

Abstract
The action of glutamate in CNS is mediated by the activation of metabotropic and ionotropic receptors. The metabotropic glutamate receptors (mGluRs) are highly enriched in prefrontal cortex (PFC) – a brain region critically involved in the regulation of cognition and emotion. Emerging evidence has suggested that mGluRs are viable drug targets for neuropsychiatric disorders associated with PFC dysfunction. However, the mGluR-mediated signalling in PFC remains unclear. To understand the physiological functions of postsynaptic group II mGluRs (mGluR2/3) in PFC neurones, we investigated the molecular and cellular mechanisms underlying the regulation of NMDA receptor channels by group II mGluRs. We found that APDC, a highly selective and potent group II mGluR agonist, reversibly increased NMDAR currents in acutely dissociated PFC pyramidal neurones. Selective group II mGluR antagonists, but not group I mGluR antagonists, blocked APDC-induced enhancement of NMDAR currents, suggesting the mediation by mGluR2/3 receptors. The APDC effect on NMDAR currents was independent of Mg2+ block or membrane voltages, and primarily targeted NR2A subunits containing NMDARs. While changing protein kinase A levels was without effect, inhibiting protein kinase C (PKC) or dialysis with Ca2+ chelators largely blocked the mGluR2/3 modulation of NMDAR currents. In contrast, inhibiting protein tyrosine kinases, cyclin-dependent kinase 5, Ca2+/calmodulin-dependent kinase II or the Ca2+/calmodulin-dependent phosphatase calcineurin failed to do so. Moreover, treatment of PFC slices with APDC significantly increased the PKC activity and PKC phosphorylation of NMDA receptors. These findings suggest that activation of mGluR2/3 receptors potentiates NMDAR channel functions in PFC through a PKC-dependent mechanism. This modulation may be relevant for developing novel mGluR-related pharmacological agents for the treatment of mental illnesses.
Prefrontal cortex (PFC), which is highly associated with the control of cognition and emotion (Goldman-Rakic, 1995; Miller, 1999), is one of the most prominent brain regions affected by schizophrenia (Andreasen et al. 1997; Lewis & Lieberman 2000). PFC is composed of two major neuronal populations: glutamatergic pyramidal projection neurones and GABAergic interneurones. PFC neurones receive glutamatergic inputs from thalamus, hippocampus and other cortical areas, in addition to the recurrent glutamatergic inputs from PFC pyramidal neurones (Conde et al. 1995; Carr & Sesack, 1996). Glutamate, by activating ionotropic and metabotropic receptors, plays a central role in regulating synaptic transmission and neuronal excitability of PFC circuits under normal and pathological conditions.
The ionotropic glutamate receptors are classified as AMPA receptors, kainate receptors and NMDA receptors, based on their physiological and pharmacological properties. Each of these ligand-gated channels is an oligomeric complex composed of different subunits (Seeburg, 1993; Hollmann & Heinemann, 1994). Hypofunction of NMDA receptors has been implicated in schizophrenia (Tsai & Coyle, 2002). Administration of the non-competitive NMDA receptor antagonists, such as phencyclidine (PCP), produces behavioural symptoms that remarkably resemble schizophrenia in humans and animals, and exacerbates symptoms in schizophrenics (Javitt & Zukin, 1991; Jentsch & Roth, 1999).
The metabotropic glutamate receptors (mGluRs) are classified into three groups based on structural homology, signal transduction mechanisms and pharmacological properties (Nakanishi, 1992). These receptors modulate glutamatergic neurotransmission in an anatomically and functionally distinct manner (Schoepp & Conn, 1993; Conn & Pin, 1997), and thus provide important pharmacotherapeutic targets for psychiatric disorders associated with glutamatergic abnormalities. The group II family of mGluRs, which consists of mGluR2 and mGluR3, is primarily expressed in forebrain regions including PFC (Ohishi et al. 1993a 1993b; Petralia et al. 1996). It has been found that systemic administration of an mGluR2/3 agonist reverses behavioural impairments in the phencyclidine model of schizophrenia (Moghaddam & Adams, 1998). However, cellular mechanisms underlying the antipsychotic action of mGluR2/3 receptors remain unclear. The mGluR2/3-mediated presynaptic depression of evoked glutamate release (Baskys & Malenka, 1991; Lovinger & McCool, 1995; Cartmell & Schoepp, 2000) is thought to be one possible mechanism for the normalization of glutamatergic disruptions by group II mGluRs (Moghaddam & Adams, 1998). We sought to examine the postsynaptic interactions between mGluR2/3 and NMDA receptors in PFC neurones, which could also be important for the reversal of NMDAR hypofunction by group II mGluRs in the schizophrenia model.
Methods
Acute-dissociation of neurones
PFC neurones from young adult (3–5 weeks postnatal) rats (Sprague Dowley) or mice (CS7 BL6) were acutely dissociated using procedures similar to those previously described (Yan & Surmeier, 1997; Feng et al. 2001). All experiments were carried out with the approval of State University of New York at Buffalo Animal Care Committee. In brief, rats were anaesthetized by inhaling 2-bromo-2-chloro-1,1,1-trifluoroethane (1 ml (100 g)−1, Sigma) and decapitated; brains were quickly removed, iced and then blocked for slicing. The blocked tissue was cut in 400 μm slices with a Vibratome while bathed in a low Ca2+ (100 μm), Hepes-buffered salt solution (mm: 140 sodium isethionate, 2 KCl, 4 MgCl2, 0.1 CaCl2, 23 glucose, 15 Hepes, 1 kynurenic acid, pH 7.4, 300–305 mosmol l−1). Slices were then incubated for 1–6 h at room temperature (20–22°C) in a NaHCO3-buffered saline bubbled with 95% O2, 5% CO2 (mm): 126 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, 10 glucose, 1 pyruvic acid, 0.05 glutathione, 0.1 NG-nitro-Loarginine, 1 kynurenic acid, pH 7.4, 300–305 mosmol l−1). All reagents were obtained from Sigma Chemical Co. (St Louis, MO, USA).
Slices were then removed into the low Ca2+ buffer and regions of the PFC were dissected and placed in an oxygenated Cell-Stir chamber (Wheaton, Inc., Millville, NJ, USA) containing papain (0.4 mg ml−1, Sigma) in Hepes-buffered Hanks' balanced salt solution (HBSS, Sigma) at 35°C. After 20–40 min of enzyme digestion, tissue was rinsed three times in the low Ca2+, Hepes-buffered saline and mechanically dissociated with a graded series of fire-polished Pasteur pipettes. The cell suspension was then plated into a 35 mm Lux Petri dish, which was then placed on the stage of a Nikon inverted microscope.
Whole-cell recordings
Whole-cell recordings of currents in dissociated or cultured neurones employed standard voltage-clamp techniques (Yan et al. 1999; Wang et al. 2003). The internal solution consisted of (mm): 180 N-methyl-d-glucamine (NMG), 40 Hepes, 4 MgCl2, 0.1 BAPTA, 12 phosphocreatine, 2 Na2ATP, 0.2 Na3GTP, 0.1 leupeptin, pH 7.2–7.3, 265–270 mosmol l−1. The external solution consisted of (mm): 127 NaCl, 20 CsCl, 10 Hepes, 1 CaCl2, 5 BaCl2, 12 glucose, 0.001 TTX, 0.02 glycine, pH 7.3–7.4, 300–305 mosmol l−1. Recordings were obtained with an Axon Instruments 200B patch clamp amplifier that was controlled and monitored with an IBM PC running pCLAMP (v. 8) with a DigiData 1320 series interface (Axon instruments, Union City, CA, USA). Electrode resistances were typically 2–4 MΩ in the bath. After seal rupture, series resistance (4–10 MΩ) was compensated (70–90%). Care was exercised to monitor the constancy of the series resistance, and recordings were terminated whenever a significant increase (> 20%) occurred. The cell membrane potential was held at −60 mV. NMDA (100 μm) was applied for 2 s every 30 s. About 30 min of recordings were performed on individual cells for most experiments. Cells with unstable NMDA responses (>20% decline throughout the recording) were discarded. Drugs were applied with a gravity-fed ‘sewer pipe’ system. The array of application capillaries (ca 150 μm i.d.) was positioned a few hundred micrometres from the cell under study. Solution changes were effected by the SF-77B fast-step solution stimulus delivery device (Warner Instrument Co., Hamden, CT, USA).
The mGluR ligands (2R,4R)-4-aminopyrrolidine-2,4-dicarboxylate (APDC), 2S-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]3-(xanth-9-yl) propanoic acid (LY341495), (RS)-1-amino-5-phosphonoindan-1-carboxylic acid (APICA), 2-methyl-6-(phenylethynyl)pyridine hydrochloride (MPEP) and 7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylate ethyl ester (CPCCOEt) were from Tocris (Ballwin, MO, USA). The second messenger reagents staurosporine, bisindolylmaleimide, cpt-cAMP, myristoylated PKI[14–22], U73122, 2-aminoethoxydiphenylborane (2APB), genistein, 3-(4-chlorophenyl)1-(1,1-dimethylethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (PP2), roscovitine, KN-93 and cyclosporin A were from Calbiochem (San Diego, CA, USA). They were made up as concentrated stocks and stored at −20°C. The final DMSO concentration in all applied solutions was less than 0.1%. Stocks were thawed and diluted immediately prior to use.
Data analyses were performed with AxoGraph (Axon Instruments), Kaleidagraph (Albeck Software, Reading, PA, USA) and StatView (Abacus Concepts, Inc.). For analysis of statistical significance, Mann–Whitney U tests were performed to compare the current amplitudes in the presence or absence of agonists. anova was performed to compare the differential degrees of current modulation between groups subjected to different treatment.
Western blot analysis
For detecting activated PKC, a phospho-PKC (pan) antibody that recognizes PKCα, βI, βII, ε, η, and δ isoforms only when phosphorylated at a carboxy-terminal residue homologous to Ser660 of PKCβII was used in the Western blot analysis. PFC slices were prepared as previously described (Gu et al. 2003). Equal amounts of protein from slice homogenates were separated on 7.5% polyacrylamide gels and transferred to nitrocellulose membranes. The blots were blocked with 5% non-fat dry milk for 1 h at room temperature. Then the blots were incubated with the phospho-PKC (pan) antibody (Cell Signalling 1: 2000) for 1 h at room temperature. After being rinsed, the blots were incubated with horseradish peroxidase-conjugated antirabbit antibodies (Amersham, 1: 2000) for 1 h at room temperature. Following three washes, the blots were exposed to the enhanced chemiluminescence substrate. Then the blots were stripped for 1 h at 50°C followed by saturation in 5% non-fat dry milk and incubated with a PKC antibody (Santa Cruz 1: 2000) recognizing the α, β, γ isoforms for the detection of the total PKC. For detecting NMDA receptors that are phosphorylated by PKC, a phospho-S896.NR1 antibody (Upstate, 1: 1000) was used. Quantification was obtained from densitometric measurements of immunoreactive bands on films. Data correspond to the mean ±s.d. of 3–8 samples per condition, and were analysed by anova.
Results
Enhancement of NMDAR currents by mGluR2/3 receptors in dissociated PFC pyramidal neurones
To test the potential impact of group II mGluR receptors on NMDAR functions, we examined the effect of APDC, a potent and highly selective mGluR2/3 receptor agonist (Schoepp et al. 1999), on NMDA receptor-mediated currents in acutely isolated PFC pyramidal neurones. The glutamatergic PFC pyramidal neurones were readily distinguished from GABAergic interneurones by their distinct morphological features: a pyramidal-shaped soma and a prominent apical dendrite (Feng et al. 2001; Cai et al. 2002). Application of NMDA evoked a partially desensitizing inward current that was blocked by the NMDA receptor antagonist D-APV (50 μm) or MK-801 (10 μm) (Fig. 1A and B), confirming mediation by the NMDA receptor. Application of APDC (50 μm) caused a potent enhancement in the amplitude of NMDAR currents in isolated PFC pyramidal neurones. The time course and current traces from a representative cell is shown in Fig. 1C and D). The APDC-induced increase of NMDAR currents was reversible and had fast onset kinetics. Following recovery from the first application, a second application of APDC resulted in a similar response (94.3 ± 3.4% of first response, n= 18). As summarized in Fig. 1E, APDC (50 μm) significantly (P < 0.01, Mann–Whitney) enhanced the amplitude of currents evoked by different concentrations of NMDA (100 μm: 19.2 ± 0.6%, n= 87; 50 μm: 10.3 ± 2.0%, n= 4; 500 μm: 22.5 ± 2.4%, n= 3; 1000 μm: 27.7 ± 2.3%, n= 3). In contrast, APDC had little effect on AMPAR currents in dissociated PFC pyramidal neurones (Figs 1F (2.1 ± 2.1%, n= 5, P > 0.05, Mann–Whitney). In cultured PFC pyramidal neurones (2–3 weeks in vitro), APDC also caused a potentiation of NMDA (100 μm)-evoked currents (14.2 ± 0.8%, n= 34, P < 0.01, Mann–Whitney, data not shown), consistent with the results obtained from acutely isolated neurones.
Figure 1. The group II mGluR agonist APDC enhanced NMDA receptor currents in acutely dissociated PFC pyramidal neurones.

A, current traces evoked by different concentrations (10, 100, 1000 μm) of NMDA in a representative cell. Scale bars: 0.1 nA, 0.5 s. B, current traces evoked by NMDA (100 μm) in the absence (control) or presence of D-APV (50 μm) or MK-801 (10 μm). Scale bars: 0.1 nA, 0.5 s. C, plot of peak NMDAR current as a function of time and agonist application. APDC (50 μm) reversibly increased NMDA (100 μm)-evoked currents in the cell. D, representative current traces taken from the records used to construct C (at time points denoted by #). E, cumulative data (mean ±s.e.m.) showing the APDC-induced percentage modulation of currents evoked by different concentrations of NMDA (100 μm: n= 87; 50 μm: n= 4; 500 μm: n= 3; 1000 μm: n= 3). F, plot of peak AMPA (100 μm)-evoked currents as a function of time and agonist application. Inset, representative current traces (at time points denoted by #). Scale bars: 0.2 nA, 1 s.
To verify that mGluR2/3 receptors were mediating the modulation seen with APDC, we examined the ability of selective mGluR2/3 receptor antagonists to prevent the action of APDC. As shown in Fig. 2A and B, APDC (50 μm) produced a reversible potentiation of NMDAR currents in the dissociated PFC neurone, and this effect was blocked by LY341495 (0.5 μm), a highly selective mGluR2/3 receptor antagonist (Kingston et al. 1998). Washing off the antagonist restored the ability of APDC to enhance NMDAR currents. As summarized in Fig. 2C, APDC had significantly (P < 0.01, anova) smaller effect on NMDAR currents in the presence of different concentrations of LY341495 (0.1 μm: 21.1 ± 10.5% of control modulation, n= 3; 0.5 μm: 34.9 ± 6.7% of control modulation, n= 9; 2 μm: 37.7 ± 8.7% of control modulation, n= 4). LY341495 (0.1–2 μm) alone had little effect on basal NMDAR currents (0.9 ± 0.8%, n= 16, P > 0.05, Mann–Whitney). Another selective mGluR2/3 receptor antagonist, APICA (200 μm), also greatly attenuated the APDC enhancement of NMDAR currents (Fig. 2D), while it alone had no significant effect on basal NMDAR currents (1.3 ± 1.8%, n= 7, P > 0.05, Mann–Whitney). In contrast, the mGluR5 antagonist MPEP (5 μm) or the mGluR1 antagonist CPCCOEt (50 μm) failed to block the APDC effect (Fig. 2E). As summarized in Fig. 2F, the effect of APDC on NMDAR currents in the presence of APICA was significantly (P < 0.01, anova) smaller (31.2 ± 8.4% control modulation, n= 7), but was almost unchanged by MPEP (91.7 ± 1.4% control modulation, n= 4) or CPCCOEt (94.4 ± 1.3% control modulation, n= 6). The pharmacological profile of these responses thus identifies mGluR2/3 as the receptor underlying the APDC-induced enhancement of NMDAR currents.
Figure 2. The APDC enhancement of NMDAR currents was mediated by mGluR2/3 receptors.

A and D, plot of peak NMDAR current as a function of time and agonist application. The selective mGluR2/3 antagonist LY341495 (0.5 μm, A) or APICA (200 μm, D) blocked APDC-induced enhancement of NMDAR currents. B, representative current traces taken from the records used to construct A (at time points denoted by #). Scale bars: 0.2 nA, 0.5 s. C, cumulative data (mean ±s.e.m.) showing the percentage control modulation of NMDAR currents by APDC in the absence (n= 6) or presence of different concentrations of LY341495 (0.1 μm: n= 3; 0.5 μm: n= 9; 2 μm: n= 4). *P < 0.01, anova. E, plot of peak NMDAR current as a function of time and agonist application. The selective mGluR5 antagonist MPEP (5 μm) and mGluR1 antagonist CPCCOEt (50 μm) failed to block APDC-induced enhancement of NMDAR currents. F, cumulative data (mean ±s.e.m.) showing the percentage control modulation of NMDAR currents by APDC in the absence (n= 6) or presence of different antagonists (APICA: n= 7; MPEP: n= 4; CPCCOEt: n= 6). *P < 0.01, anova.
Characterization of the mGluR2/3 modulation of NMDAR currents
The opening of NMDAR channels is regulated by the voltage-sensitive Mg2+ block; we therefore examined whether the mGluR2/3 modulation of NMDAR currents was through changing the Mg2+ block in a voltage-dependent mechanism. Membrane potentials were held at different levels, and NMDAR currents were recorded in Mg2+-containing versus Mg2+-free solutions. As shown in Fig. 3A and B, APDC produced similar enhancement of NMDAR currents irrespective of the holding potentials (−70, −40 and −20 mV) in the cell perfused with an Mg2+-free solution. Moreover, the APDC effect on NMDAR currents was similar irrespective of the extracellular Mg2+ concentrations (0 mm, 2 mm) in the cell voltage-clamped at −40 mV. The percentage control modulation of NMDAR currents by APDC at different membrane potentials and different concentrations of Mg2+ is summarized in Fig. 3C. The lack of changes of the APDC effect suggests that mGluR2/3 receptors potentiate NMDAR currents through a mechanism independent of membrane voltages or Mg2+ block.
Figure 3. The mGluR2/3 enhancement of NMDAR currents was independent of membrane potentials or Mg2+ concentrations, and targeted both NR2A and NR2B subunit containing NMDARs.

A and B, representative current traces in the absence or presence of APDC in a PFC neurone held at different potentials (−70, −40 and −20 mV) in an Mg2+-free solution (A), or in a PFC neurone held at −40 mV in Mg2+-free versus Mg2+-rich solutions (B). Scale bars: 0.1 nA, 0.5 s. C, cumulative data (mean ±s.e.m.) showing the percentage control modulation of NMDAR currents by APDC at different potentials (n= 7) and Mg2+ concentrations (n= 6). D, plot of peak NMDAR current as a function of time and agonist application. APDC enhanced NMDAR currents to a larger extent in the presence of the NR2B antagonist ifenprodil (10 μm). Inset, cumulative data (mean ±s.e.m.) showing the percentage modulation of NMDAR currents by APDC in the absence or presence of ifenprodil (n= 9).
NMDAR functional properties are determined by subunit composition (Monyer et al. 1994; Vicini et al. 1998). The primary NMDARs in mature cortical synapses, which are composed of NR1/NR2A or NR1/NR2B, differ in deactivation kinetics and subcellular localization (Cull-Candy et al. 2001). To determine which subpopulation(s) of NMDARs is targeted by APDC, we applied the selective inhibitor of NR2B subunit, ifenprodil (Williams, 1993). Blocking NR2B subunit-containing NMDARs with ifenprodil (10 μm) reduced the amplitude of NMDAR currents by 44.6 ± 2.7% (n= 9). As shown in Fig. 3D, in the presence of ifenprodil, APDC enhanced the remaining NMDAR currents to a larger extent than the APDC effect on the whole-cell NMDAR currents (166 ± 14.6% control modulation, n= 9, Fig. 3D, inset), suggesting that mGluR2/3 receptors may primarily target NR2A subunit containing NMDA receptors.
Involvement of PKC in the mGluR2/3 potentiation of NMDAR currents
We next examined the mechanisms mediating the potentiation of NMDAR currents by group II mGluR receptors. A classical signal transduction pathway for mGluR2/3 receptors is to couple to Gi/o-type G proteins to inhibit adenylate cyclase and cAMP formation (Conn & Pin, 1997). While PKA phosphorylation of NMDAR subunits changes the channel activity (Raman et al. 1996; Tingley et al. 1997; Westphal et al. 1999), we found that the mGluR2/3 enhancement of NMDAR currents was unlikely through the inhibition of PKA. As shown in Fig. 4A, application of the membrane-permeant PKA activator cpt-cAMP (50 μm) failed to block APDC-induced increase of NMDAR currents. Dialysis with the PKA inhibitory peptide PKI[5–24] (20 μm) also failed to prevent APDC from potentiating NMDAR currents (n= 5, data not shown).
Figure 4. The mGluR2/3 enhancement of NMDAR currents was dependent on PKC, but not PKA.

A, B and C, plot of peak NMDAR currents as a function of time and drug application. Application of the broad-spectrum kinase inhibitor staurosporine (1 μm, B) or the specific PKC inhibitor bisindolylmaleimide (Bisl, 1 μm, C), but not the PKA activator cpt-cAMP (50 μm, A), largely blocked the APDC-induced enhancement of NMDAR currents. D, representative current traces taken from the records used to construct C (at time points denoted by #). Scale bars: 0.15 nA, 0.5 s. E, plot of peak NMDAR currents in neurones dialysed with internal solutions containing low (0.5 mm) or high (10 mm) concentrations of BAPTA. APDC had significantly smaller effect in the neurone loaded with high BAPTA. F, cumulative data (mean ±s.e.m.) showing the percentage control modulation of NMDAR currents by APDC in the absence (n= 12) or presence of staurosporine (STS, n= 16), bisindolylmaleimide (Bisl, n= 19), high BAPTA (n= 13), cpt-cAMP (n= 10) or the PKA inhibitor PKI[14–22] (0.1 μm, n= 9). *P < 0.005, anova.
Previous biochemical data show that an mGluR2/3 agonist increases the activity of phospholipase C (PLC) in hippocampal slices (Klein et al. 1997), and enhances the turnover of phosphoinositides in cortical slices (Mistry et al. 1998). Since triggering the phospholipid cascade could lead to the activation of PKC, we tested whether the APDC enhancement of NMDAR currents was through activated PKC.
If mGluR2/3 receptors were exerting their effect through PKC, then inhibiting the activation of PKC should eliminate the effect of APDC on NMDAR currents. As shown in Fig. 4B, the broad-spectrum kinase inhibitor staurosporine (1 μm), which has a high affinity to PKC, largely blocked the APDC-induced enhancement of NMDAR currents. A more specific PKC inhibitor, bisindolylmaleimide (Bisl, 1 μm), with high selectivity for PKCα, β, γ, δ and ε isozymes (Toullec et al. 1991), also eliminated the APDC effect, and washing off the PKC inhibitor led to recovery of APDC enhancement of NMDAR currents (Fig. 4C, D). Since conventional PKC isoforms (PKCα, β, γ) depend on Ca2+ for their activation (Tanaka & Nishizuka 1994), we dialysed neurones with a high concentration (10 mm) of BAPTA, a potent and rapid Ca2+ chelator, and examined APDC modulation of NMDAR currents under this condition. As shown in Fig. 4E, high BAPTA significantly attenuated the effect of APDC on NMDAR currents.
The ability of various inhibitors to block the APDC effect on NMDAR currents, expressed as a percentage of the modulation in the absence of these reagents, is summarized in Fig. 4F. APDC had a significantly (P < 0.005, anova) smaller effect on NMDAR currents in the presence of staurosporine (30.7 ± 6.8% of control modulation, n= 13). Similarly, the effect of APDC on NMDAR currents in the presence of bisindolylmaleimide was significantly (P < 0.005, anova) smaller (26.2 ± 7.9% of control modulation, n= 14). Dialysis with high BAPTA also significantly (P < 0.005, anova) attenuated the APDC potentiation of NMDAR currents (38.7 ± 4.0% of control modulation, n= 13). However, the effect of APDC was not affected by cpt-cAMP (98.3 ± 10.3% of control modulation, n= 10) or the membrane-permeant PKA inhibitor, myristoylated PKI[14–22] (81.8 ± 6.4% of control modulation, n= 9). Taken together, these data suggest that mGluR2/3 receptor-mediated potentiation of NMDAR currents is through a mechanism dependent on PKC activation.
Since activation of PKC is often triggered by the phospholipid cascade, we then tested whether the phospholipid pathway is involved in the mGluR2/3 regulation of NMDA receptors. Application of the phospholipase C (PLC) inhibitor U73122 (4 μm) had little effect on basal NMDAR currents. However, in the presence of U73122, APDC significantly lost the ability to potentiate NMDAR currents in half of the neurones we tested (n= 12/24). A representative example is shown in Fig. 5A and B. In the other 12 neurones, U73122 failed to alter the APDC enhancement of NMDAR currents (data not shown). To examine the involvement of intracellular Ca2+ stores, the membrane-permeant IP3 receptor antagonist 2APB (15 μm) was applied. In many of the neurones we tested (n= 10/17), 2APB caused an increase in NMDAR currents, whereas in others (n= 7/17), 2APB had little effect on basal NMDAR currents. The enhancing effect of APDC was markedly diminished by 2APB in most cases (n= 12/17). A representative example is shown in Fig. 5C. In the other five neurones, 2APB failed to affect the APDC enhancement of NMDAR currents (data not shown). As summarized in Fig. 5D, in a subset of PFC pyramidal neurones, inhibiting PLC with U73122 significantly (P < 0.01, anova) attenuated the APDC enhancement of NMDAR currents (25.1 ± 6.4% of control modulation, n= 12 out of 24 neurones), whereas blocking Ca2+ release from intracellular Ca2+ stores via IP3 receptors prevented APDC from increasing NMDAR currents (16.4 ± 5.5% of control modulation, n= 12 out of 17 neurones). Thus, these results suggest that the phospholipid pathway is involved in the mGluR2/3 regulation of NMDAR currents in a subset of PFC pyramidal neurones. In other neurones, mGluR2/3 receptors may activate PKC through an unknown mechanism to enhance NMDAR currents.
Figure 5. The mGluR2/3 enhancement of NMDAR currents was dependent on the phospholipid pathway and IP3R-mediated intracellular Ca2+ release in a subset of PFC pyramidal neurones.

A, plot of peak NMDAR currents as a function of time and drug application. Application of the PLC inhibitor U73122 (4 μm) largely blocked the APDC-induced enhancement of NMDAR currents. B, representative current traces taken from the records used to construct A (at time points denoted by #). Scale bars: 0.3 nA, 0.5 s. C, plot of peak NMDAR currents as a function of time and drug application. Application of the IP3R antagonist 2APB (15 μm) caused an increase in NMDAR currents and prevented the APDC-induced enhancement of NMDAR currents. D, cumulative data (mean ±s.e.m.) showing the percentage control modulation of NMDAR currents by APDC in the absence (n= 12) or presence of U73122 (n= 12 out of 24) or 2APB (n= 12 out of 17). *P < 0.005, anova.
Role of other signalling molecules in the mGluR2/3 potentiation of NMDAR currents
Given the PKC dependence of mGluR2/3 enhancement of NMDAR currents, we would like to know whether it is through a direct action of PKC (Tingley et al. 1997; Lan et al. 2001) or indirectly via a PKC downstream signalling molecule. Two possible candidates that can be activated by PKC and can directly regulate NMDARs are the tyrosine kinase Src (Lu et al. 1999) and the cyclin-dependent kinase 5 (cdk5, Li et al. 2001; Liu et al. 2001). Thus, we tested the role of these kinases in mGluR2/3 regulation of NMDAR currents.
As shown in Fig. 6A, bath application of the broad-spectrum tyrosine kinase inhibitor genistein (50 μm) did not affect the ability of APDC to enhance NMDAR currents. The specific Src kinase inhibitor PP2 (20 μm) also failed to block the APDC-induced increase of NMDAR currents (n= 4, data not shown). Similarly, suppressing cdk5 activity with the specific inhibitor roscovitine (50 μm, Hellmich et al. 1992) was without effect on the APDC potentiation of NMDAR currents (Fig. 6B and C). Furthermore, in mutant mice with targeted deletion of the neuronal specific activator of cdk5, p35 (Tsai et al. 1994; Chae et al. 1997), APDC caused similar enhancement of NMDAR currents as in wild-type mice (p35−/−: 19.5 ± 2.0%, n= 8; WT: 22.8 ± 2.6%, n= 10).
Figure 6. The mGluR2/3 enhancement of NMDAR currents was independent of various kinases or phosphatases.

A, B, D and E, plot of peak NMDAR currents as a function of time and drug application. The APDC-induced enhancement of NMDAR currents was not blocked by application of the tyrosine kinase inhibitor genistein (50 μm, A), the cdk5 inhibitor roscovitine (50 μm, B), the CaMKII inhibitor KN-93 (10 μm, D), or the calcineurin inhibitor cyclosporin A (20 μm, E). C, representative current traces taken from the records used to construct B (at time points denoted by #). Scale bars: 0.2 nA, 0.5 s. F, cumulative data (mean ±s.e.m.) showing the percentage control modulation of NMDAR currents by APDC in the absence (n= 9) or presence of genistein (n= 10), roscovitine (n= 14), KN-93 (n= 9) or cyclosporin A (n= 4).
If mGluR2/3 receptors couple to the phospholipid pathway in PFC neurones, the IP3-triggered increase of intracellular Ca2+ levels could activate Ca2+/calmodulin-dependent kinase II (CaMKII) or the Ca2+/calmodulin-dependent phosphatase calcineurin, which can also directly regulate NMDARs (Lieberman & Mody, 1994; Omkumar et al. 1996; Strack & Colbran, 1998). Hence, we examined the involvement of CaMKII and calcineurin in the mGluR2/3 modulation. As shown in Fig. 6D and E), application of the specific CaMKII inhibitor KN-93 (10 μm) did not block the APDC-induced enhancement of NMDAR currents. Likewise, the specific calcineurin inhibitor cyclosporin A (20 μm) also failed to alter the APDC effect. Figure 6F summarizes the percentage control modulation of NMDAR currents by APDC in the presence of various kinase inhibitors (genistein: 79.9 ± 4.0% of control modulation, n= 10; roscovitine: 89.7 ± 2.6% of control modulation, n= 14; KN-93: 82.9 ± 3.7% of control modulation, n= 9; cyclosporin A: 106.4 ± 2.5% of control modulation, n= 4; P > 0.05, anova). Taken together, these data suggest the lack of involvement of tyrosine kinases, cdk5, CaMKII or calcineurin in the mGluR2/3 modulation of NMDA receptors.
The mGluR2/3-induced increase in PKC activity and PKC phosphoylation of NMDA receptors in PFC slices
Since the electrophysiological data suggest that the APDC-induced potentiation of NMDAR currents is likely to be through a direct activation of PKC, we then examined whether activation of mGluR2/3 receptors can indeed increase PKC activity. Because the catalytic competence of many PKC isozymes depends on autophosphorylation at the carboxyl terminus on a conserved residue (Behn-Krappa & Newton, 1999), a phospho-specific pan PKC antibody that detects PKC isoforms only when phosphorylated at this residue was used to detect activated PKC. As shown in Fig. 7A, application of APDC (50 μm, 10 min) to cortical slices induced a marked increase in the activated PKC. This effect of APDC was not altered by antagonizing mGluR1/5 receptors with MPEP (5 μm) and CPCCOEt (50 μm), but was blocked by the mGluR2/3 antagonist LY341495 (1 μm). The levels of total PKC were not changed by any of these treatments. Quantification data (Fig. 7B) exhibited a 3.2 ± 0.9-fold increase of PKC activity by APDC treatment (n= 8), which was abolished by LY341495 (1.2 ± 0.3-fold, n= 4), but not by MPEP and CPCCOEt (3.0 ± 0.6-fold, n= 4). These results suggest that activation of mGluR2/3 receptors elevates the kinase activity of PKC in PFC neurones.
Figure 7. Activation of mGluR2/3 receptors increased PKC activity and PKC phosphorylation of NMDA receptors in PFC slices.
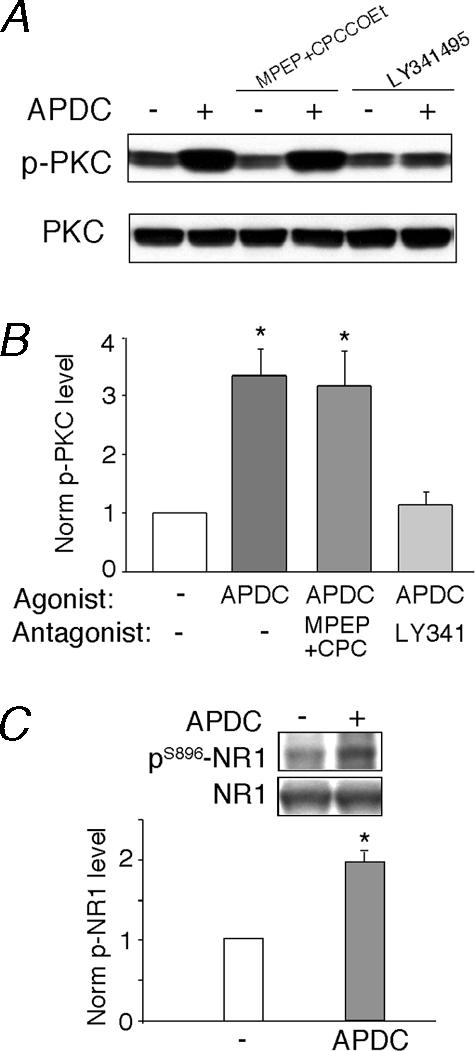
A, immunoblots of phospho-PKC and PKC. PFC slices were incubated in the absence or presence of the selective mGluR1/5 antagonists MPEP (5 μm) plus CPCCOEt (50 μm) or the mGluR2/3 antagonist LY341495 (1 μm) for 15 min, followed by a 10-min treatment with APDC (50 μm). After treatment, slice lysates were blotted with an antiphospho-PKC antibody. Following stripping out signals, membranes were reblotted with an antibody recognizing the total PKC. B, quantification of p-PKC induced by APDC under different treatments. *P < 0.001, anova, compared to the effect under control conditions (–). C, quantification of pS896-NR1 in PFC slices treated without or with APDC (50 μm, 10 min). *P < 0.01, anova. Inset, immunoblots of pS896-NR1 and total NR1.
Because NR1 subunit can be phosphorylated at Ser896 by PKC (Tingley et al. 1997), we then examined whether activation of mGluR2/3 receptors affects PKC phosphorylation of NMDA receptors. As shown in Fig. 7C, application of APDC (50 μm, 10 min) to cortical slices induced a significant (2.0 ± 0.3-fold, n= 3) increase in the phosphorylated NR1 at the PKC site (S896), suggesting that mGluR2/3 activation enhances PKC-dependent phosphorylation of NMDA receptors in PFC neurones.
Discussion
The group II mGluRs (mGluR2/3) are highly enriched in PFC neurones (Ohishi et al. 1993a, 1993b). Ultrastructural studies indicate that mGluR2 receptors are localized predominantly presynaptically at the periphery of the synaptic area, while mGluR3 receptors are found in neurones at both pre- and postsynaptic sites (Petralia et al. 1996). Electrophysiological and biochemical studies have shown that mGluR2/3 agonists inhibit evoked glutamate release in PFC via presynaptic mechanisms (Marek et al. 2000; Cartmell & Schoepp, 2000), suggesting that mGluR2/3 receptors could play a key role in reducing glutamate hyperexcitability under pathological states resulting from excessive glutamate transmission (Schoepp & Marek, 2002). Less is known about postsynaptic mGluR2/3 modulation of synaptic transmission and neuronal excitability. In this study, we demonstrated that activation of group II mGluRs significantly potentiated the currents through NMDAR channels in acutely dissociated PFC pyramidal neurones. It suggests that the postsynaptic NMDA receptor is one of the key targets of mGluR2/3 receptors in PFC. Since the schizophrenia-like behaviours induced by administration of NMDAR antagonists (Javitt & Zukin, 1991; Jentsch & Roth, 1999) have implicated NMDAR hypofunction in mental disorders, the mGluR2/3 potentiation of NMDAR channel functions in PFC could play a significant role in the reversal of behavioural impairments by group II mGluRs in the phencyclidine model of schizophrenia (Moghaddam & Adams, 1998). This modulation provides a homeostatic feedback mechanism for mGluR2/3 receptors to correct or normalize glutamatergic functions. A recent study (Wittmann et al. 2002) shows that the function of group II mGluRs is modulated by dopamine, a neurotransmitter highly involved in schizophrenia (Davis et al. 1991; Carlsson et al. 2001). It further implicates that mGluR2/3 receptors may play a key role in regulating cognitive and emotional processes in neuropsychiatric disorders.
The NMDAR channel activity can be regulated by protein phosphorylation/dephosphorylation via a variety of protein kinases/phosphatases (Lieberman & Mody, 1994; Lu et al. 1999; Westphal et al. 1999). Different groups of mGluRs, by coupling to distinct second-messenger cascades, can regulate NMDAR channels in a complex manner. Activation of group I mGluRs in cortical neurones induced an enhancement of NMDAR currents through the Pyk2/Src-family kinase pathway (Heidinger et al. 2002). In this study, we found that group II mGluRs enhanced NMDAR currents in PFC pyramidal neurones via a mechanism dependent on PKC activation. Biochemical data also confirmed that mGluR2/3 receptors elevated the level of activated PKC in PFC slices. These data suggest that postsynaptic group II mGluRs in PFC neurones may link to the phospholipid cascade, instead of coupling to the classical inhibition of PKA pathway, to regulate NMDA receptors. Consistent with this, it has been found that postsynaptic group II mGluRs induced long-term depression in PFC through the PLC/PKC/Ca2+-dependent mechanism (Otani et al. 2002). Moreover, agonists for group II mGluRs are found to potentiate the action of group I mGluRs on phosphoinositide turnover (Schoepp et al. 1996; Mistry et al. 1998) and postsynaptic Ca2+ augmentation (Cho et al. 2000). Interestingly, a recent study has found that the therapeutically effective antipsychotic drug clozapine augments NMDA-induced responses through a mechanism involving PKC activation in PFC pyramidal neurones (Jardemark et al. 2003). It supports our speculation that the group II mGluR/PKC-mediated facilitation of NMDAR currents may underlie the antipsychotic action of mGluR2/3 agonists (Moghaddam & Adams, 1998).
A previous study (Otani et al. 2002) demonstrated that group II mGluR agonists decrease synaptically evoked NMDAR-EPSPs, which is not surprising given the prominent role of group II mGluRs in presynaptic depression (Marek et al. 2000; Cartmell & Schoepp, 2000). With the opposite pre- and postsynaptic effects of group II mGluR activation on NMDAR responses, the net effect of group II mGluRs on NMDAR functions in vivo could be complicated. We hypothesize that in response to mGluR2/3 activation, the decreased glutamate release will prevent the activation of extrasynaptic NMDA receptors and reduce the number of synaptic NMDA receptors that are activated. Meanwhile, the activated synaptic NMDA receptors will have higher sensitivity or conductance due to the postsynaptic modification of channel properties. The net consequence could be a preferential facilitation of subsets of synaptic NMDA receptors and suppression of other synaptic and extrasynaptic NMDA receptors, which could lead to a higher ‘signal-to-noise’ ratio of the NMDA signalling at some synaptic sites.
Several mechanisms have been proposed to account for the PKC-mediated up-regulation of NMDAR channels. One mechanism for PKC to potentiate the NMDA response is to increase the probability of channel openings and reduce the voltage-dependent Mg2+ block of NMDAR channels (Chen & Huang, 1992). Alternatively, PKC enhances NMDA-evoked currents through activation of the non-receptor tyrosine kinase (Src) signalling cascade (Lu et al. 1999). Our data ruled out these two possibilities, suggesting that the group II mGluR/PKC regulation of NMDAR channels is through a mechanism independent of Mg2+ block or Src, as well as other signalling molecules including cdk5, CaMKII and calcineurin. Thus, mGluR2/3 may alter the biophysical properties of NMDAR channels through direct PKC phosphorylation of NR1 subunit (Tingley et al. 1993). Biochemical evidence has shown that PKC phosphorylation of NR1 decreases its affinity for calmodulin (Hisatsune et al. 1997). Hence, mGluR2/3 activation of PKC could potentiate NMDAR currents by preventing calmodulin from binding to the NR1 subunit and thereby inhibiting the inactivation of NMDARs by Ca2+/calmodulin (Ehlers et al. 1996; Zhang et al. 1998). The possible involvement of this mechanism is confirmed by our results showing the blockade of APDC effects with a high concentration of Ca2+ chelator. Another mechanism that may underlie the mGluR2/3 potentiation of NMDAR currents involves the PKC-induced rapid delivery of functional NMDARs to the surface of dendrites and spines through regulated exocytosis (Lan et al. 2001). The exact mechanism for the mGluR2/3-PKC regulation of NMDAR channels awaits to be further examined.
Acknowledgments
This work was supported by NIH grants MH63128 and AG21923 (Z.Y), NSF grant IBN-0117026 (Z.Y), and Howard Hughes Medical Institute Biomedical Research Support Program grant 53000261 (SUNY at Buffalo). We would like to thank Xiaoqing Chen for her technical support and Dr Li-Huei Tsai at Harvard Medical School for providing p35 knockout mice.
References
- Andreasen NC, O'leary DS, Flaum M, Nopoulos P, Watkins GL, Boles Ponto LL, Hichwa R. Hypofrontality in schizophrenia: distributed dysfunctional circuits in neuroleptic-naive patients. Lancet. 1997;349:1730–1734. doi: 10.1016/s0140-6736(96)08258-x. [DOI] [PubMed] [Google Scholar]
- Baskys A, Malenka RC. Agonists at metabotropic glutamate receptors presynaptically inhibit EPSCs in neonatal rat hippocampus. J Physiol. 1991;444:687–701. doi: 10.1113/jphysiol.1991.sp018901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behn-Krappa A, Newton AC. The hydrophobic phosphorylation motif of conventional protein kinase C is regulated by autophosphorylation. Curr Biol. 1999;9:728–737. doi: 10.1016/s0960-9822(99)80332-7. [DOI] [PubMed] [Google Scholar]
- Cai X, Gu Z, Zhong P, Ren Y, Yan Z. Serotonin 5-HT1A receptors regulate AMPA receptor channels through inhibiting CaMKII in prefrontal cortical pyramidal neurons. J Biol Chem. 2002;277:36553–36562. doi: 10.1074/jbc.M203752200. [DOI] [PubMed] [Google Scholar]
- Carlsson A, Waters N, Holm-Waters S, Tedroff J, Nilsson M, Carlsson ML. Interactions between monoamines, glutamate, and GABA in schizophrenia: new evidence. Annu Rev Pharmacol Toxicol. 2001;41:237–260. doi: 10.1146/annurev.pharmtox.41.1.237. [DOI] [PubMed] [Google Scholar]
- Carr DB, Sesack SR. Hippocampal afferents to the rat prefrontal cortex: synaptic targets and relation to dopamine terminals. J Comp Neurol. 1996;369:1–15. doi: 10.1002/(SICI)1096-9861(19960520)369:1<1::AID-CNE1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Cartmell J, Schoepp DD. Regulation of neurotransmitter release by metabotropic glutamate receptors. J Neurochem. 2000;75:889–907. doi: 10.1046/j.1471-4159.2000.0750889.x. [DOI] [PubMed] [Google Scholar]
- Chae T, Kwon YT, Bronson R, Dikkes P, Li E, Tsai LH. Mice lacking p35, a neuronal specific activator of cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron. 1997;18:29–42. doi: 10.1016/s0896-6273(01)80044-1. [DOI] [PubMed] [Google Scholar]
- Chen L, Huang LY. Protein kinase C reduces Mg2+ block of NMDA-receptor channels as a mechanism of modulation. Nature. 1992;356:521–523. doi: 10.1038/356521a0. [DOI] [PubMed] [Google Scholar]
- Cho K, Kemp N, Noel J, Aggleton JP, Brown MW, Bashir ZI. A new form of long-term depression in the perirhinal cortex. Nat Neurosci. 2000;3:150–156. doi: 10.1038/72093. [DOI] [PubMed] [Google Scholar]
- Conde F, Marie-Lepoivre E, Audinat E, Crepel F. Afferent connections of the medial frontal cortex of the rat. II. Cortical and subcortical afferents. J Comp Neurol. 1995;352:567–593. doi: 10.1002/cne.903520407. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol. 2001;11:327–335. doi: 10.1016/s0959-4388(00)00215-4. [DOI] [PubMed] [Google Scholar]
- Davis KI, Kahn Rs, Ko G, Davidson M. Dopamine in schizophrenia: a review and reconceptualization. Am J Psychiatry. 1991;148:1474–1486. doi: 10.1176/ajp.148.11.1474. [DOI] [PubMed] [Google Scholar]
- Ehlers MD, Zhang S, Bernhardt JP, Huganir RL. Inactivation of NMDA receptors by direct interaction of calmodulin with the NR1 subunit. Cell. 1996;84:745–755. doi: 10.1016/s0092-8674(00)81052-1. [DOI] [PubMed] [Google Scholar]
- Feng J, Cai X, Zhao JH, Yan Z. Serotonin receptors modulate GABAA receptor channels through activation of anchored protein kinase C in prefrontal cortical neurons. J Neurosci. 2001;21:6502–6511. doi: 10.1523/JNEUROSCI.21-17-06502.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman-Rakic PS. Cellular basis of working memory. Neuron. 1995;14:477–485. doi: 10.1016/0896-6273(95)90304-6. [DOI] [PubMed] [Google Scholar]
- Gu Z, Zhong P, Yan Z. Activation of muscarinic receptors inhibits β-amyloid peptide-induced signaling in cortical slices. J Biol Chem. 2003;278:17546–17556. doi: 10.1074/jbc.M209892200. [DOI] [PubMed] [Google Scholar]
- Heidinger V, Manzerra P, Wang XQ, Strasser U, Yu SP, Choi DW, Behrens MM. Metabotropic glutamate receptor 1-induced upregulation of NMDA receptor current: mediation through the Pyk2/Src-family kinase pathway in cortical neurons. J Neurosci. 2002;22:5452–5461. doi: 10.1523/JNEUROSCI.22-13-05452.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmich MR, Pant HC, Wada E, Battey JF. Neuronal cdc2-like kinase: a cdc2-related protein kinase with predominantly neuronal expression. Proc Natl Acad Sci U S A. 1992;89:10867–10871. doi: 10.1073/pnas.89.22.10867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisatsune C, Umemori H, Inoue T, Michikawa T, Kohda K, Mikoshiba K, Yamamoto T. Phosphorylation-dependent regulation of N-methyl-D-aspartate receptors by calmodulin. J Biol Chem. 1997;272:20805–20810. doi: 10.1074/jbc.272.33.20805. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- Jardemark KE, Ninan I, Liang X, Wang RY. Protein kinase C is involved in clozapine's facilitation of N-methyl-D-aspartate- and electrically evoked responses in pyramidal cells of the medial prefrontal cortex. Neuroscience. 2003;118:501–512. doi: 10.1016/s0306-4522(02)00976-4. [DOI] [PubMed] [Google Scholar]
- Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–1308. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Roth RH. The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology. 1999;20:201–225. doi: 10.1016/S0893-133X(98)00060-8. [DOI] [PubMed] [Google Scholar]
- Kingston AE, Ornstein PL, Wright RA, Johnson BG, Mayne NG, Burnett JP, Belagaje R, Wu S, Schoepp DD. LY341495 is a nanomolar potent and selective antagonist of group II metabotropic glutamate receptors. Neuropharmacology. 1998;37:1–12. doi: 10.1016/s0028-3908(97)00191-3. [DOI] [PubMed] [Google Scholar]
- Klein J, Iovino M, Vakil M, Shinozaki H, Loffelholz K. Ontogenetic and pharmacological studies on metabotropic glutamate receptors coupled to phospholipase D activation. Neuropharmacology. 1997;36:305–311. doi: 10.1016/s0028-3908(97)00024-5. [DOI] [PubMed] [Google Scholar]
- Lan JY, Skeberdis VA, Jover T, Grooms SY, Lin Y, Araneda RC, Zheng X, Bennett MV, Zukin RS. Protein kinase C modulates NMDA receptor trafficking and gating. Nat Neurosci. 2001;4:382–390. doi: 10.1038/86028. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Lieberman JA. Catching up on schizophrenia: Natural history and neurobiology. Neuron. 2000;28:325–334. doi: 10.1016/s0896-6273(00)00111-2. [DOI] [PubMed] [Google Scholar]
- Li BS, Sun MK, Zhang L, Takahashi S, Ma W, Vinade L, Kulkarni AB, Brady RO, Pant HC. Regulation of NMDA receptors by cyclin-dependent kinase-5. Proc Natl Acad Sci U S A. 2001;98:12742–12747. doi: 10.1073/pnas.211428098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman DN, Mody I. Regulation of NMDA channel function by endogenous Ca2+-dependent phosphatase. Nature. 1994;369:235–239. doi: 10.1038/369235a0. [DOI] [PubMed] [Google Scholar]
- Liu F, Ma XH, Ule J, Bibb JA, Nishi A, Demaggio AJ, Yan Z, Nairn AC, Greengard P. Regulation of cyclin-dependent kinase 5 and casein kinase 1 by metabotropic glutamate receptors. Proc Natl Acad Sci U S A. 2001;98:11062–11068. doi: 10.1073/pnas.191353898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM, Mccool BA. Metabotropic glutamate receptor-mediated presynaptic depression at corticostriatal synapses involves mGluR2 or 3. J Neurophysiol. 1995;73:1076–1083. doi: 10.1152/jn.1995.73.3.1076. [DOI] [PubMed] [Google Scholar]
- Lu WY, Xiong ZG, Lei S, Orser BA, Dudek E, Browning MD, Macdonald JF. G-protein-coupled receptors act via protein kinase C and Src to regulate NMDA receptors. Nat Neurosci. 1999;2:331–338. doi: 10.1038/7243. [DOI] [PubMed] [Google Scholar]
- Marek GJ, Wright RA, Schoepp DD, Monn JA, Aghajanian GK. Physiological antagonism between 5-hydroxytryptamine (2A) and group II metabotropic glutamate receptors in prefrontal cortex. J Pharmacol Exp Ther. 2000;292:76–87. [PubMed] [Google Scholar]
- Miller EK. The prefrontal cortex: complex neural properties for complex behavior. Neuron. 1999;22:15–17. doi: 10.1016/s0896-6273(00)80673-x. [DOI] [PubMed] [Google Scholar]
- Mistry R, Golding N, Challiss RA. Regulation of phosphoinositide turnover in neonatal rat cerebral cortex by group I- and II-selective metabotropic glutamate receptor agonists. Br J Pharmacol. 1998;123:581–589. doi: 10.1038/sj.bjp.0701626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddam B, Adams BW. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science. 1998;281:1349–1352. doi: 10.1126/science.281.5381.1349. [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Nakanishi S. Molecular diversity of glutamate receptors and implications for brain function. Science. 1992;258:597–603. doi: 10.1126/science.1329206. [DOI] [PubMed] [Google Scholar]
- Ohishi H, Shigemoto R, Nakanishi S, Mizuno N. Distribution of the messenger RNA for a metabotropic glutamate receptor, mGluR2, in the central nervous system of the rat. Neuroscience. 1993a;53:1009–1018. doi: 10.1016/0306-4522(93)90485-x. [DOI] [PubMed] [Google Scholar]
- Ohishi H, Shigemoto R, Nakanishi S, Mizuno N. Distribution of the mRNA for a metabotropic glutamate receptor (mGluR3) in the rat brain: an in situ hybridization study. J Comp Neurol. 1993b;335:252–266. doi: 10.1002/cne.903350209. [DOI] [PubMed] [Google Scholar]
- Omkumar RV, Kiely MJ, Rosenstein AJ, Min KT, Kennedy MB. Identification of a phosphorylation site for calcium/calmodulin dependent protein kinase II in the NR2B subunit of the N-methyl-D-aspartate receptor. J Biol Chem. 1996;271:31670–31678. doi: 10.1074/jbc.271.49.31670. [DOI] [PubMed] [Google Scholar]
- Otani S, Daniel H, Takita M, Crepel F. Long-term depression induced by postsynaptic group II metabotropic glutamate receptors linked to phospholipase C and intracellular calcium rises in rat prefrontal cortex. J Neurosci. 2002;22:3434–3444. doi: 10.1523/JNEUROSCI.22-09-03434.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petralia RS, Wang YX, Niedzielski AS, Wenthold RJ. The metabotropic glutamate receptors, mGluR2 and mGluR3, show unique postsynaptic, presynaptic and glial localizations. Neuroscience. 1996;71:949–976. doi: 10.1016/0306-4522(95)00533-1. [DOI] [PubMed] [Google Scholar]
- Raman IM, Tong G, Jahr CE. Beta-adrenergic regulation of synaptic NMDA receptors by cAMP-dependent protein kinase. Neuron. 1996;16:415–421. doi: 10.1016/s0896-6273(00)80059-8. [DOI] [PubMed] [Google Scholar]
- Schoepp DD, Conn PJ. Metabotropic glutamate receptors in brain function and pathology. Trends Pharmacol Sci. 1993;14:13–20. doi: 10.1016/0165-6147(93)90107-u. [DOI] [PubMed] [Google Scholar]
- Schoepp DD, Jane DE, Monn JA. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- Schoepp DD, Marek GJ. Preclinical pharmacology of mGluR2/3 receptor agonists: novel agents for schizophrenia? Current Drug Targets – CNS Neurol Disorders. 2002;1:215–225. doi: 10.2174/1568007024606177. [DOI] [PubMed] [Google Scholar]
- Schoepp DD, Salhoff CR, Wright RA, Johnson BG, Burnett JP, Mayne NG, Belagaje R, Wu S, Monn JA. The novel metabotropic glutamate receptor agonist 2R,4R-APDC potentiates stimulation of phosphoinositide hydrolysis in the rat hippocampus by 3,5-dihydroxyphenylglycine: evidence for a synergistic interaction between group 1 and group 2 receptors. Neuropharmacology. 1996;35:1661–1672. doi: 10.1016/s0028-3908(96)00121-9. [DOI] [PubMed] [Google Scholar]
- Seeburg PH. The TINS/TIPS lecture: the molecular biology of mammalian glutamate recptor channels. Trends Neurosci. 1993;16:359–365. doi: 10.1016/0166-2236(93)90093-2. [DOI] [PubMed] [Google Scholar]
- Strack S, Colbran RJ. Autophosphorylation-dependent targeting of calcium/calmodulin-dependent protein kinase II by the NR2B subunit of the N-methyl-d-aspartate receptor. J Biol Chem. 1998;273:20689–20692. doi: 10.1074/jbc.273.33.20689. [DOI] [PubMed] [Google Scholar]
- Tanaka C, Nishizuka Y. The protein kinase C family for neuronal signaling. Annu Rev Neurosci. 1994;17:551–567. doi: 10.1146/annurev.ne.17.030194.003003. [DOI] [PubMed] [Google Scholar]
- Tingley WG, Ehlers MD, Kameyama K, Doherty C, Ptak JB, Riley CT, Huganir RL. Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-D-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J Biol Chem. 1997;272:5157–5166. doi: 10.1074/jbc.272.8.5157. [DOI] [PubMed] [Google Scholar]
- Tingley WG, Roche KW, Thompson AK, Huganir RL. Regulation of NMDA receptor phosphorylation by alternative splicing of the C-terminal domain. Nature. 1993;364:70–73. doi: 10.1038/364070a0. [DOI] [PubMed] [Google Scholar]
- Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F, et al. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem. 1991;266:15771–15781. [PubMed] [Google Scholar]
- Tsai G, Coyle JT. Glutamatergic mechanisms in schizophrenia. Annu Rev Pharm Toxicol. 2002;42:165–179. doi: 10.1146/annurev.pharmtox.42.082701.160735. [DOI] [PubMed] [Google Scholar]
- Tsai LH, Delalle I, Caviness VS, Jr, Chae T, Harlow E. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature. 1994;371:419–423. doi: 10.1038/371419a0. [DOI] [PubMed] [Google Scholar]
- Vicini S, Wang JF, Li JH, Zhu WJ, Wang YH, Luo JH, Wolfe BB, Grayson DR. Functional and pharmacological differences between recombinant N-methyl-D-aspartate receptors. J Neurophysiol. 1998;79:555–566. doi: 10.1152/jn.1998.79.2.555. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhong P, Gu Z, Yan Z. Regulation of NMDA receptors by dopamine D4 signaling in prefrontal cortex. J Neurosci. 2003;23:9852–9861. doi: 10.1523/JNEUROSCI.23-30-09852.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal RS, Tavalin SJ, Lin JW, Alto NM, Fraser ID, Langeberg LK, Sheng M, Scott JD. Regulation of NMDA receptors by an associated phosphatase-kinase signaling complex. Science. 1999;285:93–96. doi: 10.1126/science.285.5424.93. [DOI] [PubMed] [Google Scholar]
- Williams K. Ifenprodil discriminates subtypes of the N-methyl-D-aspartate receptor: selectivity and mechanisms at recombinant heteromeric receptors. Mol Pharmacol. 1993;44:851–859. [PubMed] [Google Scholar]
- Wittmann M, Marino MJ, Conn PJ. Dopamine modulates the function of group II and group III metabotropic glutamate receptors in the substantia nigra pars reticulata. J Pharmacol Exp Ther. 2002;302:433–441. doi: 10.1124/jpet.102.033266. [DOI] [PubMed] [Google Scholar]
- Yan Z, Hsieh-Wilson L, Feng J, Tomizawa K, Allen PB, Fienberg AA, Nairn AC, Greengard P. Protein phosphatase 1 modulation of neostriatal AMPA channels: regulation by DARPP-32 and spinophilin. Nat Neurosci. 1999;2:13–17. doi: 10.1038/4516. [DOI] [PubMed] [Google Scholar]
- Yan Z, Surmeier DJ. D5 dopamine receptors enhance Zn2+-sensitive GABAA currents in striatal cholinergic interneurons through a PKA/PP1 cascade. Neuron. 1997;19:1115–1126. doi: 10.1016/s0896-6273(00)80402-x. [DOI] [PubMed] [Google Scholar]
- Zhang S, Ehlers MD, Bernhardt JP, Su CT, Huganir RL. Calmodulin mediates calcium-dependent inactivation of N-methyl-D-aspartate receptors. Neuron. 1998;21:443–453. doi: 10.1016/s0896-6273(00)80553-x. [DOI] [PubMed] [Google Scholar]