

Abstract
Synaptic transmission between pairs of excitatory neurones in layers V (N = 38) or IV (N = 6) of somatosensory cortex was examined in a parasagittal slice preparation obtained from young Wistar rats (14–18 days old). A combined experimental and theoretical approach reveals two characteristics of short-term synaptic depression. Firstly, as well as a release-dependent depression, there is a release-independent component that is evident in smaller postsynaptic responses even following failure to release transmitter. Secondly, recovery from depression is activity dependent and is faster at higher input frequencies. Frequency-dependent recovery is a Ca2+-dependent process and does not reflect an underlying augmentation. Frequency-dependent recovery and release-independent depression are correlated, such that at those connections with a large amount of release-independent depression, recovery from depression is faster. In addition, both are more pronounced in experiments performed at physiological temperatures. Simulations demonstrate that these homeostatic properties allow the transfer of rate information at all frequencies, essentially linearizing synaptic responses at high input frequencies.
Information transfer in neural networks is determined by the dynamics of synapses in response to sequences of presynaptic action potentials. Recordings from interconnected neurones reveal that synaptic transmission is indeed dynamic, i.e. synapses in the neocortex typically undergo activity-dependent changes in response to bursts of action potentials (Magleby, 1987; Thomson et al. 1993; Markram et al. 1997; O'Donovan & Rinzel, 1997; Reyes et al. 1998; Tarczy-Hornoch et al. 1998). Specifically, connections between neocortical pyramidal neurones often display a gradual decrease in postsynaptic response, a phenomenon known as synaptic depression.
However, the amplitudes of the postsynaptic response are determined not only by the input pattern, but also by the probabilistic nature of neurotransmitter release, which results in trial-to-trial fluctuations (Kuno, 1964; Jack et al. 1981; Korn et al. 1984). A quantal hypothesis for probabilistic synaptic transmission was developed for the frog neuromuscular junction (Del Castillo & Katz, 1954) and has later been extended to other preparations (reviewed in Redman, 1990). According to this hypothesis, the connection between two neurones is composed of several release sites that always contain at least one vesicle available for release. At the arrival of an action potential, each site releases at most one vesicle with a fixed probability, independent of the release of vesicles from other sites, and from preceding release at the same site (spatial and temporal independence). The postsynaptic response is proportional to the number of vesicles released.
At depressing synapses, it is thought that depletion of the releasable pool of vesicles (‘synaptic resource’) accounts for the reduced postsynaptic response during a burst, i.e. synaptic depression is release dependent. Particularly, in cases of failure to release transmitter during an individual trial, there is no vesicle depletion and therefore no reduction in availability of synaptic resources for subsequent release. This consideration indicates that synapses with low probability of release would be unable to reliably code presynaptic activity because based on vesicle depletion release would occur too infrequently to cause depression. Such a scenario would restrict information transfer related to presynaptic firing rate.
However, there is evidence that synaptic depression can also be release independent (Dobrunz et al. 1997; Thomson & Bannister, 1999; Brody & Yue, 2000; reviewed in Zucker & Regehr, 2002). Release-independent depression (RID) is observed if a decrease in the probability of release occurs even if the second synaptic response is preceded by a transmission failure. In contrast to the scenario above, such a mechanism would allow depression to occur even at synapses with low release probability and therefore would constitute a homeostatic mechanism potentially extending the range of information transfer considerably.
We therefore investigated these questions in a study where experimental observations were combined with the modelling of synaptic dynamics, since at this stage of investigation it is not possible to measure information transfer directly.
To gain a detailed understanding of the factors involved in governing depression and recovery from depression, we recorded from connected excitatory cell pairs in rat somatosensory cortex. This approach allows the delivery of precise presynaptic stimulus patterns and the measurement of the associated postsynaptic responses (EPSCs) under excellent signal-to-noise conditions. We investigated the synaptic dynamics by measuring both the depression caused by a burst of action potentials and the recovery gauged by an action potential delivered with a variable delay after the burst. By carefully quantifying depression, we found that at these connections, there is release-independent as well as release-dependent depression. In addition, at certain connections, recovery from depression is frequency dependent, the rate of which accelerates during periods of increased activity. We show that release-independent depression and frequency-dependent recovery are tightly correlated such that synapses, which significantly depress independently of vesicle depletion, also recover faster from depression under conditions of high frequency firing.
To match these experimental observations, we developed a model, which replicates both the average dynamics of neocortical synaptic transmission, as well as the fluctuations in synaptic release. The two properties of synaptic depression as well as frequency-dependent recovery from depression were incorporated into the model. Release-independent depression was implemented as a decrease in the probability of transmitter release to subsequent action potentials, which is independent of whether release actually occurred. Recovery from depression could be brought about by an activity-dependent decrease of the time constant of recovery from depression. Frequency-dependent recovery from depression has previously been observed at a number of invertebrate and vertebrate synapses (reviewed in Zucker & Regehr, 2002).
We show that these mechanisms enable a depressing synapse to transmit information about presynaptic firing rates, even at high input frequencies, and results in a synapse that can code rate information at all frequencies.
Methods
Slice preparation and whole-cell recording
Wistar rats, 14–18 days old, were killed by decapitation and parasagittal cortical slices were prepared as described in Simkus & Stricker (2002). The Department for Veterinary Affairs of the Canton of Zürich, Switzerland, approved the techniques used. Whole-cell recordings were made under visual control using infra-red differential interference contrast (IR-DIC) optics. The artificial cerebrospinal fluid (ACSF) contained (mm): NaCl, 125; KCl, 2.5; NaHCO3, 25; NaH2PO4, 1.25; CaCl2, 2.0; MgCl2, 1.0; glucose, 25.0; gassed with 95% O2 and 5% CO2, pH 7.4. In most cases, 10 µm bicuculline methiodide was added to block GABAA-mediated inhibitory transmission. All experiments, except where otherwise stated, were conducted at 36 ± 1°C. The intracellular solution contained (mm): potassium gluconate, 115; KCl, 20; HEPES, 10; phosphocreatine, 10; Mg-ATP, 4; Na-GTP, 0.3; pH 7.2; osmolarity, 305 mosmol l−1. In a number of experiments, the Ca2+ dependence of the mechanisms governing synaptic depression was examined. In these cases the presynaptic neurone was re-patched after the control recording with a pipette, which then contained 0.5–5 mm ethylene glycol-bis (2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA). To examine whether Na+ current boosting of the postsynaptic response occurred at these connections, the intracellular solution contained 5 mm QX-314 (Alamone Laboratories, Jerusalem, Israel) and the appropriate reduction in potassium gluconate made to maintain iso-osmolarity.
Recordings were obtained between connected pairs of layer V or IV cells. Access resistance was 10.1 ± 2.4 mΩ and was uncompensated. The presynaptic neurone was recorded in current-clamp mode (Axoclamp 2B, Axon Instruments), and action potentials were initiated (Fig. 1B) by injection of rectangular current pulses (5 ms, 0.5–1.5 nA, Fig. 1A). The postsynaptic neurone was held in voltage clamp at −70 mV and excitatory postsynaptic currents (EPSCs) recorded using an Axopatch 200B amplifier (Axon Instruments; Fig. 1C). The choice of postsynaptic voltage clamp was made for the following reason. Firing the cells at stimulus frequencies > 25 Hz would have contaminated the subsequent EPSP amplitudes by the decay phase of the previous responses. In voltage clamp, however, the current decays are markedly faster and allow the measurement of the peak amplitude even at these frequencies. Note that we do not claim to perfectly clamp postsynaptic currents, which is impossible in these distributed structures. The output was amplified (100–500 times) following a sample-and-hold step to ensure that the full resolution of the AD converter could be utilized (custom-built sample-and-hold amplifier integrated with a 8-pole Bessel filter, design JCSMR, ANU, Australia). The signal was filtered at 2 kHz and digitized at 5 kHz using an ITC-18 computer interface (Instrutech Corp, Port Washington, NY, USA). Data were acquired using software developed with IGOR Pro 4.0 (Wavemetrics, OR, USA). Each trial in an experiment involved generating a burst of action potentials (mostly 20) in the presynaptic neurone at a particular frequency (range 3–50 Hz). The frequency during each burst was randomly chosen from a set of a few predetermined frequencies (i.e. 3, 5, 10, 20, 25, 30, 40, 50) to avoid steady-state behaviour. Each burst of action potentials was then followed by a recovery interval (300–1200 ms; also randomized) before a single action potential was evoked to gauge recovery from depression. The trials were repeated every 10–15 s, and 50–900 trials were recorded for each of the frequencies studied (Fig. 1A and B).
Figure 1. Experimental protocol.

A, sample trials of 20 rectangular current pulse injections (5 ms, 1 nA at 10, 50 and 20 Hz, respectively) applied to the presynaptic neurone with a trial-to-trial interval of 10–15 s. The sequence of trials was randomized for frequency, and the repetitions for each input pattern were between 50 and 900. Recovery was gauged with a delayed current pulse (500 ms in this example). B, single burst of action potentials recorded in the presynaptic neurone in response to a current pulse sequence (A). C, the postsynaptic neurone was held in voltage clamp at −70 mV and excitatory postsynaptic currents (EPSCs) were recorded. For clarity, only sample EPSC trials in response to a burst of action potentials at 10 Hz are shown. In D–E, the analytical procedures are indicated. In D, the analysis starts with sorting trials according to stimulus frequency and delay of the action potential to gauge recovery. E, measuring E1 and E2 in each trial allows determination of the release dependence of depression (RD), construction of the PDFs and comparison of 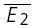 for the 20 trials with the smallest E1 (
for the 20 trials with the smallest E1 (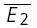 small E1) and those with the largest (
small E1) and those with the largest (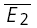 bigE1). F, the average of all trials for a particular input pattern are used to determine the total amount of depression, and the characteristics of recovery.
bigE1). F, the average of all trials for a particular input pattern are used to determine the total amount of depression, and the characteristics of recovery.
Data analysis
Averages are presented as mean ± 1 s.d., error bars in figures represent ± 1 s.e.m. For statistical comparison, a level of significance of < 0.05 was accepted and the following abbreviations are used: pr for Pearson's r, pKS for Kolmogorov-Smirnov, pχ for χ2 statistics, pA for significance calculated based on analysis of covariance (ANCOVA), pt for Student's t test and ppt for Student's paired t test.
Extracting the amplitude of postsynaptic responses
In some cases, the amplitudes were measured manually according to the technique described in Stricker et al. (1996) and an estimate of the baseline noise was obtained (averaged over all experiments σn = 0.9 ± 0.2 pA). In other cases, the response amplitudes in single trials were determined automatically by a custom Matlab routine (MathWorks Inc., Natick, MA, USA). In this case the experimental recordings were low-pass filtered (Gaussian filter with σ= 0.2 ms) to remove high frequency fluctuations, and the baseline and peak of each synaptic response determined. The detection method was verified by comparing the amplitude of the average EPSC of all trials, with the average of the automatically determined amplitudes.
Since the experiments required a prolonged recording period (> 3 h), the stability of synaptic dynamics was assessed by comparing the mean response for the first and second half of all trials. Only experiments with stable dynamics were used (i.e. where pt of the normalized averages > 0.05). In some cases, a drift (always run-down) in the average EPSC amplitude was sometimes observed (due to changes in access resistance). In these cases, stationary subsets of data were used for most analyses. However, for modelling, consistent drift in the data (< 50%) was corrected by normalizing the responses according to the slope of the linear regression of the amplitudes over time. To compare recordings under different conditions (e.g. different frequencies of stimulation), the magnitudes were normalized according to the average amplitude of the first EPSC.
Detecting and quantifying release-independent depression
RID is best demonstrated at connections with a low probability of release and a significant number of failures in response to the first action potential. Under conditions of release-dependent depression, the average, and amplitude distribution, of second EPSCs following a failure should equal those of all first EPSCs. To examine such differences in the amplitude distributions, amplitude probability density functions were calculated by kernelling each amplitude with a Gaussian as described in Stricker et al. (1994). Cumulative probability densities were determined, and significant differences tested using Kolmogorov-Smirnov statistics. However, when the initial release probability is high, there are at most few or no trials, where failures in response to the first action potential occur, and adequate statistics cannot be obtained. RID can still be demonstrated in the absence of transmission failures by examining the trials based on the amplitude of the first EPSC (smaller or larger than the mean response). If depression is release dependent, the average amplitude of the second EPSC in those 20 trials with the smallest first EPSC (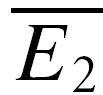 smallE1) should be larger than that of the 20 trials with the largest first EPSC (
smallE1) should be larger than that of the 20 trials with the largest first EPSC (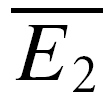 bigE1; Fig. 1F). This will not hold true for RID, and significant differences were tested using a Student t test. It should be noted that due to the small sample size, the two averages
bigE1; Fig. 1F). This will not hold true for RID, and significant differences were tested using a Student t test. It should be noted that due to the small sample size, the two averages 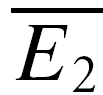 smallE1 and
smallE1 and 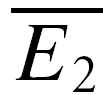 bigE1 typically constitute a biased sample and therefore deviations from the predictions of RD (see below) are likely to be observed.
bigE1 typically constitute a biased sample and therefore deviations from the predictions of RD (see below) are likely to be observed.
A further method based on the amplitudes of all first and second EPSCs was devised to quantify the relative contribution of release-dependent and -independent depression. This measure is subsequently called RD and is proportional to the correlation coefficient between the first and second EPSCs, i.e.
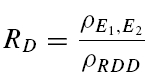 |
where E1 and E2 are random variables representing the first and second EPSCs. ρE1,E2 is the correlation coefficient, and ρRDD is the correlation coefficient for pure release-dependent depression (RDD; see Appendix for details). The idea is that if transmitter release is purely release dependent, then E2 is correlated with E1 (Matveev & Wang, 2000), In contrast, at a connection with pure RID, no correlation is expected between E1 and E2, and therefore RD equals 0. The measure quantifies the degree of RDD and (for a sufficiently large sample size) holds values between 0 ≤RD≤ 1, where 0 indicates pure RID and 1 pure RDD.
An illustration of RD is obtained in a plot of E2versusE1 (Waldeck et al. 2000; see Fig. 2B and D). For pure RDD, the slope of the regression line is ρRDD and the y-intercept 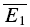 (the average of second responses that follow a failure equals the simple average of all first responses). For pure RID, the slope is 0 by definition and the y-intercept is
(the average of second responses that follow a failure equals the simple average of all first responses). For pure RID, the slope is 0 by definition and the y-intercept is 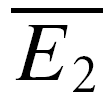 (the average of all the second responses). RD is the relative y-intercept normalized by
(the average of all the second responses). RD is the relative y-intercept normalized by 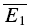 (see eqn (20) in the Appendix).
(see eqn (20) in the Appendix).
Figure 2. Release-dependent and -independent components of depression.

A, a layer V connection expressing prominent release-dependent depression (RDD). EPSCs were recorded in 148 successive trials, in which the presynaptic neurone was stimulated at a frequency of 20 Hz. Left: average (20 trials) depicting the first two EPSCs, in which the first EPSCs were the smallest (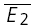 smallE1). Right: average EPSCs, in which the first EPSCs were the largest (
smallE1). Right: average EPSCs, in which the first EPSCs were the largest (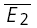 bigE1). Second EPSCs that follow small first EPSCs (left) are significantly larger than second EPSCs that follow large first EPSCs (right). B, plot of second versus first EPSCs for the same connection as in A. Filled circles: experimental data; continuous line: linear regression (r = −0.40, pr < 0.05); dashed lines: theoretical line for pure release-independent depression (RID) (horizontal line) and RDD (oblique line). The y-intercept of the RDD line is the average of first EPSCs (
bigE1). Second EPSCs that follow small first EPSCs (left) are significantly larger than second EPSCs that follow large first EPSCs (right). B, plot of second versus first EPSCs for the same connection as in A. Filled circles: experimental data; continuous line: linear regression (r = −0.40, pr < 0.05); dashed lines: theoretical line for pure release-independent depression (RID) (horizontal line) and RDD (oblique line). The y-intercept of the RDD line is the average of first EPSCs (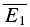 = −27.3 pA) and that of the RID line, the average of the second EPSCs (
= −27.3 pA) and that of the RID line, the average of the second EPSCs (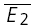 = − 19.6 pA) The regression line overlaps the pure RDD line, and the y-intercept of the regression is clearly closer to
= − 19.6 pA) The regression line overlaps the pure RDD line, and the y-intercept of the regression is clearly closer to 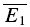 than
than 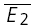 . RD = 1.03 ± 0.33, suggesting a strong component of RDD. The open squares indicate
. RD = 1.03 ± 0.33, suggesting a strong component of RDD. The open squares indicate 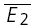 smallE1 and
smallE1 and 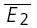 bigE1 as shown in A. C, a layer IV connection with dominant RID. The response was recorded in 191 successive trials, in which the presynaptic neurone was stimulated at a frequency of 3 Hz. Left,
bigE1 as shown in A. C, a layer IV connection with dominant RID. The response was recorded in 191 successive trials, in which the presynaptic neurone was stimulated at a frequency of 3 Hz. Left, 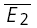 smallE1 and right,
smallE1 and right, 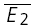 bigE1. The amplitudes of the second EPSCs are similar in both cases, largely independent of the first response. D, plot of second versus first EPSCs for the same connection as in C. Filled circles: experimental data; continuous line: linear regression (r = −0.12, pr = 0.09); dashed lines: theoretical line for pure RID (horizontal line) and RDD (oblique line). The y-intercept of the RDD line is the average of first EPSCs (
bigE1. The amplitudes of the second EPSCs are similar in both cases, largely independent of the first response. D, plot of second versus first EPSCs for the same connection as in C. Filled circles: experimental data; continuous line: linear regression (r = −0.12, pr = 0.09); dashed lines: theoretical line for pure RID (horizontal line) and RDD (oblique line). The y-intercept of the RDD line is the average of first EPSCs (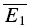 = −14.7 pA) and that of the RID line, the average of the second EPSCs (
= −14.7 pA) and that of the RID line, the average of the second EPSCs (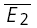 = −8.0 pA). The y-intercept of the regression line clearly closer to
= −8.0 pA). The y-intercept of the regression line clearly closer to 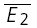 than to
than to 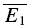 (RD = 0.21 ± 0.14), suggesting a strong component of RID. The open squares indicate
(RD = 0.21 ± 0.14), suggesting a strong component of RID. The open squares indicate 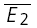 smallE1 and
smallE1 and 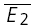 bigE1 as shown in C. E, additional evidence for release-independent depression in a layer IV connection. Probability density functions of the amplitudes of the first EPSC (continuous line, N = 554) and for the second EPSC when the first EPSC was a failure (dashed line, N = 75). The frequency of stimulation was 20 Hz. F, cumulative probability densities for the connection in E. The densities for E1 (continuous line) and E2 following a failure (dashed line) are significantly different (pKS < 0.005). The inset shows the average time course for the first response (continuous black line), the second response, given the first was a transmission failure (dashed line), and all second responses (grey line).
bigE1 as shown in C. E, additional evidence for release-independent depression in a layer IV connection. Probability density functions of the amplitudes of the first EPSC (continuous line, N = 554) and for the second EPSC when the first EPSC was a failure (dashed line, N = 75). The frequency of stimulation was 20 Hz. F, cumulative probability densities for the connection in E. The densities for E1 (continuous line) and E2 following a failure (dashed line) are significantly different (pKS < 0.005). The inset shows the average time course for the first response (continuous black line), the second response, given the first was a transmission failure (dashed line), and all second responses (grey line).
Determining if a junction shows pure RID, RDD or intermediary forms is based on the following procedures. Using ANCOVA, we stringently test if RD is significantly different from both hypotheses, namely pure RID (RD = 0; no correlation) and RDD (RD = 1; maximal correlation). Only if both hypotheses can be rejected do we conclude that RD has taken an intermediate value and therefore consists of both RID and RDD. In addition, we also test whether the slope of the linear regression as well as the offset are different from that of RDD and/or RID.
Detecting and quantifying frequency-dependent recovery from depression
Frequency-dependent recovery (FDR) can be directly observed in the averaged data. It is present when the amplitude of the EPSC after a high frequency burst is greater than that of the EPSC evoked with the same delay following a burst at lower frequency. For the quantification of FDR, we calculated the normalized recovery amplitude (Rrec) for each stimulus frequency as
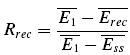 |
where 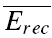 is the mean amplitude of the EPSC induced with a delay (400–600 ms) after the burst (Fig. 1C).
is the mean amplitude of the EPSC induced with a delay (400–600 ms) after the burst (Fig. 1C). 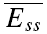 is the mean EPSC amplitude in steady state (taken as the average of the last 4 EPSCs in the burst of 20 action potentials). The values will fall between 0 and 1, with a smaller number indicating faster recovery. To quantify the degree of FDR, we calculated the ratio between Rrec at 10 and 20 Hz (RFDR). RFDR > 1 indicates FDR, whereas RFDR = 1 indicates no FDR. It would also be possible to define recovery as 1 −Rrec so that a bigger value indicates faster recovery. RFDR would then be the ratio between Rrec at 20 and 10 Hz. However, in this case, connections with very little recovery (at an interval of 400–600 ms there was on occasion very little recovery) would have a very small denominator (close to zero) and therefore, RFDR would be disproportionately large, i.e. in certain instances tend towards infinity. Equally, the measure described above is sensitive to the amount of recovery, and it would not be appropriate to use this measure for long recovery intervals where (
is the mean EPSC amplitude in steady state (taken as the average of the last 4 EPSCs in the burst of 20 action potentials). The values will fall between 0 and 1, with a smaller number indicating faster recovery. To quantify the degree of FDR, we calculated the ratio between Rrec at 10 and 20 Hz (RFDR). RFDR > 1 indicates FDR, whereas RFDR = 1 indicates no FDR. It would also be possible to define recovery as 1 −Rrec so that a bigger value indicates faster recovery. RFDR would then be the ratio between Rrec at 20 and 10 Hz. However, in this case, connections with very little recovery (at an interval of 400–600 ms there was on occasion very little recovery) would have a very small denominator (close to zero) and therefore, RFDR would be disproportionately large, i.e. in certain instances tend towards infinity. Equally, the measure described above is sensitive to the amount of recovery, and it would not be appropriate to use this measure for long recovery intervals where (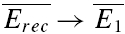 ).
).
Estimating the initial probability of transmitter release
We attempted to determine the initial release probability for each of the connections used in this study. This is usually done using quantal analysis. However, layer V pairs are not amenable to quantal analysis; the amplitude distributions of first responses are unimodal and overdispersed with more variance than would be expected from a binomial release process (A. Cowan & C. Stricker, unpublished observations). To overcome this problem, we attempted to use the extended moment analysis as described in Courtney (1978), where the probability (pr, also U0 in the model) is determined by
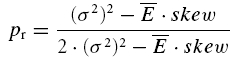 |
where σ is the standard deviation of the sample and skew is calculated as follows
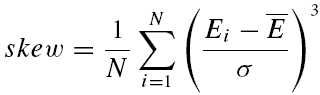 |
where i is the trial number and N is the total number of trials. The quantal content (m) is
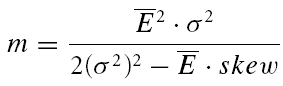 |
In cases of overdispersion (too much variance when compared to a classical release process) (σ2)2 becomes the dominant factor in both equations and renders this measure very insensitive. For these reasons, we are unable to provide a measure for release probability per se. However, we resorted to using the skew of the sample as an indication of pr. For a purely binomial process of transmitter release, a negative skew indicates that pr > 0.5. Such a distribution has an asymmetric tail extending towards EPSCs of smaller magnitude (remember, however, that since inward currents are measured, the tail extends towards less negative values). A skew of zero corresponds to pr= 0.5, whereas a positive skew indicates pr < 0.5 (such a distribution is characterized by an asymmetric tail extending towards large EPSCs). Correlations between the skew and various parameters of synaptic depression suggest a correlation of these parameters with initial release probability.
To examine whether synaptic depression in these connections was a result of a change in pr, or could be accounted for by other factors (a change in the number of release sites (n) or the quantal size (q), we constructed plots of the inverse of the normalized coefficient of variation squared (CV−2) versus normalized mean amplitude (as reviewed in Korn & Faber, 1991).
Augmentation
To test if augmentation was present at the connections studied, we used a protocol similar to that described in Stevens & Wesseling (1999). Following a sequence of 100 action potentials at 10 Hz, action potentials were evoked in the presynaptic neurone at a rate of 0.5 Hz for the next 60 s. The amplitudes of the resultant EPSCs were measured and normalized to the final amplitude (i.e. the average value for the last 30 s, as in Stevens & Wesseling, 1999). The time course for the decay of augmentation was then determined by fitting a single exponential to the normalized EPSCs for the first 15 EPSCs following the high frequency burst.
Model
The model implementation is based on the classical quantal hypothesis, to which we add the dynamics of synaptic transmission. These are formalized in previously published deterministic (Tsodyks & Markram, 1997) and probabilistic (Fuhrmann et al. 2002) models, with extensions made to accommodate the experimental findings presented below.
The modelled connection is composed of n release sites each holding at most one vesicle. In response to an action potential, each available vesicle can be released with a constant probability, USE. After release, the site can be refilled within each time step dt with the probability dt/τrec, where τrec is the recovery time constant of the refilling process. In contrast to the classical quantal hypothesis, in this model the release sites may be depleted of vesicles, thus no vesicles would be available for transmission.
The processes of transmitter release and recovery from depression can be described by a single differential equation, determining the probability Pv, of vesicle availability at time t,
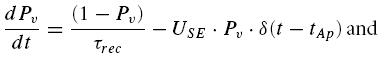 |
and
where Pr(tAp) is the release probability at each release site at the time of an action potential, tAp. It is the product of PV(tAp) and USE (see Zucker, 1973; Quastel, 1997). The postsynaptic response is proportional to the number of vesicles released.
Based on the experimental findings, we formalize the mechanism of depression as composed of two components. The first component is release dependent (RDD), which results from the depletion of synaptic resources, as formulated in eqn (5) above. The second component is RID, which is mediated by a decrease in the probability of release to subsequent action potentials, and which is independent of whether release actually occurred. It is formalized by USE being a dynamic variable, decreasing with subsequent action potentials in the sequence
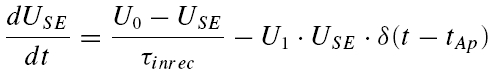 |
where U0 is the initial probability of release, U1 a constant (0 < U1 < 1) determining the decrease in USE per action potential, and τinrec is the recovery time constant of RID. The reason for this choice of implementation is discussed later (Results and Discussion). Thus, between bursts of action potentials (15 s), USE increases towards U0, and when an action potential occurs (tAp), it decreases by U1·USE(tAp).
To account for FDR, we suggest that τinrec itself is activity dependent. Its dynamics evolve according to
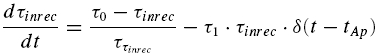 |
where τ0 is the initial value of the recovery time constant for RID, τ1 is a constant determining the step-like decrease in τinrec per action potential (0 < τ1 < 1), and ττinrec is the relaxation time constant of τinrec. Similar to the idea above, between bursts of action potentials, τinrec increases towards τ0.
The mean synaptic response 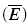 can be calculated as
can be calculated as
where ASE =nq is the maximal response produced when all the sites release their vesicles. Note that even though a uni-vesicular hypothesis is implied here, the formalism does not preclude multivesicular release, because ASE is simply a scaling factor and as such has no biological meaning. The variable R(t) is the fraction of vesicles available at time t and is calculated according to the following equation, which is analogous to eqn (5):
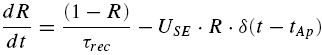 |
In order to study the functional implications of RID and FDR (see Fig. 13), we explore the total postsynaptic current (Isyn) produced by the activity of a population of K = 1500 presynaptic neurones, connected to the target neurone by uniform synapses, since the discharge frequency of a neurone is proportional to the total postsynaptic current (Granit et al. 1966); i.e.
Figure 13. Functional implications of FDR.

Simulated net postsynaptic current (Isyn, calculated according to eqn (10)) generated by a population of 1500 presynaptic neurones, firing randomly (Poisson) and undergoing synchronous transitions (indicated by arrows) from a low firing rate to a higher firing rate. Left: transition from 5 to 50 Hz. Right: transition from 30 to 50 Hz. A, model with pure RDD and no FDR. At low firing rates (left), the connections can code both a transition (by a transient increase in response), and average firing rate (by the mean current). At high firing rates (right), the connections cannot code the firing rates, as the input current does not change. However, it can still code (with the transient response) a change in rate. Model parameters: U0 = 0.4, U1 = 0, τrec = 500 ms, τ0 = 1000 ms, τ1 = 0.2, ττinrec = 2000 ms and ASE = −200 pA. B, model with pure RID and FDR. At low firing rates (left) the synapses can code both a transition (by a complex transient), and the firing rate. At high firing rates (right), the synapses retain their ability to code firing rates. Model parameters: U0 = 0.4, U1 = 0.2, τrec = 5 ms, τ0 = 1000 ms, τ1 = 0.2, ττinrec = 2000 ms and ASE =−200 pA.
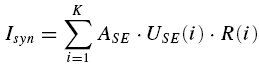 |
Fitting the model to the data
There are seven parameters (ASE, τrec, U0, U1, τ0, τ1, and ττinrec) in these equations, which determine the dynamics of transmission. While ASE is a scaling parameter, and the ratio between U0 and U1 is determined by 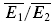 , there are five free parameters remaining. The fitting of the model to the data is done in two steps. First, we choose the few parameter sets (approx. 50) which can best describe the dynamics of the EPSCs, and which are in the biologically plausible range. This is done by minimizing the sum of squared errors between the mean experimental and predicted responses to bursts of action potentials for each stimulus condition (eqn (9)). Second, the fit is chosen which also best describes the relation between experimental
, there are five free parameters remaining. The fitting of the model to the data is done in two steps. First, we choose the few parameter sets (approx. 50) which can best describe the dynamics of the EPSCs, and which are in the biologically plausible range. This is done by minimizing the sum of squared errors between the mean experimental and predicted responses to bursts of action potentials for each stimulus condition (eqn (9)). Second, the fit is chosen which also best describes the relation between experimental 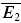 bigE1 and
bigE1 and 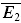 smallE1.
smallE1.
Results
The properties of synaptic depression were examined in 38 pairs of pyramidal neurones in layer V of the somatosensory cortex. In layer IV, four pairs between star pyramidal cells, one spiny stellate–star pyramid connection, and a star pyramid–spiny stellate connection were also examined. We included layer IV cells since these showed largely the same characteristics as in layer V and because in some of them, we were able to identify a significant number of transmission failures. All connections reported here failed to show any sign of facilitation at any frequency between 5 and 50 Hz, i.e. the average amplitudes of the first few EPSCs in the sequence were smaller than the preceding ones. The recordings were a subset of the connections recorded (∼80%; 20% of connections showed a facilitatory response). For neurones in layer V, the average resting membrane potential was −62.5 ± 4.0 mV, the action potential height 95.2 ± 8.7 mV, half-width 1.2 ± 0.3 ms and input resistance was 92.2 ± 30.1 mΩ. The EPSCs averaged −38.2 ± 30.1 pA, had a rise time (10–90%) of 1.2 ± 0.3 ms and a half-width of 6.4 ± 1.7 ms. The CV was 0.4 ± 0.2 and the skew of the amplitude distribution −0.3 ± 0.6. Paired-pulse depression (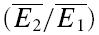 ) was 0.45 ± 0.15 (range: 0.35–0.77).
) was 0.45 ± 0.15 (range: 0.35–0.77).
Release dependence of synaptic depression
Release-dependent depression (RDD) was identified at some of the connections. At these connections, as depicted for example in Fig. 2A, 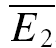 smallE1 (the average amplitude of E2 for the 20 trials where E1 was the smallest, see ‘Detecting and quantifying release-independent depression’ above) was significantly larger than
smallE1 (the average amplitude of E2 for the 20 trials where E1 was the smallest, see ‘Detecting and quantifying release-independent depression’ above) was significantly larger than 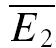 bigE1 (average E2 for 20 traces where E1 was the largest, Fig. 2A), indicating that there was some degree of release-dependent depression. In Fig. 2B, the second EPSC amplitude (E2) is plotted as a function of the first (E1). For guidance, the theoretical lines of pure RDD (oblique dashed line; for details see Appendix) and pure RID (horizontal dashed line) are depicted. It can be seen that the regression line for the data (continuous line) and the one for RDD (oblique dashed) are almost identical. The normalized factor indicating release dependence (RD, eqn (20)) was 1.03 ± 0.33, suggesting that depression at this connection was dominated by RDD. ANCOVA verified that the regression line is not significantly different from pure RDD (pA= 0.43 for RDD; pA < 0.05 for RID).
bigE1 (average E2 for 20 traces where E1 was the largest, Fig. 2A), indicating that there was some degree of release-dependent depression. In Fig. 2B, the second EPSC amplitude (E2) is plotted as a function of the first (E1). For guidance, the theoretical lines of pure RDD (oblique dashed line; for details see Appendix) and pure RID (horizontal dashed line) are depicted. It can be seen that the regression line for the data (continuous line) and the one for RDD (oblique dashed) are almost identical. The normalized factor indicating release dependence (RD, eqn (20)) was 1.03 ± 0.33, suggesting that depression at this connection was dominated by RDD. ANCOVA verified that the regression line is not significantly different from pure RDD (pA= 0.43 for RDD; pA < 0.05 for RID).
In contrast, at some connections, synaptic depression was found to be highly release independent (Fig. 2C and D). This was deduced from the facts that, at these synapses, 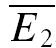 bigE1 and
bigE1 and 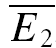 smallE1 were not significantly different from each other (Fig. 2C) and that RD was close to zero. In Fig. 2D, the linear regression analysis confirms that depression in this example is largely release independent. It is evident that the y-intercept of the regression line is indeed closer to
smallE1 were not significantly different from each other (Fig. 2C) and that RD was close to zero. In Fig. 2D, the linear regression analysis confirms that depression in this example is largely release independent. It is evident that the y-intercept of the regression line is indeed closer to 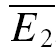 than to
than to 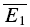 . RD was small (0.21 ± 0.14), and ANCOVA confirms that depression at this connection is significantly different from pure RDD (pA < 0.0005), but not from pure RID (pA= 0.45).
. RD was small (0.21 ± 0.14), and ANCOVA confirms that depression at this connection is significantly different from pure RDD (pA < 0.0005), but not from pure RID (pA= 0.45).
Additional evidence for the existence of RID is depicted in a layer IV connection in Fig. 2E and F, where the amplitude distribution of the first EPSCs, and that of the second EPSCs, given that the preceding EPSC was a failure, are shown (Fig. 2E). In Fig. 2F, the cumulative probabilities of the two distributions are significantly different (pKS < 1e−9). The average synaptic response that follows a failure is also smaller than 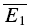 , but slightly bigger than
, but slightly bigger than 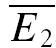 (inset).
(inset).
Our results indicate that most of the recorded connections exhibit at least some degree of RID. This can be inferred from the results of ANCOVA, where six of the layer V connections and five of the layer IV connections showed pure RID. Sixteen layer V connections were significantly different from both RID and RDD, indicating that they express both forms of depression, whereas three of the layer V connections were not significantly different from either pure RID or RDD. One layer IV connection (star pyramid–star pyramid), and seven layer V connections were highly release dependent, i.e. not significantly different from RDD but significantly different from RID. The remaining six layer V pairs were used in other experimental protocols (augmentation, recovery times, see below). The mean value of RD was 0.45 ± 0.35.
We investigated whether the existence of some degree of release-independent depression in most connections could be an artefact of our recording conditions. We do not believe this is the case for a number of reasons. Firstly, the degree of release dependence remained stable throughout long recording periods. For example, in Fig. 3, a plot of E2versusE1 for the first and last hour of a 4 h recording period is shown, during which the degree of release dependence remained unchanged (RD = 0.45 ± 0.15 in Fig. 3A, 0.53 ± 0.22 in Fig. 3B, pt= 0.7).
Figure 3. The properties of synaptic depression are stable.

A, plot of second versus first EPSCs for a layer V pair for the first hour of a prolonged recording period. RD = 0.45 ± 0.15 (N = 228, r = −0.42, pr < 0.05). B, plot of second versus first EPSCs for the same connection after 3 h of recording. The RD of 0.53 ± 0.22 (N = 238, r =−0.38, pr < 0.05) is not different (pt= 0.7) from that in A and in both cases the linear regression line is significantly different from a horizontal line. The open squares indicate 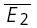 smallE1 and
smallE1 and 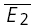 bigE1.
bigE1.
A potential artefact that could result in release-independent depression would be a change in the properties of synaptic depression as a result of wash-out during whole cell recording. In eight experiments, we evoked action potentials in the presynaptic neurone from cell-attached mode. RD for these experiments (0.35 ± 0.27) was not significantly different from the average of all those recorded in whole-cell mode (0.45 ± 0.35, pt= 0.4).
Since in most of our experiments we included bicuculline in the ACSF to block GABAA-mediated transmission, and bicuculline has been shown to block Ca2+-dependent K+ channels (see for example Debarbieux et al. 1998), we investigated whether RID was still observed in the absence of bicuculline. In 15 experiments, RD corresponded to 0.54 ± 0.35, which was not significantly different from when the ACSF contained bicuculline (pt= 0.3).
A process that resulted in a positive correlation between E1 and E2 could potentially counterbalance the negative correlation of release-dependent depression, and result in an apparent lack of correlation. Such a process could include active postsynaptic boosting (Stuart & Sakmann, 1995) in imperfectly voltage-clamped neurones. Such a process would preferentially affect the largest E1. However, subsets of data with only large or small E1 resulted in the same RD value as the complete data sets (ppt > 0.5, data not shown). As a further test for this possibility we examined connections under control recording conditions and then after re-patching the postsynaptic neurone with an intracellular solution containing 5 mm QX-314. In four experiments, the presence of postsynaptic QX-314 did not change RD(control: 0.48 ± 0.22; QX-314: 0.42 ± 0.10, ppt= 0.6) or E0/E1(control: 0.71 ± 0.15; QX-314: 0.65 ± 0.11, ppt= 0.5, data not shown).
We also examined whether RID was an artefact of the analysis technique, for example, whether quantal variance or the noise variance could mask a correlation between E1 and E2. However, we found that there was no correlation between the connections with a large CV and the expression of RID. In fact, there was the opposite correlation between large CV and the expression of RDD, as might be expected for connections with a higher initial release probability (see below, pr= 0.002, data not shown). There was no correlation between the noise variance and RD (pr= 0.6).
The site of expression
Depression could be a result of a change in release probability (Pr,eqn (5)), a change in the number of release sites (n), a change in quantal size (q), or a combination of these factors. To determine the mechanism of depression, a CV−2 analysis was performed since depression resulting from changes in either n or q will be indicated by data points significantly above the 45 deg line (dashed lines in Fig. 4A and B), whereas changes in pr alone are observed as data points on or below this line (for details see Korn & Faber, 1991). The plots of this analysis are illustrated in Fig. 4 for two types of connections, namely those which showed a high degree of RDD (RD > 0.65; Fig. 4A) and those, where the depression was predominantly RID (RD < 0.35; Fig. 4B). In both cases, the points followed the 45 deg line, thus suggesting that the most likely cause for depression in both cases is a change in the probability of release.
Figure 4. Synaptic depression results from a change in release probability.

A, plot of normalized CV−2 versus normalized mean amplitude for those neurones with RD > 0.65 (i.e. dominated by release-dependent depression). The points represent the average data for the first 3 and the last (20th) EPSCs. Open circles: 10 Hz burst; filled squares: 20 Hz burst; filled circles: 50 Hz burst. For guidance 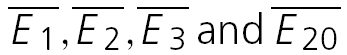 , are labelled for the 20 Hz burst. B, plot of normalized CV−2versus normalized mean amplitude for those neurones with RD < 0.35 (i.e. dominated by release-independent depression). Note that the dispersion of the data points is less than in A due to the fact that the EPSC during the sequence converges to a steady-state of similar amplitude for all frequencies of stimulation.
, are labelled for the 20 Hz burst. B, plot of normalized CV−2versus normalized mean amplitude for those neurones with RD < 0.35 (i.e. dominated by release-independent depression). Note that the dispersion of the data points is less than in A due to the fact that the EPSC during the sequence converges to a steady-state of similar amplitude for all frequencies of stimulation.
Based on these results we suggest that synaptic depression in the recorded connections is controlled by two mechanisms: one, which is release dependent (RDD) and the other, which is release independent (RID). In different synapses, the relative contribution of each of these components may vary, such that a synapse can express either pure RDD, or pure RID, or a mixture of the two forms. Experimentally, the whole range was observed (0 ≤RD≤ 1). To replicate the observed features, the release-dependent component of depression is formulated in eqn (5), by the depletion of vesicles available for synaptic transmission. The release-independent component is formulated in eqn (6) by the activity-dependent decrease in the probability of release (USE). It is independent of whether vesicles were actually released in response to preceding action potentials. In terms of parameters, a synapse with very small τrec (relative to the scale of τinrec) will be dominated by RID, due to fast replenishment of synaptic resources. RDD could result from an opposite relation between these parameters, or from very small step changes in the probability of release (U1 approaching zero).
Properties of RID and RDD
Dependence on initial release probability
It appears that the type of synaptic depression (RID or RDD) is dependent on the initial release probability of the synapses. Firstly, there is a negative correlation between the values of RD and the skew of the amplitude distribution (Fig. 5A, r =−0.54, pr < 0.005 Courtney, 1978). Since a negative skew indicates a high initial release probability (U0 > 0.5), this correlation is suggestive that RDD is more prominent for connections with a high initial probability. At the same time, we observed that the degree of paired-pulse depression (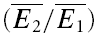 ) is inversely correlated with skew (Fig. 5B, r = 0.55, pr < 0.005). Since a high initial release probability is expected to result in a large depression of the second pulse, this finding is consistent with the above observation and, thereby, serves as an internal check for consistency of using skew as an indirect measure of initial release probability. Secondly, for the same data set, RD is correlated with the ratio
) is inversely correlated with skew (Fig. 5B, r = 0.55, pr < 0.005). Since a high initial release probability is expected to result in a large depression of the second pulse, this finding is consistent with the above observation and, thereby, serves as an internal check for consistency of using skew as an indirect measure of initial release probability. Secondly, for the same data set, RD is correlated with the ratio 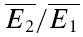 (r =−0.41, pr= 0.02, data not shown). Taken together, these correlations suggest that strongly depressing synapses (which have a relatively high initial probability of release, i.e. U0 > 0.5) are more likely to express more RDD. Conversely, pure RID is most likely to be observed at synapses with low initial release probability and weak depression.
(r =−0.41, pr= 0.02, data not shown). Taken together, these correlations suggest that strongly depressing synapses (which have a relatively high initial probability of release, i.e. U0 > 0.5) are more likely to express more RDD. Conversely, pure RID is most likely to be observed at synapses with low initial release probability and weak depression.
Figure 5. Features of RDD and RID.

A, the release dependence of depression (RD) is correlated with release probability. Here the skew of the amplitude distribution is used as a coarse indicator of release probabilities. The continuous line indicates the linear regression (r = −0.54, pr < 0.005). B, the amount of depression (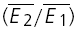 ) is also correlated with the skew of the amplitude distributions. The continuous line indicates the linear regression (r = 0.55, pr < 0.005). C, temperature dependence. The response of a layer V connection maintained at 30°C to the first two action potentials in a sequence. Stimulation frequency was 10 Hz. E2 is plotted as a function of E1 for each trial. Continuous line: linear regression of the data. Dashed lines: theoretical line for pure RID (horizontal line) and pure RDD (oblique line). The connection displays a strong component of RDD (RD = 0.88 ± 0.11). The open squares indicate
) is also correlated with the skew of the amplitude distributions. The continuous line indicates the linear regression (r = 0.55, pr < 0.005). C, temperature dependence. The response of a layer V connection maintained at 30°C to the first two action potentials in a sequence. Stimulation frequency was 10 Hz. E2 is plotted as a function of E1 for each trial. Continuous line: linear regression of the data. Dashed lines: theoretical line for pure RID (horizontal line) and pure RDD (oblique line). The connection displays a strong component of RDD (RD = 0.88 ± 0.11). The open squares indicate 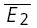 smallE1 and
smallE1 and 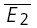 bigE1. D, same connection as in C, but at 36°C, showing a strong component of RID (RD = 0.02 ± 0.11). The open squares indicate
bigE1. D, same connection as in C, but at 36°C, showing a strong component of RID (RD = 0.02 ± 0.11). The open squares indicate 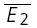 smallE1 and
smallE1 and 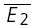 bigE1.
bigE1.
Temperature dependence of RID
We found that RID is more prominent at physiological temperature and that the mechanism of depression is highly temperature dependent. In seven experiments, connections were recorded at 30°C rather than 36°C. In the example shown in Fig. 5C and D, the same connection was recorded at both temperatures. At the higher temperature, the dominant mechanism of depression is release independent, as shown in Fig. 5D (RD = 0.02 ± 0.11, significantly different from pure RDD, but not RID). At 30°C, the depression is largely release dependent (RD = 0.88 ± 0.11, Fig. 5C), significantly different from pure RID, but not RDD. The effect of the change in temperature was reversible (data not shown). The mean RD for all seven experiments at 30°C was 0.77 ± 0.22; this was significantly higher than RD for experiments at 36°C (0.45 ± 0.35; pt= 0.03). In parallel, the total amount of paired-pulse depression (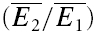 ) actually decreases at the lower temperature (
) actually decreases at the lower temperature (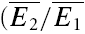 = 0.45 ± 0.15 at 36°C; 0.64 ± 0.07 at 30°C, pt= 0.05). The apparent Q10(30–36°C) of RD was 2.6 ± 0.2.
= 0.45 ± 0.15 at 36°C; 0.64 ± 0.07 at 30°C, pt= 0.05). The apparent Q10(30–36°C) of RD was 2.6 ± 0.2.
Ca2+ dependence of depression
To investigate the Ca2+ dependence of synaptic depression, we re-patched the presynaptic neurone with electrodes containing the calcium buffer EGTA at increasing concentrations (0.5–5 mm, N = 5 for each concentration; Ohana & Sakmann, 1998; Rozov et al. 2001). In the presence of EGTA, the average EPSC amplitude decreased in comparison to the control recordings (77 ± 8% of control 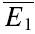 after re-patching the presynaptic neurone with intracellular solution containing 5 mm EGTA). The amount of depression tended to increase, on average (Fig. 6A); however, the effect was variable with depression increasing at some and decreasing at other connections (thus the increase in the variance in Fig. 6A). RD, however, consistently decreased at EGTA concentrations ≥ 1 mm (Fig. 6B). Using 1 mm EGTA, RD was 0.15 ± 0.18 (ppt= 0.02; the control RD for the same 5 neurones was 0.41 ± 0.22). This finding is consistent with the correlation between RD and skew (i.e. U0), since EGTA is expected to decrease the initial release probability.
after re-patching the presynaptic neurone with intracellular solution containing 5 mm EGTA). The amount of depression tended to increase, on average (Fig. 6A); however, the effect was variable with depression increasing at some and decreasing at other connections (thus the increase in the variance in Fig. 6A). RD, however, consistently decreased at EGTA concentrations ≥ 1 mm (Fig. 6B). Using 1 mm EGTA, RD was 0.15 ± 0.18 (ppt= 0.02; the control RD for the same 5 neurones was 0.41 ± 0.22). This finding is consistent with the correlation between RD and skew (i.e. U0), since EGTA is expected to decrease the initial release probability.
Figure 6. The effect of buffering presynaptic Ca2+on synaptic depression.

A, the amount of depression (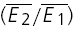 ) plotted as a function of EGTA concentration. With increasing concentration of EGTA, depression increases on average but this effect is variable; in some examples depression was reduced (note the increase in the error). B, plot of RD as a function of EGTA concentration. With increasing concentration, the release dependence of depression decreases.
) plotted as a function of EGTA concentration. With increasing concentration of EGTA, depression increases on average but this effect is variable; in some examples depression was reduced (note the increase in the error). B, plot of RD as a function of EGTA concentration. With increasing concentration, the release dependence of depression decreases.
Frequency independence of RD.
At connections stimulated at more than one frequency (5, 10 and 20 Hz; in some cases 25, 30, 40 and 50 Hz were also examined), there was no change in RD with stimulus frequency (ppt= 0.44 for 5 and 20 Hz, data not shown). However, we did not extensively examine the effects of higher input frequencies because of confounding effects produced by changes in action potential shape during the relative refractory period.
Frequency-dependent recovery from depression (FDR)
In this section, we present another phenomenon observed at the recorded connections. It relates to the mean EPSCs of a connection, evoked after a recovery interval and averaged over many repetitions of the same input pattern (Fig. 1C). In previous studies, the steady-state response at high frequencies (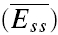 ) was shown to decrease in a manner inversely proportional to frequency (Abbott et al. 1997; Tsodyks & Markram, 1997). Indeed, some connections (Fig. 7A and B) exhibited such an inverse relationship. However, at other connections (Fig. 7C and D) the steady-state response at higher stimulus rates is frequency independent. This convergence to a similar steady state occurred for rates ≥ 10 Hz. For example in Fig. 7C, the magnitude of the steady-state response of a layer V connection is similar at 10 and 20 Hz. The convergence during steady-state to a constant value is shown in Fig. 7D.
) was shown to decrease in a manner inversely proportional to frequency (Abbott et al. 1997; Tsodyks & Markram, 1997). Indeed, some connections (Fig. 7A and B) exhibited such an inverse relationship. However, at other connections (Fig. 7C and D) the steady-state response at higher stimulus rates is frequency independent. This convergence to a similar steady state occurred for rates ≥ 10 Hz. For example in Fig. 7C, the magnitude of the steady-state response of a layer V connection is similar at 10 and 20 Hz. The convergence during steady-state to a constant value is shown in Fig. 7D.
Figure 7. Connections with and without apparent frequency-dependent recovery (FDR).

A, a layer V connection with no apparent FDR. The average response (N = 40) to action potentials triggered at three different frequencies (5, 10 and 20 Hz). In all cases, a further action potential was triggered 500 ms after the burst. The amplitudes of the steady-state response (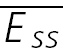 ) and the recovery EPSC (
) and the recovery EPSC (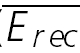 ) are smaller, with increasing input frequency. B,
) are smaller, with increasing input frequency. B, 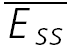 of the connection in (A) is plotted as a function of frequency. There is no convergence to a steady-state of similar amplitude at different frequencies. The dashed line indicates the change proportional to 1/f. C, a layer V connection with FDR. The average response (N = 100) recorded at 5, 10 and 20 Hz. In all cases, a further action potential was triggered 600 ms after the burst.
of the connection in (A) is plotted as a function of frequency. There is no convergence to a steady-state of similar amplitude at different frequencies. The dashed line indicates the change proportional to 1/f. C, a layer V connection with FDR. The average response (N = 100) recorded at 5, 10 and 20 Hz. In all cases, a further action potential was triggered 600 ms after the burst. 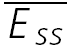 is the same at 10 and 20 Hz, but
is the same at 10 and 20 Hz, but 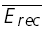 is larger when the preceding burst was 20 Hz. D,
is larger when the preceding burst was 20 Hz. D, 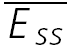 of the connection in C. There is convergence to a similar
of the connection in C. There is convergence to a similar 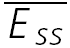 at frequencies = 10 Hz. Dashed line indicates the analytically computed value of
at frequencies = 10 Hz. Dashed line indicates the analytically computed value of 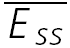 for high input frequencies.
for high input frequencies.
Additionally, we observed that at higher rates (≥ 10 Hz), the synaptic response to an additional action potential 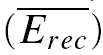 , stimulated at a fixed recovery interval (600 ms), increases with the frequency of the preceding burst (Fig. 7C). Finally, there is a transient decrease in the magnitude of subsequent synaptic responses, which then recovers (see EPSCs 6–9 at 20 Hz in Fig. 7C). These three observations cannot be explained by the classical model of depressing synapses, where recovery is characterized by a single time constant (Abbott et al. 1997; Tsodyks & Markram, 1997). They could be explained, however, if there is an activity-dependent mechanism for recovery from depression, which is recruited at higher frequencies. We term this phenomenon frequency-dependent recovery (FDR; Dittman & Regehr, 1998; Stevens & Wesseling, 1998; Wang & Kaczmarek, 1998; Weis et al. 1999; Sakaba & Neher, 2003).
, stimulated at a fixed recovery interval (600 ms), increases with the frequency of the preceding burst (Fig. 7C). Finally, there is a transient decrease in the magnitude of subsequent synaptic responses, which then recovers (see EPSCs 6–9 at 20 Hz in Fig. 7C). These three observations cannot be explained by the classical model of depressing synapses, where recovery is characterized by a single time constant (Abbott et al. 1997; Tsodyks & Markram, 1997). They could be explained, however, if there is an activity-dependent mechanism for recovery from depression, which is recruited at higher frequencies. We term this phenomenon frequency-dependent recovery (FDR; Dittman & Regehr, 1998; Stevens & Wesseling, 1998; Wang & Kaczmarek, 1998; Weis et al. 1999; Sakaba & Neher, 2003).
Analysis of the model indicates that the convergence of EPSCs to a steady-state value at high input frequencies requires that depression is strongly dominated by one of the two forms of depression (RDD or RID), which recovers in a frequency-dependent manner (see below). Since most connections exhibit some degree of RID, we investigated whether FDR was associated with release-dependent or -independent depression (Fig. 8). For this purpose, we used RFDR (see Methods) to estimate the relative degree of FDR in all the recorded connections. We found that the expression of FDR was negatively correlated with RD, indicating that connections with prominent RID also exhibited more FDR (r =−0.51, pr= 0.005). Such a relationship can also be inferred from Fig. 4A and B, where the amplitude of the 20th EPSC following different frequencies of stimulation is distributed over a wider range along the diagonal for those neurones with RD≥ 0.65 (Fig. 4A, see left-most symbols for each frequency) than for those with RD≤ 0.35 (Fig. 4B).
Figure 8. Correlation between RFDR and RD.

The ratio of the normalized recovery amplitude following a burst at 10 and 20 Hz (RFDR) is plotted against RD(determined at 20 Hz) of layer V connections (filled circles) and 3 layer IV connections (open squares). There was a significant correlation between the two parameters as indicated by the linear regression for layer V connections (r =−0.51, pr < 0.005).
We therefore formulate FDR in eqn (7) by an activity-dependent recovery from the release-independent component of depression. The time constant associated with this recovery is called τinrec. In Fig. 9 the simple model with no FDR (A, i.e. with constant τinrec), and the extended model (B), in which the recovery time constant is frequency dependent are fitted to the synaptic response shown in Fig. 7C. The magnitudes of both experimental and predicted EPSCs to sequential action potentials are depicted. It is clear that the simple model cannot account for the similar steady state responses of a synapse stimulated at 10 and 20 Hz, nor for the increasing magnitude of the recovery response, and clearly not for the shape of the curves. In contrast, the extended model replicates both the steady-state responses of the connection, and the ratio between the recovery response and the steady-state response, at different frequencies. Moreover, the model exhibits the transient decrease in the amplitude of synaptic responses before reaching the steady state, as seen in the recordings (Fig. 9B and 20 Hz).
Figure 9. Modelling FDR.

The magnitudes of EPSCs of a layer V pyramidal–pyramidal connection expressing FDR (same as in Fig. 7C and D) at three frequencies of stimulation (5, 10 and 20 Hz) with the recovery EPSC (open circle) are shown with fits of two alternative models. A, simple model with no FDR (ττinrec constant; with pure RID). The experimental EPSC amplitudes (filled circles) and the modelled one (line) are plotted as a function of the number in the sequence. In all cases the last EPSC is the response to gauge recovery (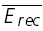 ). Model parameters: U0 = 0.1, U1 = 0.4, τrec = 1 ms, τ0 = 700 ms, τ1 = 0, ττinrec = 300 ms and ASE =−1130 pA. B, full model with FDR (τinrec activity dependent; again with pure RID). Model parameters: U0 = 0.4, U1 = 0.4, τrec = 1 ms, τ0 = 2000 ms, τ1 = 0.4, ττinrec = 500 ms and ASE = −282 pA. Dashed horizontal line indicates no response. C, steady-state response (
). Model parameters: U0 = 0.1, U1 = 0.4, τrec = 1 ms, τ0 = 700 ms, τ1 = 0, ττinrec = 300 ms and ASE =−1130 pA. B, full model with FDR (τinrec activity dependent; again with pure RID). Model parameters: U0 = 0.4, U1 = 0.4, τrec = 1 ms, τ0 = 2000 ms, τ1 = 0.4, ττinrec = 500 ms and ASE = −282 pA. Dashed horizontal line indicates no response. C, steady-state response (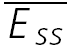 ) as a function of frequency for models with both RDD and RID for various values of τrec (continuous lines). Dashed lines represent the analytically calculated relation between
) as a function of frequency for models with both RDD and RID for various values of τrec (continuous lines). Dashed lines represent the analytically calculated relation between 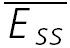 and input frequency (f), expected at high input frequencies (1/f). Model parameters: U0 = 0.3, U1 = 0.1, τ0 = 1800 ms, τ1 = 0.5, ττinrec = 1800 ms and ASE = −100 pA, τrec = 200, 300, 400, 500, 600 ms. D,
and input frequency (f), expected at high input frequencies (1/f). Model parameters: U0 = 0.3, U1 = 0.1, τ0 = 1800 ms, τ1 = 0.5, ττinrec = 1800 ms and ASE = −100 pA, τrec = 200, 300, 400, 500, 600 ms. D, 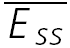 of a connection with pure RID and FDR.
of a connection with pure RID and FDR. 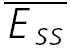 (continuous line) of the connection converges to an analytically calculated constant value at high input frequencies (dashed line). Model parameters: U0 = 0.4, U1 = 0.4, τrec = 0 ms, τ0= 2000 ms, τ1 = 0.4, ττinrec = 500 ms and ASE =−62 pA.
(continuous line) of the connection converges to an analytically calculated constant value at high input frequencies (dashed line). Model parameters: U0 = 0.4, U1 = 0.4, τrec = 0 ms, τ0= 2000 ms, τ1 = 0.4, ττinrec = 500 ms and ASE =−62 pA.
As mentioned above, the convergence of EPSCs to a steady-state of similar amplitude at high input frequencies depends on two conditions. First, the synapse must exhibit RID. Second, the recovery from the release-independent depression (τinrec) must be activity dependent. In Fig. 9D, the steady-state response of a synapse, which satisfies these conditions (continuous line), is depicted as a function of frequency. In contrast, for synapses with a mixture of RID and RDD, the steady-state response amplitudes decline in inverse proportion to the input frequency (Fig. 9C). If a synapse shows pure RDD, the steady-state amplitude should be inversely proportional to the input frequency (1/f, Tsodyks & Markram, 1997). Indeed, as the release-dependent component becomes more pronounced (longer τrec), the response converges to that predicted by 1/f at lower input frequencies (Fig. 9C).
Further experimental evidence for FDR is provided in Fig. 10, where the normalized recovery amplitude is plotted against the delay before the final action potential for a connection where there was no increase in recovery rate with increasing frequency (Fig. 10A; note that 1 −Rrec is plotted so that recovery increases with time), and for an alternative example, where there was strong FDR (Fig. 10B). In every case, the data points were adequately fitted by a single exponential (pχ < 0.05). The apparent recovery time constants (τa) for frequencies of 10, 20 and 50 Hz were not significantly different for the first example (580–660 ms). However, τa increased approximately 4-fold over this frequency range for the connection with FDR (Fig. 10B; from 1240 ± 184 ms at 10 Hz to 321 ± 112 ms at 50 Hz).
Figure 10. Time course of recovery from depression.

A, a connection showing no FDR. The time constant for recovery following a burst at 10 Hz (580 ± 78 ms, open circles) is not significantly different from that at 20 Hz (654 ± 81 ms, filled squares) or at 50 Hz (609 ± 73 ms, filled circles). B, a connection showing prominent FDR. The time constant for recovery following a burst at 10 Hz (1243 ± 184 ms, open circles) is significantly slower than that at 20 Hz (525 ± 76 ms, filled squares) and that at 50 Hz (321 ± 112 ms, filled circles). Note that 1 −Rrec (where Rrec is recovery amplitude) is plotted here so that recovery increases with time and the time constant of recovery is more easily demonstrated.
Averaged over all connections the normalized recovery amplitude (Rrec) following a burst at 10 Hz was 0.59 ± 0.12, with τa = 760 ± 150 ms. There was no significant difference in either of these parameters for those connections that demonstrated FDR and those that did not (pt= 0.7 and 0.3, respectively). Following a burst at 20 Hz, the average normalized recovery amplitude was 0.43 ± 0.14 (pt < 0.001 when compared with 10 Hz recovery amplitude). Thus, RFDR (ratio between normalized recovery amplitude at 10 and 20 Hz, see Methods) was 1.37 ± 0.25. We examined whether there was a difference in τa between connections that demonstrated FDR and those that did not, and chose an arbitrary cut-off of RFDR < 1.1 for no RFDR(since this value was not significantly different from RFDR = 1) and RFDR > 1.2 for the sample that demonstrated RFDR. The τa following a 20 Hz burst for those connections with RFDR > 1.2 (528 ± 137 ms, N = 7) was significantly faster than those with RFDR < 1.1 (691 ± 126 ms, N = 6, pt= 0.05).
Since two processes seem to participate in the overall recovery from depression, we needed to find evidence that FDR was not caused by a process associated with eliciting a series of 20 action potentials at an intermediate frequency. Augmentation (Zengel et al. 1980; Stevens & Wesseling, 1999) is such a mechanism, which could superimpose on the depression and thereby mimic FDR. Despite the fact that we chose a subset of neurones that showed no signs of facilitation within the first few action potentials, we investigated whether augmentation was still present. When the presynaptic burst of action potentials was increased to 100 pulses at 10 Hz, we could discern a small amount of augmentation. This was maximally 40% greater than the initial EPSC amplitude (Fig. 11A) and decayed with a τAug of 10.9 ± 5.3 s, which is much longer than τa at the same frequency (0.76 ± 0.15 s; pt < 0.0002). In order to further corroborate that augmentation was not causing an apparent FDR, we checked the differential sensitivity to Ca2+ buffering by EGTA. Surprisingly, in our study presynaptic EGTA at a concentration of 5 mm did not affect augmentation (Fig. 11B). Under this condition, τAug was 7.3 ± 2.4 s, which was not different from that with the control intracellular solution (N = 5, ppt= 0.2). However, the inability of EGTA to block augmentation was significantly different from the effect on FDR, which was abolished at this concentration (see below).
Figure 11. Augmentation.

A, the recovery from augmentation is plotted as the normalized amplitude (NAmpl) versus time following a sequence of 100 pulses at 10 Hz. Note that a normalized amplitude is used (see Methods and Stevens & Wesseling, 1999) not Rrec. The recovery time constant is 11 ± 5 s, the maximum increase in amplitude is 40% of control (N = 5). B, in the presence of 5 mm presynaptic EGTA, the recovery from augmentation is not significantly different from that in control for the same 5 connections. The recovery time constant from augmentation is 8 ± 2 s.
Properties of recovery
Temperature dependence of recovery
As was to be expected for a kinetic process, the recovery decreased when the temperature was 30°C rather than 36°C. The normalized recovery amplitude following a burst at 10 Hz was 0.84 ± 0.40 (N = 7) as compared to recordings at 36°C (0.59 ± 0.12, pt= 0.05). Following a burst at 20 Hz it was 0.73 ± 0.31 (N = 7, 36°C: 0.43 ± 0.14, pt= 0.005). Thus, RFDR at 30°C was 1.15 ± 0.21, significantly different from the value at 36°C (1.37 ± 0.25; pt= 0.02). The apparent Q10(30–36°C) was 1.4 ± 0.1, significantly smaller than that for RD(2.6 ± 0.2, pt= 0.001).
Ca2+ dependence of recovery
Previous studies have found that FDR-like phenomena are caused by an increase in intracellular Ca2+ (Dittman & Regehr, 1998; Wang & Kaczmarek, 1998; Sakaba & Neher, 2003). We tested this hypothesis directly by buffering Ca2+ concentration in the presynaptic neurone with 0.5–5 mm EGTA in the intracellular solution. Consistent with this hypothesis, recovery was prolonged. Even with 0.5 mm EGTA, the normalized recovery amplitudes at 10 and 20 Hz were significantly greater than for the same five connections in the absence of EGTA (10 Hz: 0.76 ± 0.08 versus 0.54 ± 0.13, ppt= 0.005; 20 Hz: 0.62 ± 0.19 versus0.39 ± 0.13, ppt= 0.02, Fig. 12). In addition to slowing the recovery, buffering intracellular Ca2+ abolished the difference in recovery following bursts of 10 and 20 Hz at concentrations ≥ 2 mm so that RFDR under these conditions was not significantly different from unity (Fig. 12), suggesting that the expression of FDR requires intracellular Ca2+.
Figure 12. Effect of EGTA on the characteristics of recovery from depression.

The normalized recovery amplitude (Rrec,eqn (2)) following a burst at 10 Hz (open circles) and at 20 Hz (filled squares) is plotted against EGTA concentration in the presynaptic neurone. RFDR, the ratio between Rrec at 10 and 20 Hz, is plotted (open squares). For EGTA concentration ≥ 2 mm, there is no significant difference in Rrec and therefore RFDR is not different from unity. For clarity, only one-sided error bars are displayed.
Functional implications of FDR and RID
We simulated the implications of FDR and RID in order to gain insight into the capacity of the synapse to code rate as well as temporal information. The simulation describes the net postsynaptic current generated by the activity of a population of 1500 presynaptic neurones (as described by eqn (10)), each firing a sequence of action potentials according to Poisson statistics. The synapses all have the same characteristics, and all neurones have the same average firing frequency. After reaching steady-state, transitions from one frequency to another (indicated by arrow in Fig. 13) are induced. Consistent with earlier reports (Tsodyks & Markram, 1997), the analysis shows that at low firing rates, a synapse with pure RDD can transmit information about average firing rate and rate changes. However, at higher frequencies, it loses its ability to code information about firing rate, but can still signal transient changes (Fig. 13A).
In contrast, a synapse exhibiting pure RID and FDR operates in a very different manner (Fig. 13B): it can transmit information about the average firing rate and the rate transitions both at low and at high frequencies.
Discussion
Two characteristics of synaptic depression at connections in somatosensory cortex are described in this paper that may provide important functional properties to networks of connected neurones. The first, RID, is a depression, which occurs even after failure to release transmitter. The second, frequency-dependent recovery from depression (FDR), allows a faster recovery of transmission with increased input frequency. These characteristics are correlated and the functional significance of their expression was addressed.
Components of synaptic depression
Depression at excitatory neocortical connections is composed of release-dependent and -independent components. Forms of RID have been recognized in previous studies at invertebrate (Hatt & Smith, 1976; Gover et al. 2002) and vertebrate synapses (Dobrunz et al. 1997; Bellingham & Walmsley, 1999; Thomson & Bannister, 1999; Brody & Yue, 2000; Kraushaar & Jonas, 2000; Waldeck et al. 2000).
Reliability and range of RD.
We base our finding of RID on three measures. Firstly, at connections with failures on the first response, the second EPSC is still depressed for the subset of EPSCs with preceding failures (Fig. 2E and F). This indicates that RID is presynaptic and is also activity dependent, i.e. caused by the preceding action potential and lasting for up to 300 ms. Secondly, even at connections without failures, when EPSCs are sorted into two categories depending on the size of the first EPSC, depression on the second EPSC is not bigger for large first responses (Fig. 2C). Thirdly, the measure RD exploits the expected correlations between two successive EPSCs quantitatively and can be derived explicitly (see Appendix).
The reliability of RD can be inferred from the following arguments. RD is specific to a junction, is maintained during long recording sessions, does not ‘wash-out’, is the same for different recording modes (cell-attached and whole-cell), is restored to the same value when conditions are changed back to control (temperature) and is not affected by the presence of bicuculline and, thus, is not caused by a potential block of Ca2+-dependent K+ channels (SK-type; see for example Debarbieux et al. 1998). It is not dependent on artefacts induced by large quantal variance or recording noise.
Range of RD
The relative contribution of the components of depression varies at different synapses (Fig. 2B and D), and the whole range of possibilities from purely release dependent to purely release independent was observed. The average value of RD of 0.45 ± 0.35 suggests that, in our data set, at least 50% of the depression is release independent.
Potential causes of RID
One potential cause for an apparent RID could be an underlying facilitation, which obscured the release dependence of the dominant synaptic depression. We are certainly not disputing the fact that facilitation can occur at excitatory neocortical connections but believe that, by carefully choosing a subset of connections with evident depression only (∼80%), the influence of this form of plasticity on our results was small. The most convincing argument against an underlying facilitation is presented in Fig. 2E and F. If facilitation was an underlying mechanism, it should show up in the subset of EPSCs that follow a failure. However, even in this case we observed depression rather than facilitation. Furthermore, the facilitatory process would require a positive correlation between the amplitudes of the second and first EPSC; only in this way could it reduce the negative correlation in release-dependent depression. This is very unlikely since we have not seen such a correlation in any of our data sets and it would require that the amplitude of the second EPSC is larger even when the preceding EPSC was large, a scenario severely challenging vesicular depletion.
We can rule out a postsynaptic mechanism (i.e. Na+ current boosting of poorly clamped EPSCs) resulting in a counterbalancing positive correlation along similar lines. Firstly, Fig. 2E and F indicates that RID occurs when E1 is a failure and is therefore clearly presynaptic. Secondly, a mechanism involving active dendritic properties would preferentially influence larger E1, which was not observed. Finally, the experiments using QX-314 postsynaptically to block dendritic boosting showed that there was no significant involvement of postsynaptic Na+ currents in the observed EPSCs.
Site of expression
The rate of transmitter release is the product of release probability (Pr) and the number of quantal release units (n). Depression could theoretically result from a decrease in either or both of the parameters with, for a depletion model, the assumption that n represented the vesicular pool (Stevens & Wesseling, 1999; Kraushaar & Jonas, 2000). To investigate whether Pr changes during the sequence of action potentials, a CV−2 analysis revealed that both RDD and RID appear to result from a change in Pr, given that the change in CV−2 is largely identical to the change in normalized mean amplitude. If, in contrast, the change in the number of release sites and/or the quantal size occurred, one would expect such a plot to deviate horizontally and above the diagonal. An alternative explanation would be that changes in Pr, n and q are so finely balanced that the CV−2 remains proportional to the change in amplitude despite such changes. We cannot rule this alternative out without further experiments, but such an explanation appears highly unlikely. The change in Pr does not identify whether the change is in the probability that a vesicle is available for release (Pv), which is a release-dependent process, or in the probability that an available vesicle can actually be released (USE), a release-independent process.
We provide evidence that RD is correlated with the skewness of the amplitude distribution (Fig. 5A), a simple measure which might reflect the initial release probability of the connection. The correlation was such that there is more release-dependent depression in those connections with a higher release probability. Similarly, RD is negatively correlated with the paired-pulse ratio. These correlations are to some extent intuitive: vesicle depletion is caused by release, and release is more likely to occur when there is a high release probability at the synapse. The fact that there is also more depression at these connections might suggest that RDD is an additional mechanism to RID, which only occurs or is apparent at synapses with a high release probability.
The temperature dependence of RD (Fig. 5C and D) is such that release-independent depression is more pronounced when recording near physiological temperature. This could result from either an increase in RID or a decrease in RDD, indicating that both forms of depression might be temperature sensitive, but in opposite directions. An increased RID could result from an increase in, for example, channel inactivation, whereas a decreased RDD could be due to faster replenishment of the vesicular pool, as indicated in Results. At lower temperatures, it has been shown that release becomes less reliable (Hardingham & Larkman, 1998). This observation requires that at the lower temperature the correlation between RD and skew is changed to allow RDD to dominate, while the probability of release decreases. At this stage, we are unable to comment on which mechanism(s) might be at work, since our major aim was to examine mechanisms of depression at physiological temperature. The Q10(30–36°C) value for RD is 2.6 ± 0.2 and indicates a higher activation energy requirement than for recovery from depression (see below). This value is in the range of voltage-gated channels and is about double the value for aqueous diffusion (Hille, 2001). It is consistent with the potential mechanism relying on a channel, for example a K+ (Huguenard et al. 1991), a Ca2+-activated K+, a Ca2+ channel (Coulter et al. 1989) or a ligand-gated ion channel (Dreyer et al. 1976).
Ca2+ sensitivity of RID.
In an attempt to investigate the mechanism(s) underlying RID, we examined its dependence on residual Ca2+ using EGTA as a chelator (Fig. 6). If RID is Ca2+ dependent, then a Ca2+-dependent reduction in release probability could be a likely cause. We could not, however, demonstrate a Ca2+ dependence of RID. This inability might result from several factors. Firstly, it might be that EGTA is too slow to cause an efficient Ca2+ concentration clamp at the synapse and/or that the concentration achieved in the terminals was not high enough or was saturated by Ca2+ influx. Secondly, since RDD is clearly Ca2+ dependent (most likely due to the effect that buffering residual Ca2+ has on the probability of release), and, at the same time, buffering slowed recovery from depression (see below), depression tended to increase and become more release independent. The decrease in the parameter RD that was observed could indicate that only RDD was Ca2+ dependent, or that both types of depression are Ca2+ dependent, however, with a greater Ca2+ sensitivity of RDD.
Mechanisms generating RID
A number of potential mechanisms for the generation of RID have been suggested: inactivation of presynaptic Ca2+ channels (Thomson & Bannister, 1999), change in action potential shape (reviewed in Zucker & Regehr, 2002), extracellular Ca2+ depletion (Borst & Sakmann, 1999), desensitization of the release machinery to Ca2+ (Bellingham & Walmsley, 1999), voltage-dependent inactivation of release (Dobrunz et al. 1997; Kraushaar & Jonas, 2000) and axonal conduction failure (Hatt & Smith, 1976; Brody & Yue, 2000).
At mammalian central synapses, there is little evidence from in vivo recordings or slice preparations that conduction failure occurs under physiological conditions (Cox et al. 2000; Koester & Sakmann, 2000) even though studies of cultures have shown that axonal propagation might not always be reliable (Lüscher et al. 1994; Debanne et al. 1997). It is also unlikely that changes in action potential shape during the burst of presynaptic activity play a significant role in the RID observed in our experiments, since RID was seen at low firing rates (< 5 Hz), where the interspike intervals were much longer than the relative refractory period. We also think that desensitization is an unlikely candidate for the depression observed for the following reasons. Firstly, stimulus frequencies in the range of 10 Hz are sufficiently low for AMPA receptors to recover appreciably and, secondly, such a scenario would argue for a negative correlation between consecutive EPSCs (Matveev & Wang, 2000; Scheuss et al. 2002), a fact that is challenged by the observation that many connections do not show any sign of correlation at all. In addition, the same line of thought also rules out a major involvement of presynaptic autoreceptors, which could be activated by the release of glutamate. We therefore conclude that the mechanism(s) of depression occur(s) prior to the exocytotic step.
RID could be caused by three different scenarios. Firstly, if Ca2+ indeed causes this form of depression, activation of Ca2+-dependent K+ channels might hyperpolarize the terminals and thereby limit the Ca2+ influx on subsequent action potentials. A similar mechanism has been observed at terminals in the posterior pituitary (Bielefeldt & Jackson, 1993). Secondly, along a similar line, Ca2+-dependent inactivation of Ca2+ channels could provide a reduced Ca2+ influx on subsequent action potentials (Forsythe et al. 1998). We think that this mechanism is unlikely since recovery from inactivation is of the order of minutes rather than the ∼600 ms observed in our data. And, thirdly, if inactivation of the release machinery were the cause of this depression (Kraushaar & Jonas, 2000; Waldeck et al. 2000), it would require that those terminals with pronounced RID have an altered release machinery either by biochemical composition or, for example, by phosphorylation. Detailed investigation of the mechanism underlying the form of RID reported in our study remains for future investigation.
Recovery from depression
The second feature of depression we observed was that at certain synapses, a steady-state response is attained, which is largely independent of input frequency within a physiological range (≤ 50 Hz, Fig. 7C and D). This is in contrast to synapses in which the rate of recovery from depression is constant, and the steady-state response is inversely related to the presynaptic firing frequency (1/f, Abbott et al. 1997; Tsodyks & Markram, 1997). This feature of synaptic depression is explained by a frequency-dependent mechanism of recovery from depression (FDR).
FDR has been reported at invertebrate and mammalian synapses (reviewed in Zucker & Regehr, 2002). FDR at the giant synapse of mouse auditory brainstem (Wang & Kaczmarek, 1998; Sakaba & Neher, 2001, 2003) was shown to be regulated by Ca2+ influx via voltage-gated Ca2+ channels (although the data of Weis et al. 1999 does not support this hypothesis). This neuronal pathway needs to relay high frequency input in very precise temporal output and, thus, it is crucial that these synapses recover rapidly from depression. At this synapse, however, FDR occurs at frequencies > 100 Hz, a much higher rate than observed in our study. FDR at cerebellar climbing fibre–Purkinje cell synapses also depends on residual presynaptic calcium (Dittman & Regehr, 1998; Dittman et al. 2000).
In our study, FDR occurs in paired recordings at neocortical synapses performed at physiological temperature. Moreover, FDR occurs at frequencies as low as 10 Hz, definitely in the physiological firing range of excitatory cortical neurones in vivo (Brecht & Sakmann, 2002). The apparent FDR can be identified clearly in the different recovery rates following bursts at 10 and 20 Hz, and we quantified FDR by comparing the normalized recovery amplitudes (Rrec) at these frequencies. RFDR ranges between 0.9 and 1.9 indicating that at some connections recovery is about twice as fast at the higher stimulus frequency whereas at others there is no significant FDR.
FDR and augmentation
An apparent FDR could theoretically be the result of augmentation, which is also frequency dependent (Magleby & Zengel, 1976; Swandulla et al. 1991). With 100 pulses at 10 Hz, we could demonstrate that augmentation is 40% and decays with a τAug of 10.9 ± 5.3 s, which is consistent with other reports at neocortical synapses (Klein, 1994). Under our conditions and following 20 action potentials, augmentation (Magleby & Zengel, 1976) would only contribute about 8% to the increase in EPSC amplitude and therefore cannot explain the 200% increase in recovery amplitude observed. Furthermore, augmentation was not sensitive to 5 mm EGTA in the patch-pipette, whereas FDR was completely abolished at the same concentration. In addition, if augmentation were the underlying mechanism, it would have to precisely counterbalance the amount of depression since at different frequencies a steady-state of similar amplitude can still be observed. We therefore think that augmentation cannot explain our data and propose that FDR is a genuine mechanism operating at these synapses.
Ca2+ sensitivity
The inability to block augmentation at the chosen concentrations of EGTA could result from at least two factors. Firstly, the concentration reached in the terminal(s) is very likely smaller than that in the pipette solution and therefore might be saturated by the increase in background Ca2+. We think that this is unlikely since at the same time FDR was blocked. It suggests that the Kd value for the Ca2+ dependence of recovery is smaller than that for augmentation. Secondly, augmentation might be caused by smaller changes in background Ca2+ than could be effectively buffered by the EGTA concentrations used. It might be that very much higher concentrations, which we did not test, would be effective.
As a preliminary investigation into the mechanisms underlying FDR, we investigated its temperature and Ca2+ dependence. FDR was temperature dependent, but had a lower Q10 than the one for RD. A value of 1.4 ± 0.1 for Q10 is suggestive of aqueous diffusion (Hille, 2001) and therefore consistent with the idea that background Ca2+ is the causative agent.
The sensitivity of FDR to presynaptic EGTA also provides evidence that there are no significant underlying postsynaptic mechanisms influencing the time course of recovery. Potentially, AMPA receptor desensitization could have the opposite effect to FDR in reducing recovery at higher frequencies. However, in the presence of EGTA, RFDR was blocked but recovery was the same at 10 and 20 Hz, which would not have been the case if there were significant desensitization.
Correlation between RID and FDR
We noted that FDR is correlated with RID (Fig. 8), such that connections, which express pronounced RID, also show FDR. Such a correlation could occur in two ways. Firstly, FDR and RID could be causally related. This could be brought about if FDR consists of a frequency-dependent increase in the recovery rate from RID (τinrec; as implemented in the model). Secondly, FDR could be a frequency-dependent increase in the recovery rate of RDD (τrec). In this scheme, recovery from RDD would be sufficiently fast for it to only provide a negligible component of the depression. Then only RID would be observed and therefore it would appear to be correlated with FDR. The latter explanation appears more unlikely, since in this case the apparent recovery rate (combination of τrec and τinrec) following a burst at 10 Hz would be faster for those connections with dominant RID (as τrec would be sufficiently fast for negligible RDD to occur). This was not observed in our set of data.
Model of synaptic depression
Models replicating the dynamics of mean synaptic responses have previously been proposed (Abbott et al. 1997; Tsodyks & Markram, 1997) as well as models based on vesicle depletion (Quastel, 1997; Maass & Zador, 1999; Kraushaar & Jonas, 2000; Matveev & Wang, 2000; Fuhrmann et al. 2002). With the exception of Kraushaar & Jonas (2000), these models do not in general account for RID and FDR and therefore they cannot adequately describe most of our experimental data.
The model described here extends a previous version (Fuhrmann et al. 2002) and reproduces the responses of neocortical connections to sequences of action potentials, both in terms of the average response dynamics, and the response fluctuations. A synapse with short τrec (relative to τinrec) will exhibit prominent RID, because replenishment of the vesicular pool (i.e. recovery from RDD) will not be rate-limiting. RDD results from the opposite relation between the recovery time constants, or from negligible changes in release probability with subsequent action potentials (U1→ 0, eqn (6)).
Since we noted that FDR and RID are correlated, FDR is formalized as a gradual acceleration of recovery from the release-independent component of depression, i.e. a mechanism counterbalancing the decrease in the probability of release. However, in theory, the independence of the steady-state response from input frequency and FDR could be expressed at connections with pure RDD (as discussed above). We cannot exclude the possibility, although we consider this less likely for the reasons given above.
Functional consequences of RID and FDR
The presence of RID has important consequences for information transfer (Fig. 13). If there were no RID, synapses with low probability of release would be unable to reliably code presynaptic activity because, based on vesicle depletion alone, release would occur too infrequently to cause depression. However, since RID is independent of release, it allows reliable coding at such synapses.
The functional significance of FDR is that at high frequencies of stimulation it enables information transmission by preventing the synapse from depressing too much, and therefore linearizing the dependence of the postsynaptic depolarization on the presynaptic frequency.
Our data suggest that there are different operating modes of synapses. At one extreme, dominated by RDD, the synapse cannot transmit rate information at high input rates due to the decrease in the amplitude of responses in proportion to the input frequency. This mode favours temporal information processing, as a transient response to a frequency change occurs at all frequencies. At the other extreme, if pure RID and FDR are expressed, rate information is transmitted even at high frequencies. Our data indicates that there is a continuum between the two extreme operating modes. Since different neuronal connections are dominated either by RDD or RID, it may be that different connections are optimized for relaying either rate or temporal information.
The underlying factors determining whether a synapse expresses RID and/or RDD are at present unknown. It could be that the type of depression is determined by the postsynaptic neurone (target specificity) or that there is presynaptic specificity. Presynaptic neurones whose response to depolarization consists of a brief burst of action potentials may only express RDD since continuous firing would quickly end up in no transmission. Similarly, tonically firing neurones may then have RID synapses. Functionally, the expression of both components of depression may be important in neural networks. The response times of many systems can be optimized by combining an appropriate mixture of signals of the first derivative (the rate change in the current context) and steady state (Wilts, 1960).
Acknowledgments
We are grateful to Dr S. J. Redman for comments on the manuscript. This study was supported by the Swiss National Science Foundation (5002-52085, 5002-57809, 31-59309-99), Bonizzi-Theler Foundation, Zürich, Jubiläumsstiftung der Schweizerischen Mobiliar Gesellschaft, Bern, Switzerland, and Israeli Academy of Science.
Appendix
Quantifying the release dependence of depression (RD)
E1 and E2 are random variables representing the first and second EPSCs. The correlation coefficient is
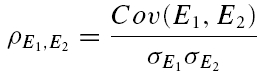 |
where
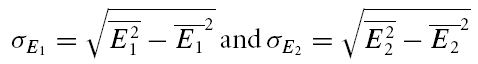 |
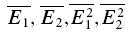 ,are the expectation values. The covariance is
,are the expectation values. The covariance is
Intuitively, if there is no correlation between E1 and E2(ρE1,E2= 0), the synaptic depression is purely release independent. As the correlation between E1 and E2 becomes more negative (Fig. 2B), there is more RDD. The maximal negative correlation between E1 and E2 occurs for pure RDD. ρE1,E2 can take values between 0 (pure RID) and ρRDD (pure RDD). We define RD as the correlation coefficient between E1 and E2, normalized by ρRDD (eqn (1)), where 0 ≤RD≤ 1. In practice, if the sample size is small, RD might fall outside these boundaries.
To compute ρRDD, we assume that the frequency of stimulation approaches infinity; thus, there is no recovery from depletion between action potentials. We can then derive ρRDD using the formalism for a binomial process applied to a depressing synapse (eqn (5)). Based on this idea, at the arrival of the first action potential, there are n vesicles available for release. In the case of pure RDD, following the release of n1 vesicles in response to the first action potential, there are only n − n1=n2 vesicles left. According to the binomial theorem in our notation
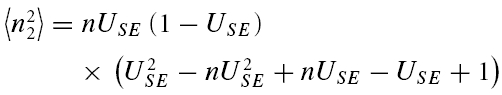 |
The postsynaptic response to the release of ni vesicles is proportional to ni. Substitution of 〈n1〉, 〈n2〉, 〈n12〉, 〈n12〉 and 〈n1n2〉 into eqn (11), eqn (12) and eqn (13) gives
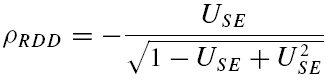 |
which is equivalent to
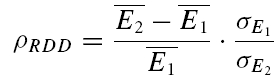 |
when substituting the expectations given above. This expression can be evaluated from experimental recordings directly.
For a graphical illustration of RD, we note that  (average E2 for a given E1) is linearly related to E1 with E1= ASE.n1/n, i.e.
(average E2 for a given E1) is linearly related to E1 with E1= ASE.n1/n, i.e.
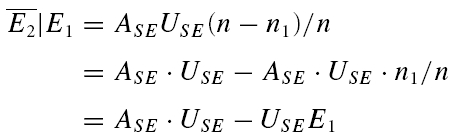 |
We plot E2 against E1 and calculate the linear regression with the y-intercept
and slope
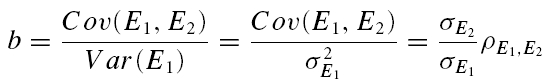 |
Therefore, a can be rewritten as a function of the correlation coefficient
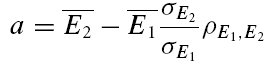 |
For pure RID, there is no correlation between E1 and E2 and therefore a= 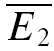 . For pure RDD, the y-intercept is equal to
. For pure RDD, the y-intercept is equal to 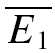 (see and eqn (8) and eqn (16)). Therefore, RD is the y-intercept of the regression line normalized within the range of 0–1, i.e.
(see and eqn (8) and eqn (16)). Therefore, RD is the y-intercept of the regression line normalized within the range of 0–1, i.e.
 |
References
- Abbott LF, Varela JA, Sen K, Nelson SB. Synaptic depression and cortical gain control. Science. 1997;275:220–224. doi: 10.1126/science.275.5297.221. [DOI] [PubMed] [Google Scholar]
- Bellingham MC, Walmsley B. A novel presynaptic inhibitory mechanism underlies paired pulse depression at a fast central synapse. Neuron. 1999;23:159–170. doi: 10.1016/s0896-6273(00)80762-x. [DOI] [PubMed] [Google Scholar]
- Bielefeldt K, Jackson MB. A calcium-activated potassium channel causes frequency-dependent action-potential failures in a mammalian nerve terminal. J Neurophysiol. 1993;70:284–298. doi: 10.1152/jn.1993.70.1.284. [DOI] [PubMed] [Google Scholar]
- Borst JGG, Sakmann B. Depletion of calcium in the synaptic cleft of a calyx-type synapse in the rat brainstem. J Physiol. 1999;521:123–133. doi: 10.1111/j.1469-7793.1999.00123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brecht M, Sakmann B. Dynamic representation of whisker deflection by postsynaptic potentials in morphologically reconstructed spiny stellate and pyramidal cells in the barrels and septa of layer 4 in rat somatosensory cortex. J Physiol. 2002;543:49–70. doi: 10.1113/jphysiol.2002.018465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody DL, Yue DT. Release-independent short-term synaptic depression in cultured hippocampal neurons. J Neurosci. 2000;20:2480–2494. doi: 10.1523/JNEUROSCI.20-07-02480.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulter DA, Huguenard JR, Prince DA. Calcium currents in rat thalamocortical relay neurons: Kinetic properties of the transient, low-threshold current. J Physiol. 1989;414:587–604. doi: 10.1113/jphysiol.1989.sp017705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney KR. Extended moment analysis for binomial parameters of transmitter release. J Theor Biol. 1978;73:285–292. doi: 10.1016/0022-5193(78)90191-1. [DOI] [PubMed] [Google Scholar]
- Cox CL, Denk W, Tank DW, Svoboda K. Action potentials reliably invade axonal arbors of rat neocortical neurons. Proc Natl Acad Sci U S A. 2000;97:9724–9728. doi: 10.1073/pnas.170278697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debanne D, Guérineau NC, Gähwiler BH, Thompson SM. Action-potential propagation gated by an axonal IA-like K+ conductance in hippocampus. Nature. 1997;389:286–289. doi: 10.1038/38502. [DOI] [PubMed] [Google Scholar]
- Debarbieux F, Brunton J, Charpak S. Effect of bicuculline on thalamic activity: a direct blockade of IAHP in reticularis neurons. J Neurophysiol. 1998;79:2911–2918. doi: 10.1152/jn.1998.79.6.2911. [DOI] [PubMed] [Google Scholar]
- Del Castillo J, Katz B. Quantal components of the end-plate potential. J Physiol. 1954;124:560–573. doi: 10.1113/jphysiol.1954.sp005129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittman JS, Kreitzer AC, Regehr WG. Interplay between facilitation, depression, and residual calcium at three presynaptic terminals. J Neurosci. 2000;20:1374–1385. doi: 10.1523/JNEUROSCI.20-04-01374.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittman JS, Regehr WG. Calcium dependence and recovery kinetics of presynaptic depression at the climbing fiber to Purkinje cell synapse. J Neurosci. 1998;18:6147–6167. doi: 10.1523/JNEUROSCI.18-16-06147.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrunz LE, Huang EP, Stevens CF. Very short-term plasticity in hippocampal synapses. Proc Natl Acad Sci U S A. 1997;94:14843–14847. doi: 10.1073/pnas.94.26.14843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyer F, Walther C, Peper K. Junctional and extrajunctional acetylcholine receptors in normal and denervated frog muscle fibres. Pflugers Arch. 1976;366:1–9. doi: 10.1007/BF02486555. [DOI] [PubMed] [Google Scholar]
- Forsythe ID, Tsujimoto T, Barnes-Davies M, Cuttle MF, Takahashi T. Inactivation of presynaptic calcium current contributes to synaptic depression at a fast central synapse. Neuron. 1998;20:797–807. doi: 10.1016/s0896-6273(00)81017-x. [DOI] [PubMed] [Google Scholar]
- Fuhrmann G, Segev I, Markram H, Tsodyks MV. Coding of temporal information by activity-dependent synapses. J Neurophysiol. 2002;87:140–148. doi: 10.1152/jn.00258.2001. [DOI] [PubMed] [Google Scholar]
- Gover TD, Jiang X-Y, Abrams TW. Persistent, exocytosis-independent silencing of release sites underlies homosynaptic depression at sensory synapses in Aplysia. J Neurosci. 2002;22:1942–1955. doi: 10.1523/JNEUROSCI.22-05-01942.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granit R, Kernell D, Lamarre Y. Algebraical summation in synaptic activation of motoneurones firing within the ‘primary range’ to injected current. J Physiol. 1966;187:379–399. doi: 10.1113/jphysiol.1966.sp008097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham NR, Larkman AU. The reliability of excitatory synaptic transmission in slices of rat visual cortex in vitro is temperature dependent. J Physiol. 1998;507:249–256. doi: 10.1111/j.1469-7793.1998.249bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatt H, Smith DO. Synaptic depression related to presynaptic axon conduction block. J Physiol. 1976;259:367–393. doi: 10.1113/jphysiol.1976.sp011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Ionic Channels of Excitable Membranes. Sunderland, MA USA: Sinauer Assoc., Inc; 2001. [Google Scholar]
- Huguenard JR, Coulter DA, Prince DA. A fast transient potassium current in thalamic relay neurons: Kinetics of activation and inactivation. J Neurophysiol. 1991;66:1304–1315. doi: 10.1152/jn.1991.66.4.1304. [DOI] [PubMed] [Google Scholar]
- Jack JJB, Redman SJ, Wong K. The components of synaptic potentials evoked in cat spinal motoneurones by impulses in single group Ia afferents. J Physiol. 1981;321:65–96. doi: 10.1113/jphysiol.1981.sp013972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein M. Synaptic augmentation by 5-HT at rested Aplysia sensorimotor synapses: Independence of action potential prolongation. Neuron. 1994;13:159–166. doi: 10.1016/0896-6273(94)90466-9. [DOI] [PubMed] [Google Scholar]
- Koester HJ, Sakmann B. Calcium dynamics associated with action potentials in single nerve terminals of pyramidal cells in layer 2/3 of the young rat neocortex. J Physiol. 2000;529:625–646. doi: 10.1111/j.1469-7793.2000.00625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn H, Faber DS. Quantal analysis and synaptic efficacy in the CNS. Trends Neurosci. 1991;14:439–445. doi: 10.1016/0166-2236(91)90042-s. [DOI] [PubMed] [Google Scholar]
- Korn H, Faber DS, Burnod Y, Triller A. Regulation of efficacy at central synapses. J Neurosci. 1984;4:125–130. doi: 10.1523/JNEUROSCI.04-01-00125.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraushaar U, Jonas P. Efficacy and stability of quantal GABA release at a hippocampal interneuron-principal neuron synapse. J Neurosci. 2000;20:5594–5607. doi: 10.1523/JNEUROSCI.20-15-05594.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuno M. Quantal components of excitatory synaptic potentials in spinal motoneurones. J Physiol. 1964;175:81–99. doi: 10.1113/jphysiol.1964.sp007504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Streit J, Quadroni R, Lüscher H-R. Action potential propagation through embryonic dorsal root ganglion cells in culture. I. Influence of the cell morphology and propagation properties. J Neurophysiol. 1994;72:622–633. doi: 10.1152/jn.1994.72.2.622. [DOI] [PubMed] [Google Scholar]
- Maass W, Zador AM. Dynamic stochastic synapses as computational units. Neural Comput. 1999;11:903–917. doi: 10.1162/089976699300016494. [DOI] [PubMed] [Google Scholar]
- Magleby KL. Short-term changes in synaptic efficacy. In: Edelman GM, Gall WE, Cowan WM, editors. Synaptic Function. New York: John Wiley & Sons; 1987. pp. 21–56. [Google Scholar]
- Magleby KL, Zengel JE. Augmentation: a process that acts to increase transmitter release at the frog neuromuscular junction. J Physiol. 1976;257:449–470. doi: 10.1113/jphysiol.1976.sp011378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Lübke J, Frotscher M, Roth A, Sakmann B. Physiology and anatomy of synaptic connections between thick tufted pyramidal neurones in the developing rat neocortex. J Physiol. 1997;500:409–440. doi: 10.1113/jphysiol.1997.sp022031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matveev V, Wang X-J. Implications of all-or none synaptic transmission and short-term depression beyond vesicle depletion: a computational study. J Neurosci. 2000;20:1575–1588. doi: 10.1523/JNEUROSCI.20-04-01575.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donovan MJ, Rinzel J. Synaptic depression: a dynamic regulator of synaptic communication with varied functional roles. Trends Neurosci. 1997;20:431–433. doi: 10.1016/s0166-2236(97)01124-7. [DOI] [PubMed] [Google Scholar]
- Ohana O, Sakmann B. Transmitter release modulation in nerve terminals of rat neocortical pyramidal cells by intracellular calcium buffers. J Physiol. 1998;513:135–148. doi: 10.1111/j.1469-7793.1998.135by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quastel DMJ. The binomial model in fluctuation analysis of quantal neurotransmitter release. Biophys J. 1997;72:728–753. doi: 10.1016/s0006-3495(97)78709-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redman SJ. Quantal analysis of synaptic potentials in neurons of the central nervous system. Physiol Rev. 1990;70:165–198. doi: 10.1152/physrev.1990.70.1.165. [DOI] [PubMed] [Google Scholar]
- Reyes AD, Luján R, Rozov A, Burnashev N, Somogyi P, Sakmann B. Target-cell-specific facilitation and depression in neocortical circuits. Nat Neurosci. 1998;1:279–285. doi: 10.1038/1092. [DOI] [PubMed] [Google Scholar]
- Rozov A, Burnashev N, Sakmann B, Neher E. Transmitter release modulation by intracellular Ca2+ buffers in facilitating and depressing nerve terminals of pyramidal cells in layer 2/3 of the rat neocortex indicates a target cell-specific difference in presynaptic calcium dynamics. J Physiol. 2001;531:807–826. doi: 10.1111/j.1469-7793.2001.0807h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaba T, Neher E. Calmodulin mediates rapid recruitment of fast-releasing synaptic vesicles at a calyx-type synapse. Neuron. 2001;32:1119–1131. doi: 10.1016/s0896-6273(01)00543-8. [DOI] [PubMed] [Google Scholar]
- Sakaba T, Neher E. Direct modulation of synaptic vesicle priming by GABAB receptor activation at a glutamatergic synapse. Nature. 2003;424:775–778. doi: 10.1038/nature01859. [DOI] [PubMed] [Google Scholar]
- Scheuss V, Schneggenburger R, Neher E. Separation of presynaptic and postsynaptic contributions to depression by covariance analysis of successive EPSCs at the calyx of Held synapse. J Neurosci. 2002;22:728–739. doi: 10.1523/JNEUROSCI.22-03-00728.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simkus CRL, Stricker C. Analysis of mEPSCs recorded in layer II neurons of rat barrel cortex. J Physiol. 2002;545:509–520. doi: 10.1113/jphysiol.2002.022095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Wesseling JF. Activity-dependent modulation of the rate at which synaptic vesicles become available to undergo exocytosis. Neuron. 1998;21:415–424. doi: 10.1016/s0896-6273(00)80550-4. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Wesseling JF. Augmentation is a potentiation of the exocytotic process. Neuron. 1999;22:139–146. doi: 10.1016/s0896-6273(00)80685-6. [DOI] [PubMed] [Google Scholar]
- Stricker C, Daley D, Redman SJ. Statistical analysis of synaptic transmission: Model discrimination and confidence limits. Biophys J. 1994;67:532–547. doi: 10.1016/S0006-3495(94)80513-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stricker C, Field AC, Redman SJ. Statistical analysis of amplitude fluctuations in EPSCs evoked in rat CA1 pyramidal neurones in vitro. J Physiol. 1996;490:419–441. doi: 10.1113/jphysiol.1996.sp021155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart GJ, Sakmann B. Amplification of EPSPs by axosomatic sodium channels in neocortical pyramdial neurons. Neuron. 1995;15:1065–1076. doi: 10.1016/0896-6273(95)90095-0. [DOI] [PubMed] [Google Scholar]
- Swandulla D, Hans M, Zipser K, Augustine GJ. Role of residual calcium in synaptic depression and posttetanic potentiation: Fast and slow calcium signaling in nerve terminals. Neuron. 1991;7:915–926. doi: 10.1016/0896-6273(91)90337-y. [DOI] [PubMed] [Google Scholar]
- Tarczy-Hornoch K, Martin KAC, Jack JJB, Stratford KJ. Synaptic interaction between smooth and spiny neurones in layer 4 of cat visual cortex in vitro. J Physiol. 1998;508:351–363. doi: 10.1111/j.1469-7793.1998.351bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson AM, Bannister AP. Release-independent depression at pyramidal inputs onto specific cell targets: Dual recordings in slices of rat cortex. J Physiol. 1999;519:57–70. doi: 10.1111/j.1469-7793.1999.0057o.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson AM, Deuchars J, West DC. Large, deep layer pyramid-pyramid single axon EPSPs in slices of rat motor cortex display paired pulse and frequency-dependent depression, mediated presynaptically and self-facilitation, mediated postsynaptically. J Neurophysiol. 1993;70:2354–2369. doi: 10.1152/jn.1993.70.6.2354. [DOI] [PubMed] [Google Scholar]
- Tsodyks MV, Markram H. The neural code between neocortical pyramidal neurons depends on neurotransmitter release probability. Proc Natl Acad Sci U S A. 1997;94:719–723. doi: 10.1073/pnas.94.2.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldeck RF, Pereda AE, Faber DS. Properties and plasticity of paired-pulse depression at a central synapse. J Neurosci. 2000;20:5312–5320. doi: 10.1523/JNEUROSCI.20-14-05312.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L-Y, Kaczmarek LK. High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature. 1998;394:384–388. doi: 10.1038/28645. [DOI] [PubMed] [Google Scholar]
- Weis S, Schneggenburger R, Neher E. Properties of a model of Ca2+-dependent vesicle pool dynamics and short term synaptic depression. Biophys J. 1999;77:2418–2429. doi: 10.1016/S0006-3495(99)77079-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilts CH. Principles of Feedback Control. Reading MA, USA: Addison-Wesley; 1960. [Google Scholar]
- Zengel JE, Magleby KL, Horn JP, McAfee DA, Yarowsky PJ. Facilitation, augmentation, and potentiation of synaptic transmission at the superior cervical ganglion of the rabbit. J General Physiol. 1980;76:213–231. doi: 10.1085/jgp.76.2.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS. Changes in the statistics of transmitter release during facilitation. J Physiol. 1973;229:787–810. doi: 10.1113/jphysiol.1973.sp010167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]