

Abstract
SQUAMOSA and APETALA1 are floral meristem identity genes from snapdragon (Antirrhinum majus) and Arabidopsis, respectively. Here, we characterize the floral meristem identity mutation proliferating inflorescence meristem (pim) from pea (Pisum sativum) and show that it corresponds to a defect in the PEAM4 gene, a homolog of SQUAMOSA and APETALA1. The PEAM4 coding region was deleted in the pim-1 allele, and this deletion cosegregated with the pim-1 mutant phenotype. The pim-2 allele carried a nucleotide substitution at a predicted 5′ splice site that resulted in mis-splicing of pim-2 mRNA. PCR products corresponding to unspliced and exon-skipped mRNA species were observed. The pim-1 and pim-2 mutations delayed floral meristem specification and altered floral morphology significantly but had no observable effect on vegetative development. These floral-specific mutant phenotypes and the restriction of PIM gene expression to flowers contrast with other known floral meristem genes in pea that additionally affect vegetative development. The identification of PIM provides an opportunity to compare pathways to flowering in species with different inflorescence architectures.
The transition from the vegetative to the reproductive phase in plants commences when a signal from the leaves evokes a response in the shoot apical meristem that results in the development of flowers. The genes regulating the cascade of processes that occur in the shoot apex after this switch to reproductive growth have been well studied in the herbaceous species snapdragon (Antirrhinum majus) and Arabidopsis (Simpson et al., 1999; Theissen, 2001). For example, in snapdragon, the floral meristem identity gene SQUAMOSA (SQUA) is required for the transition to flowering and floral organ specification. This was determined by the phenotypes of squa null mutants, which typically produce reiterated inflorescences in place of flowers (Huijser et al., 1992). Flower formation, when it occurs, includes a wide range of floral abnormalities, especially in the two outer whorls (Huijser et al., 1992), suggesting that SQUA also functions in first- and second-whorl organ specification in snapdragon.
In Arabidopsis, a corresponding role in floral development is carried out by APETALA1 (AP1). Flowers on Arabidopsis plants carrying strong ap1 mutant alleles retain many inflorescence-like characteristics; first-whorl organs are converted into bract or leaf-like organs bearing axillary flowers, which then repeat the pattern of the first flower (Irish and Sussex, 1990; Mandel et al., 1992; Bowman et al., 1993). The addition of a second mutation, cauliflower (cal), to an ap1 mutant background completely transforms the aberrant flowers into proliferating inflorescences, although the cal mutation has no effect in a wild-type (AP1) background (Bowman et al., 1993). This double mutant phenotype implicates CAL in the acquisition of floral meristem identity and suggests that it has a redundant role with AP1 in this process. The functional redundancy of AP1 and CAL reflects their close molecular relationship; both are members of the same MADS-box gene subclade (Kempin et al., 1995; Theissen et al., 2000).
AP1 is transcribed in response to light treatments (Hempel et al., 1997) and the flowering time gene, CONSTANS (Simon et al., 1996); thus, it acts as a molecular marker for floral determination (Hempel et al., 1997). It is also transcriptionally activated by another floral meristem identity gene, LEAFY (LFY; Parcy et al., 1998; Wagner et al., 1999). Although LFY acts non-cell autonomously in floral specification, AP1 activates target genes in a mainly cell autonomous manner (Sessions et al., 2000). Transgenic experiments demonstrated that target genes of AP1, such as APETALA3 (Hill et al., 1998), are activated via the formation of ternary and quaternary complexes of MADS-box proteins in Arabidopsis (Honma and Goto, 2001). Corroborating in vitro experiments with SQUA showed that it binds to promoter motifs in multimeric complexes, together with other MADS-box proteins, including DEF and GLO (Egea-Cortines et al., 1999). It was suggested that the combinations of proteins within these complexes provides regulatory specificity during floral development (Egea-Cortines et al., 1999; Honma and Goto, 2001). Thus, a detailed picture of a hierarchy of genes regulating floral meristem specification and development is emerging. Identification of homologous mutations in crop species will help to indicate the extent to which gene activities uncovered in these model species have diversified or been conserved.
The phenotype of the proliferating inflorescence meristem (pim) mutant from pea (Pisum sativum) is similar to that of squa and ap1 mutants, and it was suggested that PIM may represent a floral meristem identity gene (Singer et al., 1994). A good candidate for the gene corresponding to PIM is PEAM4, a MADS-box gene that is closely related to AP1, CAL, and SQUA. PEAM4 has been shown to rescue floral organ defects in the ap1-1 mutant of Arabidopsis when expressed in transgenic plants under the control of the 35S promoter (Berbel et al., 2001). PEAM4 expression was altered in the pea floral homeotic mutants calix carpellaris and frondosus (Berbel et al., 2001), but the allelic relationship between these mutations, pim, and PEAM4 has not been investigated. In this paper, we describe two pim mutations that result in delayed floral meristem specification and first- and second-whorl floral abnormalities, and we show that both pim mutants carry altered PEAM4 alleles, one of which results in aberrant transcript splicing.
RESULTS
Wild-Type Pea Inflorescence Structure
The inflorescence of pea has been described as a raceme (Hole and Hardwick, 1976) and a panicle (Tucker, 1989). Current interpretations agree that the main shoot apex is converted into a primary inflorescence on floral induction, and this primary inflorescence bears morphologically distinct secondary inflorescences that terminate in a hairy stub after producing one or two flowers (Singer et al., 1994; Ferrándiz et al., 1999). The primary inflorescence bears compound leaves and is indistinguishable from the vegetative shoot from which it is derived, apart from the production of secondary inflorescences (Makasheva, 1983). This inflorescence architecture is illustrated schematically in Figure 1A. Pea flowers are typical of the Papilionoideae, with five green sepals fused at the base, forming a cup, and five colored petals differentiated into three petal types. The standard is the largest and uppermost and there are two wings laterally and two fused petals that form the keel (Fig. 1B). Enclosed within the keel are 10 stamens, nine fused and one free, which surround the single, central carpel (Tucker, 1989; Ferrándiz et al., 1999).
Figure 1.

Wild-type and mutant inflorescences. A, Schematic diagram of a wild-type pea plant. The center line with an arrow represents the abbreviated (//) main axis of the pea plant with its indeterminate apical meristem. At first, the apical meristem is vegetative and produces leaves (ellipses). On floral induction, the apical meristem is converted to an indeterminate primary inflorescence apex that bears secondary inflorescences (/) in the leaf axils. These in turn bear one or two flowers (●), and terminate in a stub (▾). B, Secondary inflorescence from a wild-type pea bearing two flowers. These are typical pea flowers with wild-type anthocyanin pigmentation, showing standard (st), two wings (w), and two fused petals forming the keel (k). Within the keel are the 10 stamens and a central carpel. Also visible are some of the five sepals (se), which form a cup surrounding the petals. The stub that terminates the secondary inflorescence (inf) is not visible behind the pedicel (pd). C, Young (at anthesis) secondary inflorescences from wild-type (WT), pim-1, and pim-2 flowers from plants with a white-flowered (anthocyanin absent) background. In the pim mutants, each flower is replaced by additional secondary inflorescences (inf) that bear abnormal flowers. Flowers are surrounded by leafy bracts (br) but are able to produce some petals, stamens, and carpels. Mosaic organs are also produced (mo). D, Secondary inflorescences from the same genotypes approximately 3 weeks later. A terminal stub is visible on the wild-type inflorescence (sb). The carpel of each wild-type flower has developed into a pod (p), and the proliferation of the pim mutant inflorescences has continued.
pim Mutations Delay Floral Meristem Specification and Alter Floral Morphology
A spontaneous, recessive mutant was identified in Minnesota and named pim-1 after its severe floral abnormalities (Singer et al., 1994). A second, spontaneous mutant with a similar phenotype was identified in Tasmania. This latter mutation segregated in accordance with a 3:1 ratio (P > 0.5) from a cross to its wild-type progenitor line, indicating that it was controlled by a single recessive allele. Allelism between pim-1 and the Tasmanian mutant (pim-2) was confirmed by crosses between a plant heterozygous for pim-2 and a homozygous pim-1 plant (HL 244): Five of seven F1 plants produced mutant flowers. Comparison of pim-2 plants with their isogenic wild-type siblings failed to reveal any significant differences in vegetative traits, such as length of basal internodes and the nodes where leaflet number increased; likewise, the node where the first secondary inflorescence occurred was not altered (P > 0.5 for all traits). This analysis indicated that the pim-2 mutation specifically affected flower development.
Primary and secondary inflorescences were correctly specified in both pim-1 and pim-2 mutants, but the transition from secondary inflorescence to flower production was delayed. In place of floral meristems, additional secondary-like inflorescences were produced (Fig. 1, C and D). Eventually, each of these inflorescences bore two or more abnormal flowers. pim mutants occasionally showed a form of floral reversion with a leafy shoot replacing one of the flowers on the secondary inflorescence. These leafy shoots seemed to represent a reversion to primary inflorescence, rather than vegetative development, because they bore aberrant flowers, as described below.
Floral morphology of pim mutants was aberrant in that first-whorl sepals were replaced by leafy bract-like structures, and second- and third-whorl organs were either absent or mosaic (Fig. 1, C and D). Early flowers on pim-1 plants often consisted of these bracts surrounding the reproductive whorls; petals were entirely absent. Flowers consisting of outer bracts, petals, and a cluster of central stamens were also noted, as were complex, proliferating flowers, composed of combinations of the simpler flower types. Later flowers on pim-1 and all flowers on pim-2 produced morphologically normal standard and wing petals; however, petal position was irregular, and some flowers produced more than one standard or more than two wing petals. Wild-type flowers develop a single standard and two wings. Normal stamens and a single central carpel were seen in many flowers, although fusion of the carpel margins was not always complete. Self-pollination was uncommon in both pim-1 and pim-2 plants. Flowers produced late on the primary and lateral shoots often had a simpler structure, approaching wild type in appearance, except that the five sepal-like organs of the outer whorl were larger and leafier than those of wild-type flowers. These flowers also tended to produce fewer petals and stamens than wild type. To illustrate the extent of floral abnormality, counts were made of organs found on the secondary inflorescence of the eighth flowering node of pim-1 and pim-2 mutants, and these are listed in Table I.
Table I.
Floral organs present in flowers from the secondary inflorescence at reproductive node 8 of wild-type and pim mutant plants
| Genotype
|
|||
|---|---|---|---|
| Wild type | pim-1a | pim-2a | |
| Flowersb | 1 | 2.83 ± 0.28 | 2.83 ± 0.07 |
| Leafy shoot | 0 | 1.14 ± 0.14 | 0 ± 0.00 |
| Bracts | 0 | 5.86 ± 1.34 | 1.23 ± 0.20 |
| Sepaloid bracts | 0 | 4.00 ± 0.69 | 3.18 ± 0.33 |
| Sepals | 5 | 3.00 ± 0.96 | 4.40 ± 0.45 |
| Petaloid sepals | 0 | 4.29 ± 0.67 | 2.60 ± 0.28 |
| Petals | 5 | 4.71 ± 1.30 | 5.35 ± 0.44 |
| Staminoid petals | 0 | 4.86 ± 1.29 | 1.85 ± 0.16 |
| Stamens | 10 | 3.28 ± 0.81 | 12.83 ± 0.45 |
| Carpelloid stamens | 0 | 1.20 ± 0.17 | 0.20 ± 0.06 |
| Carpels | 1 | 0.14 ± 0.14 | 1.95 ± 0.10 |
Seven pim-1 and 20 pim-2 flowers were examined. Values are mean no. of organs present at each position normally occupied by a single flower on a wild-type secondary inflorescence (±se).
The no. of flowers on distinct pedicels present at each point normally occupied by a single flower in wild-type plants.
The Relationship between pim and the PEAM4 Gene from Pea
Because the phenotypes of the pim-1 and pim-2 mutants resembled those described for squa and ap1, a pea homolog of these genes was isolated from a shoot-tip cDNA library using the snapdragon SQUA gene (Huijser et al., 1992) as a probe. A full-length cDNA of 1,207 bp (GenBank accession no. AF461740), called PEASQUA, was mapped to linkage group IV of pea, using an EcoRI RFLP that segregated in a recombinant-inbred-line population derived from the cross JI 281 × JI 399 (Hall et al., 1997). PEASQUA was found to be 99% identical to PEAM4, an independently isolated pea homolog of AP1 and SQUA (Berbel et al., 2001), although it is 10 bp longer than PEAM4 in the 5′-untranslated region and 63 bp longer in the 3′-untranslated region. There are only two single-base mismatches within the coding regions when the sequences are aligned with each other, but these do not result in differences between the amino acid sequences. Given this degree of similarity, these SQUA homologs probably represent alleles of the same gene. The absence of a farnesylation motif at the 3′ end of the open reading frame (Berbel et al., 2001) was confirmed in the PEAM4 cDNA we isolated.
DNA gel blots of pim-1, pim-2, and wild-type plants, probed with the PEAM4 cDNA minus the MADS-box region and washed at low stringency, were carried out to ascertain gene copy number and to compare the structures of the mutant and wild-type alleles, as shown in Figure 2A. There is one NcoI site in the PEAM4 cDNA, and only two strongly hybridizing bands were observed in the pim-2 and wild-type lanes on the DNA gel blot. This suggests that PEAM4 is not duplicated in the genome, unless the duplicated copy has identical flanking and internal restriction enzyme sites. There are two HindIII sites in the PEAM4 cDNA, and two strongly hybridizing bands were observed on the pim-2 and wild-type lanes of the blot. Weaker hybridizing bands were also observed, one of which is predicted to produce a weak signal because it hybridizes to only 159 bp of the probe; the others probably represent a closely related gene. Apart from the single strongly hybridizing bands in the EcoRI and EcoRV-digested lanes, which again provide support for a single-copy gene, a faintly hybridizing band can also be seen in pim-1 and pim-2 mutant lanes and wild-type lanes, which is likely to represent a closely related gene.
Figure 2.

DNA gel-blot analysis. A, EcoRI, EcoRV, HindIII, and NcoI-digested pim-1, pim-2, and wild-type (WT) genomic DNA, probed with the C-terminal fragment of the PEAM4 cDNA and washed at low stringency. B, Ethidium bromide-stained gel of the samples shown in A, before they were blotted to a filter. Marker lane (M) contained bacteriophage lambda DNA digested with EcoRI and HindIII to generate 14 fragments, 21, 9.4, 6.6, 5.0, 4.3, 3.6, 2.3, 2.0, 1.9, 1.6, 1.4, 0.9, 0.8, and 0.6 kb in size.
No hybridization signals were detected in the lanes corresponding to pim-1, although the ethidium bromide-stained gel (Fig. 2B) confirmed that all lanes were equally loaded with digested DNA. The absence of both bands in the NcoI-digested pim-1 lane indicated that a deletion of the entire PEAM4 coding region had occurred in the pim-1 mutant line. This deletion cosegregated with the pim-1 mutant phenotype (data not shown), consistent with PEAM4 corresponding to PIM. It was possible, however, that the deletion in pim-1 mutants was large, encompassing other genes besides PEAM4. To substantiate further the possible correspondence between PEAM4 and PIM, the pim-2 allele was examined. No differences could be detected between wild type and pim-2 on DNA gel blots using restriction enzymes BamHI (data not shown), EcoRI, EcoRV, HindIII, and NcoI (Fig. 2A). On an RNA gel blot probed with PEAM4, shown in Figure 3A, the hybridizing transcript from pim-2 mutant flowers was larger and less abundant than that seen in similarly aged wild-type flowers. The difference in transcript abundance was assessed on the basis that the wild-type and pim-2 mutant lanes were approximately equally loaded with RNA when the gel blot was reprobed with an rDNA probe (Fig. 3B). This indicated that the pim-2 mutation disrupted PEAM4 gene expression and further confirmed their identity.
Figure 3.
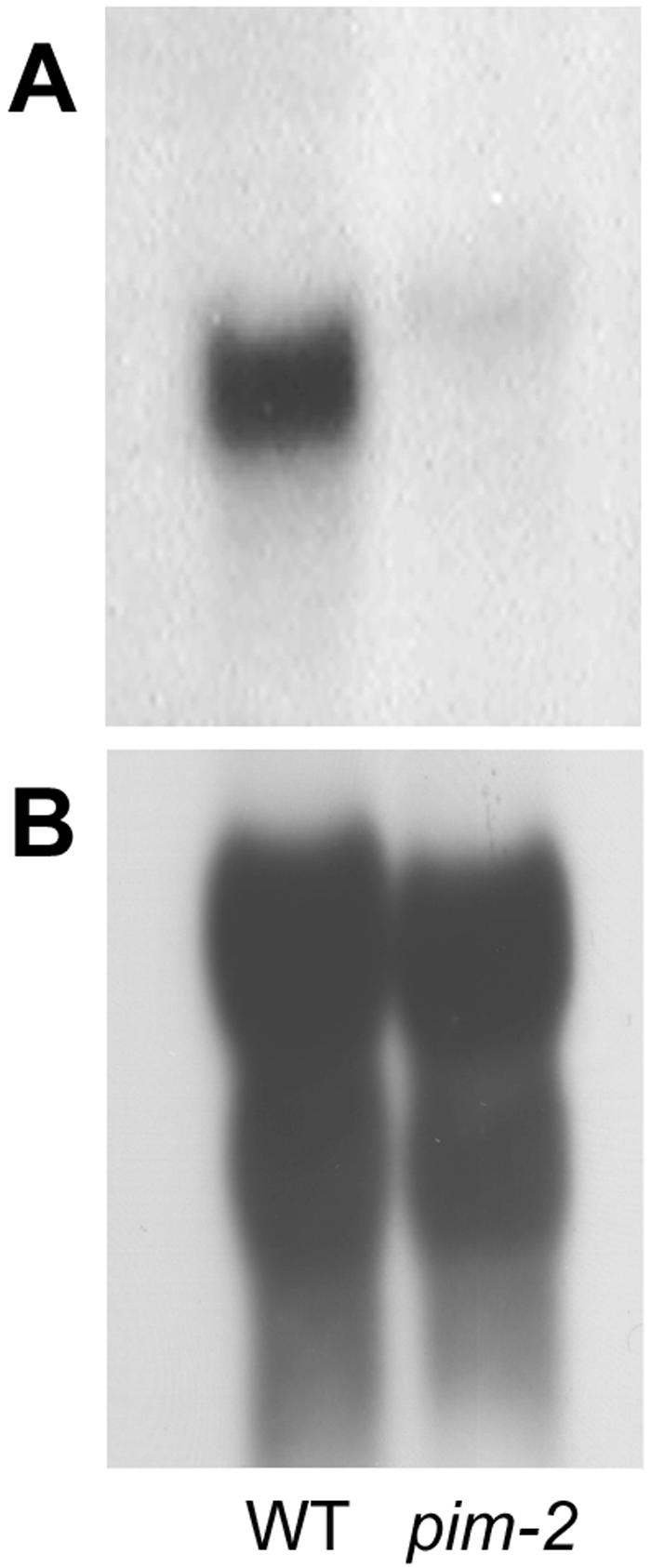
RNA gel-blot analysis. A, Total RNA from pim-2 and wild-type (WT) flowers, probed with the C-terminal fragment of the PEAM4 cDNA. B, Total RNA from pim-2 and wild-type (WT) flowers, probed with rDNA.
To characterize the aberrant transcript in more detail, nested PCR was performed on reverse-transcribed cDNA templates, produced from wild-type and pim-2 flowers, using oligonucleotides specific to the PEAM4 sequence. Only one PCR product was amplified from wild-type cDNA, but two pim-2 PCR products were amplified: One was approximately 100 bp larger than wild type, and the second was approximately 100 bp smaller (not shown). Sequence analysis revealed a 104-bp insert in the larger pim-2 PCR product that was not present in wild-type cDNA, as illustrated in Figure 4. Sequences flanking the insert were identical to those of the wild-type PCR product and the original cDNA clone. To examine the nature of this insert further, the region was amplified from wild-type and mutant genomic DNA using PCR. The aligned sequences confirmed that the insert was present in both wild-type and mutant genomic DNA PCR products (data not shown). The insert was AT rich (76%; see Fig. 4), which is characteristic of plant introns (Goodall and Filipowicz, 1989), and the position of this putative intron was consistent with the position of the fourth intron present in SQUA and AP1 genomic sequences (Huijser et al., 1992; Mandel et al., 1992). It is significant that wild-type genomic DNA sequence differed from the pim-2 sequence by a single-base change, substitution of an adenine for a guanine, at the predicted 5′ splice site (Fig. 4); thus, the presence of the 104-bp insert in pim-2 mRNA was probably a consequence of a failure in splicing. The resulting pim-2 mutant open reading frame is predicted to terminate with a stop codon three bases after the A for G substitution. The low abundance of this larger transcript relative to wild type (Fig. 3A) suggests that the unspliced transcript may be less stable than the wild-type transcript.
Figure 4.

Sequence analysis of PCR products. Alignment of sequences from PEAM4 PCR products from wild-type (WT) genomic DNA and from pim-2 and wild-type cDNA. A single guanine to adenine substitution at the 5′-splice acceptor site is highlighted in bold (arrowhead).
Sequence from the smaller PCR product amplified from pim-2 cDNA revealed a 100-bp deletion relative to wild type that removed the predicted exon between the predicted third and fourth intron positions. This mis-splicing by exon skipping (removing the third intron, intervening exon, and fourth intron) would result in a frame shift that would terminate translation at a stop codon 16 amino acids after the splice junction. Other intron sites, whose approximate positions were predicted from the conserved intron positions in SQUA and AP1, were correctly spliced in the pim-2 mutant cDNA, and no additional sequence differences were detected in the PCR products obtained using cDNA from wild-type and mutant plants.
Together, the results of this molecular analysis of PEAM4 alleles present in two independent pim mutants strongly supports the identity of PEAM4 and PIM: the pim-1 allele corresponding to a gene deletion and the pim-2 allele corresponding to a single-base change that results in aberrant transcript splicing.
Expression Pattern of PIM
The phenotype of the two pim mutants suggested a role for PIM during floral meristem development, therefore, we examined the expression pattern of PIM in shoot tips before and after flowering as shown in Figure 5. PIM expression was not detected in vegetative shoot tips, but was detected in flowering shoots of all three genotypes examined (Fig. 5A). The mutants unifoliata (uni) and stamina pistilloida (stp), which correspond to lfy and unusual floral organs (ufo) in Arabidopsis (Hofer et al., 1997; Taylor et al., 2001), were included in this analysis to investigate whether PIM expression was dependent on UNI or STP. PIM expression in flowering shoots was not dependent on UNI or STP (Fig. 5A). In both mutants, the level of PIM expression was higher than in wild type, as assessed by the approximately equal amounts of RNA loaded in each gel lane (Fig. 5B).
Figure 5.

PEAM4 expression in uni and stp mutants before and after flowering. A, Northern gel blot using total RNA from sibling uni, stp, and wild-type (WT) plants, probed with the C-terminal fragment of the PEAM4 cDNA and washed at 65°C in 0.5× SSC. The first three lanes contain RNA from plants in the vegetative phase, the last three lanes contain RNA from flowering plants. The positions of the 25S and 18S ribosomal RNA bands are shown on the right. The PEAM4 transcript is indicated (arrow). B, Ethidium bromide-stained gels of the samples shown in A, before they were blotted to a filter.
We examined PIM expression in floral tissues in more detail by RNA in situ hybridization analysis. PIM had a clearly delineated pattern of expression within developing floral primordia, as illustrated in Figure 6, and expression was not observed in vegetative tissue or mature inflorescences. PIM expression occurred throughout the entire floral primordium at stage 2 (Fig. 6A, flower F1), as defined by Ferrándiz et al. (1999). Later, during stage 4 of floral ontogeny, PIM expression was limited to the outer two whorls that were initiating sepal and common petal/stamen primordia, but expression also extended downward into the pedicel of the developing flower; the central carpel dome clearly lacked the hybridization signal (Fig. 6B, flower F1). The location of PIM expression within common primordia at stage 4 marked the identity of organs subsequently initiated during stage 5, because expression was present in petal-fated cells but absent from stamen-fated cells (Fig. 6B, flower F1). At stage 5 and later, PIM expression was restricted to sepals (Fig. 6B, flower F2) and petals (Fig. 6C). This pattern of expression confirms the observations of Berbel and colleagues (2001) and is very similar to the expression patterns of AP1 (Mandel et al., 1992) and SQUA (Huijser et al., 1992) during the development of Arabidopsis and snapdragon flowers. The absence of PIM expression in vegetative tissues (Fig. 5) was confirmed in the afila genotype, where the UNI gene is known to be highly expressed in developing leaves (Gourlay et al., 2000). In this longitudinal section, PIM expression was clearly confined to the floral primordia and absent from subtending leaves (Fig. 6D).
Figure 6.
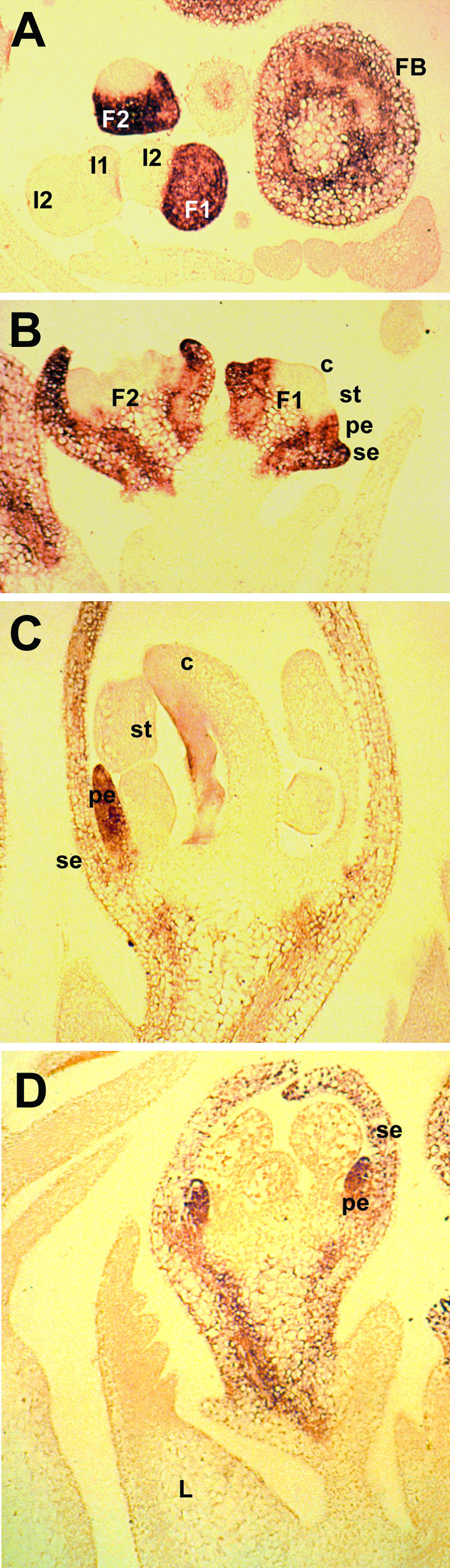
RNA in situ analysis of PEAM4 expression in developing pea flowers. A, PEAM4 expression in wild-type genotype HL 107 was confined to the flower and was not detected in vegetative or inflorescence tissue (I1 and I2 indicate the primary and secondary inflo- rescences, respectively). Expression occurred throughout young floral primordia (flower F1) at stage 2 and was also apparent in the oblique section through flower 2 (F2) and in a transverse section through the base of an older flower bud (FB) at approximately stage 7 of development. B, Stage 4 (F1) of development in genotype HL 107, showing PEAM4 expression in the petal region of the petal/stamen common primordia (pe) and the sepals (se). Expression was absent from the stamen region of the petal/stamen common primordia (st) and the carpel primordium (c). Stage 5 (F2) flower showing expression in the outer sepal whorl. Petals express PIM at this stage but were not in the plane of this section. C, Stage 7 flower bud of genotype HL 107 showing PEAM4 expression limited to sepals and petals and absent from stamens (st) and the carpel. D, Stage 7 flower bud of the afila genotype, JI 1195, showing PIM expression limited to the two outer whorls and absent from the subtending compound leaf (L). Magnification ×75 in A through D.
DISCUSSION
Comparative Flower Development
Mutations in SQUA homologs have been characterized so far in only two species, snapdragon and Arabidopsis. The identification here of PIM as a homolog of SQUA and AP1 provides an opportunity to extend our understanding of the role of these genes in floral meristem specification to a third species, pea. It also enables a more complete comparison of the pathways to flowering proposed for pea (Weller et al., 1997) with those proposed for Arabidopsis (Piñeiro and Coupland, 1998; Simpson et al., 1999) and those being investigated in snapdragon (Cremer et al., 1998).
The similarity between the pim mutant phenotype and those of squa and ap1 implies a conservation of gene activity. Mutations in SQUA and AP1 result in a reiterating inflorescence phenotype (Irish and Sussex, 1990; Huijser et al., 1992; Mandel et al., 1992; Bowman et al., 1993) that is analogous to the replacement of flowers by proliferating secondary inflorescences and primary inflorescence-like leafy shoots seen in pim mutants. Conservation of AP1 and PIM gene function is also supported by transgenic experiments in Arabidopsis. PIM (PEAM4) overexpression in an ap1 mutant partially complemented the mutation, and overexpression in a wild-type background mimicked AP1 overexpression in that it resulted in early flowering (Berbel et al., 2001).
Although the proliferating inflorescence phenotype is in common, flower formation also occurs in squa, ap1, and pim mutants, suggesting that there is a redundant factor that can provide floral meristem identity in all three species. In Arabidopsis, redundant genes providing this function have been identified. For example, the role of CAL, which is very similar in sequence to AP1, was unmasked in an ap1 cal double mutant, because the cal mutation alone has no observable mutant phenotype (Bowman et al., 1993; Kempin et al., 1995). The flowers produced by squa and pim mutants are sometimes almost normal, in that they contain all organ types. In contrast, an entire complement of normal floral organs has not been observed on single-mutant ap1 flowers; even on plants carrying weak alleles, sepals, and wild-type numbers of petals, are not seen (Irish and Sussex, 1990; Bowman et al., 1993). This suggests that, unlike snapdragon and pea, a redundant factor providing AP1 function in outer whorl organ specification is absent in Arabidopsis.
Flowers with mosaic, or altered numbers of stamens and carpels, were observed on pim mutants (Table I), suggesting that PIM may have a role in the inner whorls of the flower. The nature of this role is difficult to clarify at present, because inner whorl organ numbers and types were not consistently altered in pim-1 and pim-2 mutants (Table I), which differ in their genetic backgrounds. Increased numbers of stamens and carpels and petaloid stamens were also described in squa mutants (Huijser et al., 1992), whereas reduced numbers of stamens, petaloid stamens, and incompletely fused carpels were reported in ap1 mutants (Bowman et al., 1993). In snapdragon, Arabidopsis, and pea, the two outer whorls of mutant flowers are more strongly affected than the inner whorls (Huijser et al., 1992; Bowman et al., 1993; this work). Given that the ontogeny of a pea flower differs greatly from snapdragon and Arabidopsis flowers, in that the second- and third-organ whorls are derived from a common primordium (Tucker, 1989), it is surprising that there are not more profound differences in the corresponding mutant phenotypes.
PIM Is the Ortholog of SQUA
Identification of orthologous gene pairs is useful, not only for comparison of gene functions, but also because they provide definitive single-point comparisons in genetic map alignments between species pairs. Resolution of orthologous relationships among SQUA, PIM, AP1, and CAL based on sequence similarity is difficult because the presence of two or more SQUA-like genes in some species suggests that complex relationships exist between the subfamily members, with the possibility of multiple independent duplication events. For example, phylogeny reconstructions suggest that CAL may have originated after a gene duplication (Theissen et al., 2000). However, because CAL orthologs have not yet been identified in species outside the Brassicaceae, the relationship of CAL to other SQUA homologs remains unclear. For this reason, we use the more general term homolog when referring to members of the clade containing SQUA, AP1, and CAL. Despite this difficulty in determining orthology with Arabidopsis genes, we consider that PIM, the only representative from pea in this clade (http://www.mpiz-koeln.mpg.de/mads/madstrees.html), and SQUA, the only representative from snapdragon, are orthologous genes. This conclusion is supported by the fact that only one band was detected on DNA gel blots probed with PEAM4, therefore it is unlikely that a duplicated gene exists in pea.
Orthology relationships between genes may be reflected by their map positions, so we have compared the map positions of AP1, CAL, and PIM to clarify their relationship to each other. AP1 and CAL are 53 cM apart on the same Arabidopsis chromosome, and CAL maps very close to UFO (http://Arabidopsis. org/servlets/mapper). The pea ortholog of UFO, STP, maps to linkage group VII (Taylor et al., 2001), whereas PIM maps to linkage group IV (as marker PEASQUA; Hall et al., 1997). PIM is, thus, more like AP1 than CAL in that it is not closely linked to the pea UFO ortholog. This is consistent with the mutant phenotypes, which also suggest that PIM shares more in common with AP1 than it does with CAL.
The pim-2 Mutation Affects Transcript Splicing
The deletion of PIM in pim-1 plants suggests that pim-1 is a null allele. It is likely that pim-2 also represents a null allele, first because the incorrectly spliced pim-2 transcripts are very low in abundance and are predicted to terminate the open reading frame prematurely, and second, because the pim-1 and pim-2 mutants exhibit similar morphological defects. The similarity of the pim-1 and pim-2 mutant phenotypes, furthermore, suggests that the deletion in pim-1 is not so large as to include closely linked genes with major developmental effects. It is important to note that differences between the pim-1 and pim-2 mutant phenotypes may not be allelic differences but may result instead from the different genetic backgrounds of these two mutants.
Northern gel-blot and sequence analysis of the pim-2 allele indicate that the G to A transition results in the production of aberrant transcripts by failure to excise the fourth intron and by exon skipping. There are other cases of G to A mutations in the 5′ splice sites of Arabidopsis introns where the effects of the mutations on splicing have been studied. For example, the transition present in the Rubisco activase mutant resulted in an accumulation of differently sized splicing intermediates that were detectable by northern gel-blot analysis (Orozco et al., 1993). The higher Mr pim-2 transcripts we detected on northern gel blots were of a uniformly larger size than wild type and were thus likely to represent the intron 4-containing transcript that was also identified among the cloned pim-2 cDNA products. A similar effect was observed in the phytochrome B-103 mutant, where the major effect of the mutation was a failure to splice the intron (Bradley et al., 1995).
Another splicing behavior of the pim-2 mutant, detected only among sequenced cDNA products, was exon-skipping. Exon 4, which lies 5′ adjacent to the mutation, and both flanking introns, were excised. This was not reported for the Rubisco activase and phytochrome B-103 mutations, but was the major defect caused by the G to A mutation in the 5′ splice site of the constitutive photomorphogenic1-2 allele (Simpson et al., 1998). The pim-2 mutation, thus, provides further support for a role for exons, as well as introns, in pre-mRNA splice site definition (Simpson et al., 1998).
Both types of pim-2 defective transcripts would lead to premature truncation of the C-terminal domain of the PIM open reading frame, which is required by SQUA, DEF, and GLO proteins for the formation of ternary complexes in yeast (Saccharomyces cerevisiae; Egea-Cortines et al., 1999). The low abundance of the transcripts relative to wild type suggests that they may be subject to mRNA surveillance-mediated degradation (Hilleren and Parker, 1999). If this type of degradation occurs, it is not possible to distinguish whether the complete absence of the exon-skipping transcript on northern gel blots is because it is subject to more rapid decay than the intron-retaining transcript, or because the exon-skipped transcript is a rarer aberrant splicing product in the mRNA pool.
The Role of PIM in Pea Flower Development
PIM gene expression in developing flowers has been described recently and was found to be generally similar to the expression patterns of AP1 and SQUA (Berbel et al., 2001). The early transcription of these genes within developing floral primordia (Huijser et al., 1992; Mandel et al., 1992) reflects their common roles in floral meristem specification. Later in floral development, differences are apparent. SQUA is expressed in the developing carpel, but expression is excluded from stamen primordia (Huijser et al., 1992), whereas in this work, we confirm that PIM expression is excluded from both inner whorls (Berbel et al., 2001), as is AP1 (Mandel et al., 1992). Another difference is that SQUA is expressed in the bracts subtending flowers in snapdragon (Huijser et al., 1992), but not in Arabidopsis, where bracts are absent, nor in pea, where production of bracts in these genotypes is rare and unpredictable. However, these variations in patterns of gene expression do not seem to correlate with the minor differences in mutant phenotypes of the three species, such as the stronger effect of the ap1 mutation on outer whorl organ identity, compared with pim and squa. Differences in expression patterns or mutant phenotypes may reflect differences in wild-type development between these three species. Different requirements for farnesylation may also contribute to species differences. PIM and genes homologous to AP1 cloned from grass species (Gocal et al., 2001) do not contain a 3′-farnesylation sequence motif that is present in AP1 and other members of the clade (Berbel et al., 2001).
Peas have more complex leaves and inflorescence architecture than do Arabidopsis and snapdragon, and for this reason pea is an interesting species in which to examine the functions of homologous genes. Two other floral meristem identity genes have been identified previously. These are UNI, the ortholog of LFY (Hofer et al., 1997), and STP, the ortholog of UFO (Taylor et al., 2001). Both of these have been shown to have wider roles in vegetative development, apart from their participation in floral meristem specification. In contrast, the role of PIM is specific to the flower, because other aspects of plant development are unaffected in pim mutant plants.
Steroid-inducible activation of LFY in transgenic Arabidopsis showed that AP1 is directly transcriptionally regulated by LFY in inflorescences (Wagner et al., 1999). Although LFY was misexpressed throughout Arabidopsis plants using this inducible 35S promoter construct, AP1 transcription was activated only in the tissues and at the stage when floral fate would normally be assumed in wild type (Wagner et al., 1999): AP1 was not transcriptionally activated throughout the plant. Contrasting results were obtained by Parcy et al. (1998), who showed that activation of an AP1::GUS reporter gene occurred throughout transgenic 35S::LFY Arabidopsis seedlings before flowering. Our data suggest that tissue specificity in the activation of AP1 by LFY is conserved in peas. In pea leaves, UNI expression alone seems to be insufficient to up-regulate PIM, because afila mutant leaves, with prolonged and high levels of UNI expression (Gourlay et al., 2000), do not express PIM (see Fig. 6D).
Reports on the transcriptional activation of AP1 by LFY also vary on whether AP1 expression is reduced (Wagner et al., 1999), or almost normal (Parcy et al., 1998), in lfy mutants. In snapdragon, SQUA expression in the floricaula mutant is comparable with that of wild type (Huijser et al., 1992). Our results show that in pea, PIM expression is not reduced, but is increased, in a uni mutant background. The same result was obtained in a stp mutant background. Both of these mutations result in the production of flowers with supernumerary whorls of sepals and sepalloid organs (Hofer et al., 1997; Taylor et al., 2001). Thus, increased PIM expression relative to wild type is consistent with an increased number of first-whorl organs in the mutants. Our results clearly demonstrate that PIM expression is independent of UNI and STP during flowering.
Previous studies of uni have emphasized its unique leaf phenotype and its interactions with the leaf homeotic mutants in pea (Hofer et al., 1997; Gourlay et al., 2000; Taylor et al., 2001), rather than its role in floral specification. A detailed analysis of double mutants and their effects on flowering is now possible. These experiments and the identification of B- and C-class floral homeotic genes corresponding to APETALA3, PISTILLATA, and AGAMOUS are required to elucidate further the gene interactions in pea flower development and to determine the extent of conservation of gene function between Arabidopsis and pea.
MATERIALS AND METHODS
Plant Material and Cultivation
The pea (Pisum sativum) pim-1 and pim-2 mutations occurred spontaneously as independent events at Carleton College (Northfield, MN) and the University of Tasmania (Hobart, Australia), respectively. The pim-1 mutant does not have an isogenic wild-type line. The pim-2 mutation arose in cv Torsdag (line HL107), and phenotypic analyses of pim-2 were carried out on plants segregating in a second backcross to this line. Line HL107 was also used as the source of wild-type DNA and RNA. Seed of the pim-2 mutant line resulting from the second backcross was deposited into the Hobart germplasm collection as HL285. Sibling plants carrying uni-2171 (Hofer et al., 1997) or stp-4 (Taylor et al., 2001) mutant alleles or the corresponding wild-type alleles were used in northern gel-blot analyses. All siblings were short-statured afila tendril-less genotypes (Taylor et al., 2001). Shoot tips from plants at the vegetative phase of development were harvested 21 d after sowing, and flowers and shoot tips from flowering plants were harvested 33 d after sowing.
Plants used in the phenotypic analysis of pim-1 and pim-2, allelism tests, and gel blots were grown in Hobart in a 1:1 (v/v) mix of vermiculite and dolerite chips topped with 2 to 3 cm of pasteurized peat-sand potting mix under an 18-h photoperiod. Plants used for additional phenotypic analysis, gel blots, and RNA in situ hybridization studies were grown at the John Innes Centre in John Innes number 1 potting mix with 30% grit, under a 16-h photoperiod. All plants received liquid fertilizer weekly.
Molecular Analysis of PIM
The PEAM4 cDNA, cloned into the EcoRI and XhoI sites of pBluescript (Stratagene, La Jolla, CA), was initially identified as PEASQUA, and was isolated by screening a pea flowering-shoot-tip cDNA library (Hofer et al., 1997) with a full-length SQUA clone provided by Peter Huijser (Max Planck Institute, Köln, Germany). For analysis of transcript splicing, cDNA was produced by reverse transcription from total RNA isolated from pim-2 and wild-type (HL107) flowers just before anthesis. Two pairs of primers specific to the PEAM4 sequence were used for nested PCR: first round, (5′) GGG ACG AGC TCA AAC TCA CAC (3′) and (5′) GGA GTT CCT TCT AGT GAT AG (3′); second round, (5′) AGG AGA GCT GGA CTT CTC AAG (3′) and (5′) CTA CCA AAC ATA TAT ATA AGC (3′), using cDNA as a template. Primers flanking the insert present in the pim-2 cDNA (5′, ATG GGA GAA GAT TTG GGT ACA ATG and 5′, TTC TGA AGC TCT GAA ATG GAC TCG) were used to amplify fragments from pim-2 and wild-type genomic DNA. Amplified fragments were either subcloned into pGEM-T easy vectors (Promega, Madison, WI) for sequencing, or purified using a Concert PCR purification system (Invitrogen, Carlsbad, CA) and sequenced directly. Sequencing was carried out using ABI big dye terminator technology (Applied Biosystems, Foster City, CA).
RNA in situ hybridization was performed as described previously (Hofer et al., 1997) on 8-μm sections of wild-type flowering pea apices using digoxigenin-labeled sense and antisense probes. DNA and RNA blots and in situ hybridization analyses were performed using a modified clone that had the MADS-box region between restriction sites EcoRI and SpeI removed, to prevent cross hybridization with other MADS-box genes. Unless otherwise specified, high-stringency washes were at 65°C in 0.1× SSC and low-stringency washes were at 50°C in 2× SSC.
ACKNOWLEDGMENTS
We thank Tracey Jackson and Ian Cummings for technical assistance in Hobart, Richard Gould and Hilary Ford for horticultural assistance, and Claire Costello and Lynda Turner for technical assistance at the John Innes Centre.
Footnotes
This work was supported by the Australian Research Council (I.C.M.), the Biotechnology and Biological Sciences Research Council, UK (T.H.N.E. and M.R.K.), the Department for Environment, Food, and Rural Affairs, UK (grant no. AR0102 to J.M.I.H.), and the National Science Foundation (grant no. NSF 9977087 to S.R.S.). S.A.T. received funding from an Australian Postgraduate Award and European Union project EuDicot Map (no. B104 CT 97–2170).
Article, publication date, and citation information can be found at www.plantphysiol.org/cgi/doi/10.1104/pp.001677.
LITERATURE CITED
- Berbel A, Navarro C, Ferrándiz C, Cañas LA, Madueño F, Beltrán J. Analysis of PEAM4, the pea AP1 functional homologue, supports a model for AP1-like genes controlling both floral meristem and floral organ identity in different plant species. Plant J. 2001;25:441–451. doi: 10.1046/j.1365-313x.2001.00974.x. [DOI] [PubMed] [Google Scholar]
- Bowman JL, Alvarez J, Weigel D, Meyerowitz EM, Smyth DR. Control of flower development in Arabidopsis thaliana by APETALA1 and interacting genes. Development. 1993;119:721–743. [Google Scholar]
- Bradley JM, Whitelam GC, Harberd NP. Impaired splicing of phytochrome B pre-mRNA in a novel phyB mutant of Arabidopsis. Plant Mol Biol. 1995;27:1133–1142. doi: 10.1007/BF00020886. [DOI] [PubMed] [Google Scholar]
- Cremer F, Havelange A, Saedler H, Huijser P. Environmental control of flowering time in Antirrhinum majus. Physiol Plant. 1998;104:345–350. [Google Scholar]
- Egea-Cortines M, Saedler H, Sommer H. Ternary complex formation between the MADS-box proteins SQUAMOSA, DEFICIENS and GLOBOSA is involved in the control of floral architecture in Antirrhinum majus. EMBO J. 1999;18:5370–5379. doi: 10.1093/emboj/18.19.5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrándiz C, Navarro C, Gómez MD, Cañas LA, Beltrán JP. Flower development in Pisum sativum: from the war of the whorls to the battle of the common primordia. Dev Genet. 1999;25:280–290. doi: 10.1002/(SICI)1520-6408(1999)25:3<280::AID-DVG10>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Gocal GFW, King RW, Blundell CA, Schwartz OM, Andersen CH, Weigel DW. Evolution of floral meristem identity genes: analysis of Lolium temulentum genes related to APETALA1 and LEAFY of Arabidopsis. Plant Physiol. 2001;125:1788–1801. doi: 10.1104/pp.125.4.1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodall GJ, Filipowicz W. The AU-rich sequences present in the introns of plant nuclear pre-mRNAs are required for splicing. Cell. 1989;58:473–483. doi: 10.1016/0092-8674(89)90428-5. [DOI] [PubMed] [Google Scholar]
- Gourlay CW, Hofer JMI, Ellis THN. Pea compound leaf architecture is regulated by interactions among the genes UNIFOLIATA, COCHLEATA, AFILA and TENDRIL-LESS. Plant Cell. 2000;12:1279–1294. doi: 10.1105/tpc.12.8.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall KJ, Parker JS, Ellis THN, Turner L, Knox MR, Hofer JMI, Lu J, Ferrándiz C, Hunter PJ, Taylor JD et al. The relationship between genetic and cytogenetic maps of pea: II. Physical maps of linkage mapping populations. Genome. 1997;40:755–769. doi: 10.1139/g97-798. [DOI] [PubMed] [Google Scholar]
- Hempel FD, Weigel D, Mandel MA, Ditta G, Zambryski PC, Feldman LJ, Yanofsky MF. Floral determination and expression of floral regulatory genes in Arabidopsis. Development. 1997;124:3845–3853. doi: 10.1242/dev.124.19.3845. [DOI] [PubMed] [Google Scholar]
- Hill TA, Day CD, Zondlo SC, Thackeray AG, Irish VF. Discrete spatial and temporal cis-acting elements regulate transcription of the Arabidopsis floral homeotic gene APETALA3. Development. 1998;125:1711–1721. doi: 10.1242/dev.125.9.1711. [DOI] [PubMed] [Google Scholar]
- Hilleren P, Parker R. Mechanisms of mRNA surveillance in eukaryotes. Annu Rev Genet. 1999;33:229–260. doi: 10.1146/annurev.genet.33.1.229. [DOI] [PubMed] [Google Scholar]
- Hofer J, Turner L, Hellens R, Ambrose M, Matthews P, Michael A, Ellis N. UNIFOLIATA regulates leaf and flower morphogenesis in pea. Curr Biol. 1997;7:581–587. doi: 10.1016/s0960-9822(06)00257-0. [DOI] [PubMed] [Google Scholar]
- Hole CC, Hardwick RC. Development and control of the number of flowers per node in Pisum sativum L. Ann Bot. 1976;40:707–722. [Google Scholar]
- Honma T, Goto K. Complexes of MADS-box proteins are sufficient to convert leaves into floral organs. Nature. 2001;409:525–529. doi: 10.1038/35054083. [DOI] [PubMed] [Google Scholar]
- Huijser P, Klien J, Lönnig WE, Meijer H, Saedler H, Sommer H. Bracteomania, an inflorescence anomaly, is caused by the loss of function of the MADS-box gene squamosa in Antirrhinum majus. EMBO J. 1992;11:1239–1249. doi: 10.1002/j.1460-2075.1992.tb05168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irish VF, Sussex IM. Function of the apetala-1 gene during Arabidopsis floral development. Plant Cell. 1990;2:741–751. doi: 10.1105/tpc.2.8.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempin SA, Savidge B, Yanofsky MF. Molecular basis of the cauliflower phenotype in Arabidopsis. Science. 1995;267:522–525. doi: 10.1126/science.7824951. [DOI] [PubMed] [Google Scholar]
- Makasheva RK. The Pea. New Delhi, India: Oxonian Press; 1983. [Google Scholar]
- Mandel MJ, Gustafson-Brown C, Savidge B, Yanofsky MF. Molecular characterisation of the Arabidopsis floral homeotic gene APETALA1. Nature. 1992;360:273–277. doi: 10.1038/360273a0. [DOI] [PubMed] [Google Scholar]
- Orozco BM, McClung CR, Werneke JM, Ogren WL. Molecular basis of the ribulose-1,5-bisphosphate carboxylase/oxygenase activase mutation in Arabidopsis thaliana is a guanine-to-adenine transition at the 5′-splice junction of intron 3. Plant Physiol. 1993;102:227–232. doi: 10.1104/pp.102.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parcy F, Nilsson O, Busch MA, Lee I, Weigel D. A genetic framework for floral patterning. Nature. 1998;395:561–566. doi: 10.1038/26903. [DOI] [PubMed] [Google Scholar]
- Piñeiro M, Coupland G. The control of flowering time and floral identity in Arabidopsis. Plant Physiol. 1998;117:1–8. doi: 10.1104/pp.117.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessions A, Yanofsky MF, Weigel D. Cell-cell signaling and movement by the floral transcription factors LEAFY and APETALA1. Science. 2000;289:779–781. doi: 10.1126/science.289.5480.779. [DOI] [PubMed] [Google Scholar]
- Simpson GG, Gendall T, Dean C. When to switch to flowering. Annu Rev Cell Dev Biol. 1999;15:519–550. doi: 10.1146/annurev.cellbio.15.1.519. [DOI] [PubMed] [Google Scholar]
- Simpson CG, McQuade C, Lyon J, Brown JWS. Characterisation of exon skipping mutants of the COP1 gene from Arabidopsis. Plant J. 1998;15:125–131. doi: 10.1046/j.1365-313x.1998.00184.x. [DOI] [PubMed] [Google Scholar]
- Simon R, Igeño MI, Coupland G. Activation of floral meristem identity genes in Arabidopsis. Nature. 1996;384:59–62. doi: 10.1038/384059a0. [DOI] [PubMed] [Google Scholar]
- Singer SR, Maki SL, Mullen HJ. Specification of the floral meristem identity in Pisum sativum inflorescence development. Flowering Newslett. 1994;18:26–32. [Google Scholar]
- Taylor S, Hofer J, Murfet I. Stamina pistilloida, the pea ortholog of Fim and UFO, is required for normal development of flowers, inflorescences and leaves. Plant Cell. 2001;13:31–46. doi: 10.1105/tpc.13.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theissen G. Development of floral organ identity: stories from the MADS house. Curr Opin Plant Dev. 2001;4:75–85. doi: 10.1016/s1369-5266(00)00139-4. [DOI] [PubMed] [Google Scholar]
- Theissen G, Becker A, Di Rosa A, Kanno A, Kim JT, Münster KW, Saedler H. A short history of MADS-box genes in plants. Plant Mol Biol. 2000;42:115–149. [PubMed] [Google Scholar]
- Tucker SC. Overlapping organ initiation and common primordia in flowers of Pisum sativum (Leguminosae: Papilionoideae) Am J Bot. 1989;76:714–729. [Google Scholar]
- Wagner D, Sablowski RWM, Meyerowitz EM. Transcriptional activation of APETALA1 by LEAFY. Science. 1999;285:582–584. doi: 10.1126/science.285.5427.582. [DOI] [PubMed] [Google Scholar]
- Weller JL, Reid JB, Taylor SA, Murfet IC. The genetic control of flowering in pea. Trends Plant Sci. 1997;2:412–418. [Google Scholar]