pressure and/or renal injury. One of the networks is shown in Fig. 3. Importantly, the expression patterns of the majority of these genes were consistent with reduced blood pressure salt-sensitivity and reduced renal injury in SS-13BN/Mcw. For example, glucagon receptor in the renal medulla was down-regulated after an overnight exposure to a high-salt diet in both SS and SS-13BN/Mcw. However, it returned to the control level after 3 days in SS-13BN/Mcw while remaining suppressed in SS through 2 weeks. On the basis of the known effect of glucagon to increase glomerular filtration rate and sodium excretion, this expression pattern would be consistent with glucagon receptor contributing to the lower blood pressure in SS-13BN/Mcw rats and suggest a direct renal effect of glucagon in addition to indirect effects through the liver. Furthermore, inspecting the entire network revealed several general characteristics. For example, multiple pathways appeared to contribute to the development of salt-induced hypertension in a temporally specific manner. Several genes related to blood pressure regulation were differentially expressed on early time points, others on later time points. Without using high-throughput profiling techniques, it would have been difficult to identify network-wide characteristics of gene expression or the differential expression of many genes shown in Fig. 3 that have less well-known links to the regulation of blood pressure and renal injury. These regulatory networks are obviously not comprehensive and require additional data to complete our understanding. Interactions between various components of these networks, which most certainly exist, are not apparent. New roles of genes suggested by these networks need to be experimentally validated. Nonetheless, these studies supported the feasibility of using high-throughput expression profiling techniques in combination with genetic and functional approaches to integrate mammalian physiology.
Figure 3. A network of genes potentially related to the reduction of blood pressure salt-sensitivity in SS-13BN/Mcw rats identified by cDNA microarray (based on data from Liang et al. 2003b).

Each panel depicts the expression pattern of a gene, which is followed by a brief description of the relevant functions of this gene. Text in the boxes indicates potential functional consequences in SS-13BN/Mcw compared to SS as a result of the expression patterns. The thickness of arrow indicates the strength of support for those links based on current literature. LS, low-salt (0.4% NaCl) diet; HS, high-salt (4% NaCl) diet.
From the studies analysed above, several points become clear. First, high throughput expression profiling techniques such as DNA microarray are indispensable tools in multidimensional integration of physiology. Second, clustering expression profiles based on certain similarity measures is currently the most-often used approach to analyse regulatory networks based on microarray data. Third, as in conventional modelling, assumptions and arbitrary criteria are a necessary part of the analysis of global regulatory networks. In the meantime, noise is common in these network models due to limitations in current technologies and perhaps the nature of the networks per se. This suggests that experimental validation would be critical.
Technical considerations
Despite the demonstrated power of DNA microarray techniques, some investigators still hesitate to accept them. One of the sharpest criticisms that microarray-based studies receive from physiologists is its lack of hypotheses, and thus the term ‘fishing expedition’. In response to or in anticipation of such criticism, some investigators try to attach some sort of hypotheses to their microarray-based studies. Such hypotheses often turn out to be either too vague to be relevant or so specific that they undermine the rationale for using microarrays.
Philosophically, the lack of a hypothesis hardly qualifies as a reason to dismiss a study. Although hypothesis-driven research has been very effective and widely accepted, it is in essence an approach to achieve the objective of finding the answer to a scientific question. Mandating a hypothesis for every study is confusing approaches with objectives. If one knows that a question has a thousand potential answers and can practically examine all of them, it becomes redundant to hypothesize that answer X is the right one. In cases like this, it makes more sense to bypass hypothesis and carry out experiments that directly address the question (Fig. 4). In fact, the minimal need for a hypothesis can sometimes be an advantage of microarray-based studies. Because of the high-throughput ability provided by microarray techniques, one could carry out question-driven studies without prior knowledge of the involvement or even the function of many genes and generate un-biased, global views of biological process, which may in turn become a rich source of testable hypotheses and discoveries.
Figure 4. Question-driven researches can complement traditional hypothesis-driven researches.

This, however, does not imply that microarray-based studies can be carried out without a solid rationale. On the contrary, because of the large financial and intellectual burden that an extensive microarray study often carries, a solid rationale is even more needed than in a conventional study where trial-and-error can be more easily tolerated. Such a solid rationale begins with identifying a question that is sufficiently complicated to justify a global profiling. Questions in which the expression of only one or a few specific genes is concerned are better addressed using other techniques such as real-time PCR. It then involves carefully designing the microarray study and optimizing the entire range of microarray techniques to a point where reasonable confidence can be placed on the massive amount of data that will be generated. A large number of articles have reviewed various aspects of DNA microarray techniques ranging from array fabrication, experimental design, target preparation, to data analysis. For example, determining the threshold of differential expression requires careful consideration of issues prominent in microarray such as multiple comparisons. The readers are referred to the Chipping Forecast (1999, 2002) as well as several other review articles (Yang & Speed, 2002, Quackenbush, 2001) for further discussion on these issues.
Once one has adapted a set of techniques, the immediate concern is how reliable the results are. Assessing reliability is not an easy task, especially when a ‘gold standard’ is not available as in the case of high-throughput gene expression profiling (see Liang et al. 2003a; for further discussion). It is also self-evident that the reliability of microarray results, as with any other complex techniques, would be substantially affected by the technical details of each step of the experiment. However, several pieces of evidence suggest that the massive amount of data generated by microarray techniques can be reliable. As shown in Fig. 5A, microarray images generated by repeating the same hybridization on two arrays can be visually reproducible. In the study by Liang et al. (2002), the reproducibility of array results was further demonstrated by the significant correlation and outlier concordance between experimental results of thousands of genes and results predicted on the basis of a four-way comparison design. As shown in Fig. 5B, ln (ratio)s of dozens of randomly selected genes obtained from microarrays also significantly correlated with those obtained from Northern blots (Liang et al. 2002, 2003b; Yuan et al. 2003).
Figure 5. Reliability of cDNA microarray data.
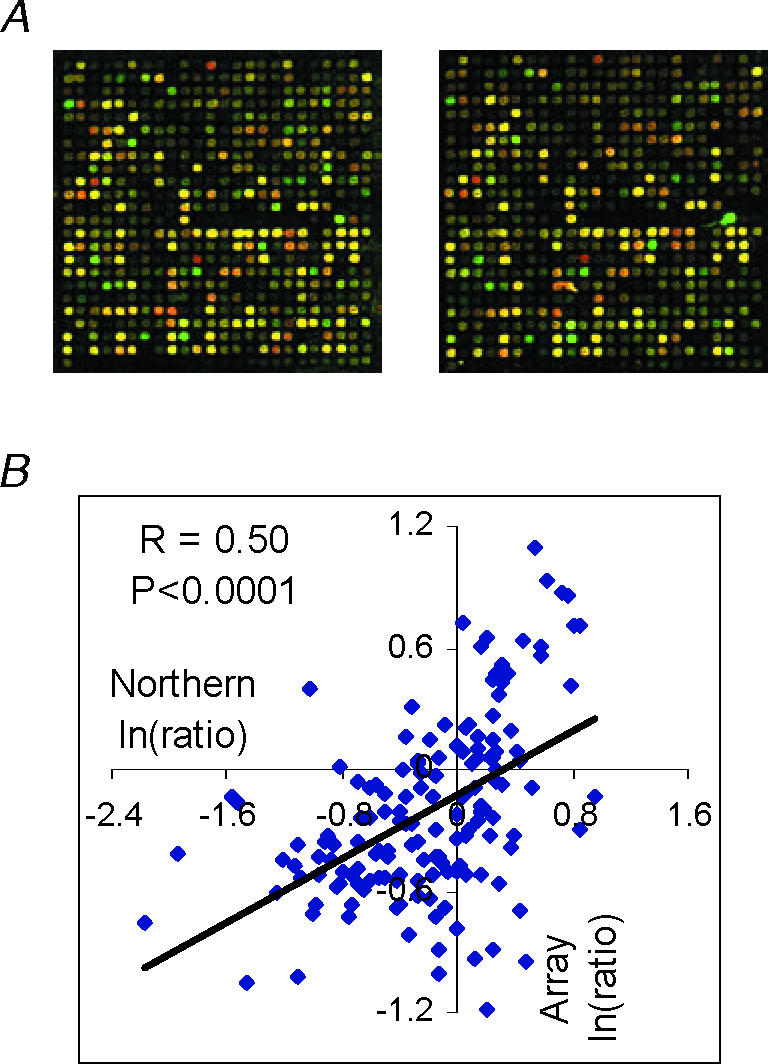
A, partial images from duplication microarray hybridizations. B, significant correlation between ln(ratio)s obtained from microarrays and Northern blots examining approximately 60 randomly selected genes under various treatment conditions (based on data from Liang et al. 2002, 2003b; Yuan et al. 2003).
It is important to recognize several limitations in cDNA microarray techniques. Perhaps the most significant one is their reduced ability to reliably detect low-abundance mRNAs compared to Northern blots or real-time quantitative PCR, although this may in part be related to the conservative data filtering criteria that are applied. There are also concerns regarding the ability of cDNA microarrays to distinguish highly similar sequences. The amount of mRNA samples needed is at the microgram range (unless target or signal amplification methods are used), which could be difficult to obtain in some circumstances. Finally, these techniques are rather complex, and initial set-up and optimization may require efforts that are considerable for an average laboratory.
Other methods for high-throughput gene expression profiling
In addition to DNA microarray, a number of other experimental techniques have been developed and utilized to screen or determine mRNA expression in a high-throughput manner. They include subtractive hybridization (Sargent & Dawid, 1983), representational difference analysis (Lisitsyn et al. 1993; Hubank & Schatz, 1994), suppression subtractive hybridization (Diatchenko et al. 1996), mRNA differential display (Liang & Pardee, 1992), and serial analysis of gene expression (SAGE, Velculescu et al. 1995). Subtractive hybridization basically involves hybridizing cDNAs generated from two mRNA populations and identifying mRNA species that are differentially expressed in the two populations by separating single-stranded and double-stranded cDNAs. Representational difference analysis and suppression subtractive hybridization are variants of subtractive hybridization that incorporate PCR-based steps to improve the efficiency of the method. These methods are mainly used for screening purposes, especially to identify mRNAs that are present in one sample, but absent in the other. mRNA differential display is a PCR-based method that distinguishes mRNA species by the size of PCR products and allows the comparison of the expression levels of genes that are covered by the primer sets. SAGE makes use of a pair of restriction endonucleases (the anchoring and the tagging enzymes) to create a short sequence tag from each mRNA molecule. The sequence of the tag is likely unique for each gene. These tags are then amplified, concatenated, and sequenced. The copy number of each unique tag sequence represents the abundance of the corresponding mRNA. SAGE is an attractive profiling technique because of its potential ability to provide truly global view of expression even for genes whose sequences are unknown.
Summary
Progress in genomics have generated new possibilities for advancing physiology (Chien, 2000; Cowley, 2003). High throughput expression profiling techniques such as DNA microarray are powerful tools that, when combined with other approaches, can be utilized by physiologists to achieve the goal of multidimensional integration of physiology. It is clear that new technologies as well as refinements of current technologies, both experimental and computational, are needed to construct global regulatory networks. Integrative physiologists are uniquely qualified for advancing and applying such technologies.
References
- Brown Laboratory. http://cmgm.stanford.edu/pbrown/
- Chien KR. Genomic circuits and the integrative biology of cardiac diseases. Nature. 2000;407:227–232. doi: 10.1038/35025196. [DOI] [PubMed] [Google Scholar]
- Chipping Forecast. Nat Genet. 2002;32(Suppl.) [Google Scholar]
- Chipping Forecast. Nat Genet. 1999;21(Suppl.) [Google Scholar]
- Cho RJ, Campbell MJ, Winzeler EA, Steinmetz L, Conway A, Wodicka L, Wolfsberg TG, Gabrielian AE, Landsman D, Lockhart DJ, Davis RW. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol Cell. 1998;2:65–73. doi: 10.1016/s1097-2765(00)80114-8. [DOI] [PubMed] [Google Scholar]
- Cowley AW., Jr The Banbury Conference. Genomics to physiology and beyond: how do we get there? Physiologist. 1997;40:205–211. [PubMed] [Google Scholar]
- Cowley AW., Jr Genomics and homeostasis. Am J Physiol Regul Integr Comp Physiol. 2003;284:R611–627. doi: 10.1152/ajpregu.00567.2002. [DOI] [PubMed] [Google Scholar]
- Cowley AW, Jr, Roman RJ, Kaldunski ML, Dumas P, Dickhout JG, Greene AS, Jacob HJ. Brown Norway chromosome 13 confers protection from high salt to consomic Dahl S rat. Hypertension. 2001;37:456–461. doi: 10.1161/01.hyp.37.2.456. [DOI] [PubMed] [Google Scholar]
- Diatchenko L, Lau YF, Campbell AP, Chenchik A, Moqadam F, Huang B, Lukyanov S, Lukyanov K, Gurskaya N, Sverdlov ED, Siebert PD. Suppression subtractive hybridization. a method for generating differentially regulated or tissue-specific cDNA probes and libraries. Proc Natl Acad Sci USA. 1996;93:6025–6030. doi: 10.1073/pnas.93.12.6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlong EE, Andersen EC, Null B, White KP, Scott MP. Patterns of gene expression during Drosophila mesoderm development. Science. 2001;293:1629–1633. doi: 10.1126/science.1062660. [DOI] [PubMed] [Google Scholar]
- Ge H, Liu Z, Church GM, Vidal M. Correlation between transcriptome and interactome mapping data from Saccharomyces cerevisiae. Nat Genet. 2001;29:482–486. doi: 10.1038/ng776. [DOI] [PubMed] [Google Scholar]
- Guyton AC, Coleman TG, Granger HJ. Circulation: overall regulation. Annu Rev Physiol. 1972;34:13–46. doi: 10.1146/annurev.ph.34.030172.000305. [DOI] [PubMed] [Google Scholar]
- Hacia JG. Resequencing and mutational analysis using oligonucleotide microarrays. Nat Genet. 1999;21(Suppl.):42–47. doi: 10.1038/4469. [DOI] [PubMed] [Google Scholar]
- Halfon MS, Michelson AM. Exploring genetic regulatory networks in metazoan development. methods and models. Physiol Genomics. 2002;10:131–143. doi: 10.1152/physiolgenomics.00072.2002. [DOI] [PubMed] [Google Scholar]
- Hubank M, Schatz DG. Identifying differences in mRNA expression by representational difference analysis of cDNA. Nucl Acids Res. 1994;22:5640–5648. doi: 10.1093/nar/22.25.5640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TR, Mao M, Jones AR, Burchard J, Marton MJ, Shannon KW, Lefkowitz SM, Ziman M, Schelter JM, Meyer MR, Kobayashi S, Davis C, Dai H, He YD, Stephaniants SB, Cavet G, Walker WL, West A, Coffey E, Shoemaker DD, Stoughton R, Blanchard AP, Friend SH, Linsley PS. Expression profiling using microarrays fabricated by an ink-jet oligonucleotide synthesizer. Nat Biotechnol. 2001;19:342–347. doi: 10.1038/86730. [DOI] [PubMed] [Google Scholar]
- Hughes TR, Marton MJ, Jones AR, Roberts CJ, Stoughton R, Armour CD, Bennett HA, Coffey E, Dai H, He YD, Kidd MJ, King AM, Meyer MR, Slade D, Lum PY, Stepaniants SB, Shoemaker DD, Gachotte D, Chakraburtty K, Simon J, Bard M, Friend SH. Functional discovery via a compendium of expression profiles. Cell. 2000;102:109–126. doi: 10.1016/s0092-8674(00)00015-5. [DOI] [PubMed] [Google Scholar]
- Ideker T, Thorsson V, Ranish JA, Christmas R, Buhler J, Eng JK, Bumgarner R, Goodlett DR, Aebersold R, Hood L. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science. 2001;292:929–934. doi: 10.1126/science.292.5518.929. [DOI] [PubMed] [Google Scholar]
- Kim SK, Lund J, Kiraly M, Duke K, Jiang M, Stuart JM, Eizinger A, Wylie BN, Davidson GS. A gene expression map for Caenorhabditis elegans. Science. 2001;293:2087–2092. doi: 10.1126/science.1061603. [DOI] [PubMed] [Google Scholar]
- Kita Y, Shiozawa M, Jin W, Majewski RR, Besharse JC, Greene AS, Jacob HJ. Implications of circadian gene expression in kidney, liver and the effects of fasting on pharmacogenomic studies. Pharmacogenetics. 2002;12:55–65. doi: 10.1097/00008571-200201000-00008. [DOI] [PubMed] [Google Scholar]
- Kitano H. Systems biology: a brief overview. Science. 2002;295:1662–1664. doi: 10.1126/science.1069492. [DOI] [PubMed] [Google Scholar]
- Kononen J, Bubendorf L, Kallioniemi A, Barlund M, Schraml P, Leighton S, Torhorst J, Mihatsch MJ, Sauter G, Kallioniemi OP. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat Med. 1998;4:844–847. doi: 10.1038/nm0798-844. [DOI] [PubMed] [Google Scholar]
- Kuruvilla FG, Shamji AF, Sternson SM, Hergenrother PJ, Schreiber SL. Dissecting glucose signalling with diversity-oriented synthesis and small-molecule microarrays. Nature. 2002;416:653–657. doi: 10.1038/416653a. [DOI] [PubMed] [Google Scholar]
- Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, Hannett NM, Harbison CT, Thompson CM, Simon I, Zeitlinger J, Jennings EG, Murray HL, Gordon DB, Ren B, Wyrick JJ, Tagne JB, Volkert TL, Fraenkel E, Gifford DK, Young RA. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- Liang M, Briggs AG, Rute E, Greene AS, Cowley AW., Jr Quantitative assessment of the importance of dye switching and biological replication in cDNA microarray studies. Physiol Genomics. 2003a;14:199–207. doi: 10.1152/physiolgenomics.00143.2002. [DOI] [PubMed] [Google Scholar]
- Liang P, Pardee AB. Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science. 1992;257:967–971. doi: 10.1126/science.1354393. [DOI] [PubMed] [Google Scholar]
- Liang M, Yuan B, Rute E, Greene AS, Olivier M, Cowley AW., Jr Insights into Dahl salt-sensitive hypertension revealed by temporal patterns of renal medullary gene expression. Physiol Genomics. 2003b;12:229–237. doi: 10.1152/physiolgenomics.00089.2002. [DOI] [PubMed] [Google Scholar]
- Liang M, Yuan B, Rute E, Greene AS, Zou AP, Soares PMC, Question GD, Slocum GR, Jacob HJ, Cowley AW., Jr Renal medullary genes in salt-sensitive hypertension: a chromosomal substitution and cDNA microarray study. Physiol Genomics. 2002;8:139–149. doi: 10.1152/physiolgenomics.00083.2001. [DOI] [PubMed] [Google Scholar]
- Lisitsyn N, Lisitsyn N, Wigler M. Cloning the differences between two complex genomes. Science. 1993;259:946–951. doi: 10.1126/science.8438152. [DOI] [PubMed] [Google Scholar]
- Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV, Chee MS, Mittmann M, Wang C, Kobayashi M, Horton H, Brown EL. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat Biotechnol. 1996;14:1675–1680. doi: 10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- Luo L, Salunga RC, Guo H, Bittner A, Joy KC, Galindo JE, Xiao H, Rogers KE, Wan JS, Jackson MR, Erlander MG. Gene expression profiles of laser-captured adjacent neuronal subtypes. Nat Med. 1999;5:117–122. doi: 10.1038/4806. [DOI] [PubMed] [Google Scholar]
- Nature Focus on Computational Biology. http://www.nature.com/nature/computationalbiology.
- Okamoto T, Suzuki T, Yamamoto N. Microarray fabrication with covalent attachment of DNA using bubble jet technology. Nat Biotechnol. 2000;18:438–441. doi: 10.1038/74507. [DOI] [PubMed] [Google Scholar]
- PhysGen. Programs for genomic applications. http://pga.mcw.edu.
- Physiome Project. http://www.physiome.org.
- Pilpel Y, Sudarsanam P, Church GM. Identifying regulatory networks by combinatorial analysis of promoter elements. Nat Genet. 2001;29:153–159. doi: 10.1038/ng724. [DOI] [PubMed] [Google Scholar]
- Quackenbush J. Computational analysis of microarray data. Nat Rev Genet. 2001;2:418–427. doi: 10.1038/35076576. [DOI] [PubMed] [Google Scholar]
- Randolph JB, Waggoner AS. Stability, specificity and fluorescence brightness of multiply-labeled fluorescent DNA probes. Nucl Acids Res. 1997;25:2923–2929. doi: 10.1093/nar/25.14.2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rat Genome Database. http://rgd.mcw.edu.
- Rat Genome Project. http://www.hgsc.bcm.tmc.edu/projects/rat.
- Sargent TD, Dawid IB. Differential gene expression in the gastrula of Xenopus laevis. Science. 1983;222:135–139. doi: 10.1126/science.6688681. [DOI] [PubMed] [Google Scholar]
- Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- Science Functional Genomics. http://www.sciencemag.org/feature/plus/sfg.
- Stathopoulos A, Van Drenth M, Erives A, Markstein M, Levine M. Whole-genome analysis of dorsal-ventral patterning in the Drosophila embryo. Cell. 2002;111:687–701. doi: 10.1016/s0092-8674(02)01087-5. [DOI] [PubMed] [Google Scholar]
- Stears RL, Getts RC, Gullans SR. A novel, sensitive detection system for high-density microarrays using dendrimer technology. Physiol Genomics. 2000;3:93–99. doi: 10.1152/physiolgenomics.2000.3.2.93. [DOI] [PubMed] [Google Scholar]
- Stoll M, Cowley AW, Jr, Tonellato PJ, Greene AS, Kaldunski ML, Roman RJ, Dumas P, Schork NJ, Wang Z, Jacob HJ. A genomic-systems biology map for cardiovascular function. Science. 2001;294:1723–1726. doi: 10.1126/science.1062117. [DOI] [PubMed] [Google Scholar]
- Tavazoie S, Hughes JD, Campbell MJ, Cho RJ, Church GM. Systematic determination of genetic network architecture. Nat Genet. 1999;22:281–285. doi: 10.1038/10343. [DOI] [PubMed] [Google Scholar]
- Van Gelder RN, Zastrow ME, Yool A, Dement WC, Barchas JD, Eberwine JH. Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proc Natl Acad Sci U S A. 1990;87:1663–1667. doi: 10.1073/pnas.87.5.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velculescu VE, Zhang L, Vogelstein B, Kinzler KW. Serial analysis of gene expression. Science. 1995;270:484–487. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]
- Vidal M. A biological atlas of functional maps. Cell. 2001;104:333–339. doi: 10.1016/s0092-8674(01)00221-5. [DOI] [PubMed] [Google Scholar]
- Yang YH, Speed T. Design issues for cDNA microarray experiments. Nat Rev Genet. 2002;3:579–588. doi: 10.1038/nrg863. [DOI] [PubMed] [Google Scholar]
- Yuan B, Liang M, Yang Z, Rute E, Taylor N, Olivier M, Cowley AW., Jr Gene expression reveals vulnerability to oxidative stess and interstitial fibrosis of the renal outer medulla to non-hypertensive elevations of angiotensin II. Am J Physiol Regul Integr Comp Physiol. 2003;284:R1219–1230. doi: 10.1152/ajpregu.00257.2002. [DOI] [PubMed] [Google Scholar]
- Zhu H, Bilgin M, Bangham R, Hall D, Casamayor A, Bertone P, Lan N, Jansen R, Bidlingmaier S, Houfek T, Mitchell T, Miller P, Dean RA, Gerstein M, Snyder M. Global analysis of protein activities using proteome chips. Science. 2001;293:2101–2105. doi: 10.1126/science.1062191. [DOI] [PubMed] [Google Scholar]
- Ziauddin J, Sabatini DM. Microarrays of cells expressing defined cDNAs. Nature. 2001;411:107–110. doi: 10.1038/35075114. [DOI] [PubMed] [Google Scholar]