

Abstract
ATP-sensitive potassium (KATP) channels comprise Kir6.2 and SUR subunits. The site at which ATP binds to mediate KATP channel inhibition lies on Kir6.2, but the potency of block is enhanced by coexpression with SUR1. To assess the structure of the ATP-binding site on Kir6.2, we used a range of adenine nucleotides as molecular measuring sticks to map the internal dimensions of the binding site. We compared their efficacy on Kir6.2–SUR1, and on a truncated Kir6.2 (Kir6.2ΔC) that expresses in the absence of SUR. We show here that SUR1 modifies the ATP-binding pocket of Kir6.2, by increasing the width of the groove that binds the phosphate tail of ATP, without changing the length of the groove, and by enhancing interaction with the adenine ring.
ATP-sensitive potassium (KATP) channels play key roles in many cells by linking cell metabolism to cellular electrical activity (Ashcroft, 1988). They are involved in glucose sensing in the pancreatic islets and brain, in the regulation of vascular smooth muscle tone, in ischaemic preconditioning and in seizure protection (Seino, 1999; Seino & Miki 2003). Metabolic regulation of KATP channel activity is thought to be mediated by changes in the intracellular concentrations of adenine nucleotides, which interact with both the pore-forming (Kir6.2) and regulatory (SURx) subunits of the channel. Binding of ATP and ADP to Kir6.2 produces channel inhibition (Tucker et al. 1997; Tanabe et al. 1999; Wang et al. 2002), whereas interaction of Mg-nucleotides with the nucleotide-binding domains (NBDs) of SUR results in stimulation of channel activity (Shyng et al. 1997; Gribble et al. 1998; Matsuo et al. 2000).
The structure of the inhibitory site on Kir6.2 is unknown, but it is likely to differ from that of classical ATP binding sites, because it has several unusual properties. In particular, Mg2+ is not required for the action of the nucleotide (Ashcroft & Kakei, 1989; Gribble et al. 1998); the site is extremely selective for the adenine base (Tucker et al. 1998); and addition of bulky groups to the end of the phosphate chain does not abolish the inhibitory effect of ATP (Ämmäläet al. 1991; Martin et al. 1998; Tanabe et al. 2000; Babenko & Bryan, 2001). Furthermore, although the inhibitory ATP-binding site lies on Kir6.2, coexpression with SUR enhances the potency of ATP block about 10-fold (Tucker et al. 1997).
We have used adenine nucleotides with different numbers of phosphate groups, and bulky additions to the phosphate tail, as molecular measuring sticks to map the gross architecture of the binding pocket of Kir6.2. We also use different modifications of the adenine ring to probe the structure of the nucleotide-binding site at the level of the adenine moiety. We make the assumption that the ability of adenine nucleotides to inhibit the KATP channel reflects differences in their ability to bind to Kir6.2, rather than differences in the ability of nucleotide binding to be transduced into channel closure.
Methods
Molecular biology
For excised patch studies, mouse Kir6.2 (GenBank D50581; Inagaki et al. 1995; Sakura et al. 1995) and rat SUR1 (GenBank L40624; Aguilar-Bryan et al. 1995) cDNAs were cloned into the pBF vector. A truncated form of Kir6.2 (Kir6.2ΔC), which lacks the C-terminal 36 amino acids and forms functional channels in the absence of SUR, was prepared as previously described (Tucker et al. 1997). An N-terminal deletion of Kir6.2 (Kir6.2ΔN) was prepared by using the polymerase chain reaction to replace residue 14 with an initiator methionine residue (Reimann et al. 1999). We refer to Kir6.2 with both N and C terminal truncations as Kir6.2ΔNΔC. Mutagenesis of the individual amino acids was performed using the altered sites II System (Promega). Capped mRNA was prepared using the mMESSAGE mMACHINE large scale in vitro transcription kit (Ambion, Austin, TX, USA) or the mRNA capping kit (Stratagene, La Jolla, CA, USA), as previously described (Gribble et al. 1997).
Oocyte collection
Xenopus laevis oocytes were isolated by partial ovariectomy from female frogs anaesthetized with 0.1% tricaine (3-aminobenzoic acid ethyl ester, 2 g l−1 added to the water). The incision was sutured and the animals were monitored during the recovery period before being returned to their tank. Once the incision had completely healed the second ovary was removed in a similar operation and the animal was then killed by decapitation whilst under anaesthesia. Immature stage V–VI oocytes were incubated for 60 min with 1.0 mg ml−1 collagenase (Sigma, type V) and manually defolliculated. Oocytes were either injected with ∼1 ng Kir6.2ΔC mRNA or coinjected with ∼0.1 ng Kir6.2 mRNA and ∼2 ng of mRNA encoding wild type or mutated SUR. The final injection volume was 50 nl oocyte−1. Isolated oocytes were maintained in Barth's solution and studied 1–7 days after injection (Gribble et al. 1997). All procedures conformed with the UK Animals (Scientific Procedures) Act 1986 and University of Oxford ethical committee guidelines.
Electrophysiology
Patch pipettes were pulled from thick-walled borosilicate glass and had resistances of 250–500 kΩ when filled with pipette solution. Macroscopic currents were recorded from giant excised inside-out patches at a holding potential of 0 mV and at 20–24°C (Gribble et al. 1997). Currents were evoked by repetitive 3 s voltage ramps from −110 mV to +100 mV and recorded using an EPC7 patch-clamp amplifier (List Electronik, Darmstadt, Germany). They were filtered at 0.2 kHz, digitized at 0.4 kHz using a Digidata 1200 Interface and analysed using pCLAMP software (Axon Instruments Inc., Union City, CA, USA).
The pipette (external) solution contained (mm): 140 KCl, 1.2 MgCl2, 2.6 CaCl2, 10 Hepes (pH 7.4 with KOH). The intracellular (bath) solution contained (mm): 110 KCl, 2 MgCl2, 1 CaCl2, 10 EGTA, 10 Hepes (pH 7.2 with KOH; final [K+]∼140 mm). The Mg-free solution contained (mm): 110 KCl, 2.6 CaCl2, 10 EDTA, 10 Hepes (pH 7.2 with KOH; final [K+]∼140 mm). Nucleotides were obtained from Sigma with the exception of purine triphosphate and methylATP (MeATP), which were custom synthesized by Jena Bioscience (07749 Jena, Germany). Solutions containing nucleotides were made up fresh each day and the pH subsequently readjusted if required. Rapid exchange of solutions was achieved by positioning the patch in the mouth of one of a series of adjacent inflow pipes placed in the bath.
Data analysis
The slope conductance was measured by fitting a straight line to the current–voltage relation between −20 and −100 mV. Conductance was measured from an average of five consecutive ramps in each solution. Responses to nucleotides were expressed relative to the conductance measured in control solution without added drugs or nucleotides. Concentration–response curves were constructed by expressing the conductance in the presence of drug (G) as a fraction of that in control solution (GC). The data were fitted to the Hill equation:
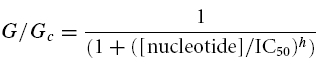 |
where [nucleotide] is the concentration of nucleotide, IC50 is the nucleotide concentration at which inhibition is half-maximal and h is the Hill coefficient. All data are presented as mean ± 1 standard error of the mean. Statistical significance was tested using Student's t test.
Results
In the presence of Mg-nucleotides, Kir6.2–SUR1 activity will be determined by the balance between their stimulatory and inhibitory effects, the former being mediated by SUR1 and the latter by Kir6.2 (Tucker et al. 1997). To evaluate the relative contributions of Kir6.2 and SUR to the nucleotide inhibition of KATP channel activity, we therefore carried out all experiments on Kir6.2-SUR1 in Mg2+-free solutions. Although neither KATP channel subunit trafficks to the surface membrane in the absence of its partner, deletion of the last 26–36 residues from the C-terminus of Kir6.2 (Kir6.2ΔC) removes an ER retention signal, and enables the surface expression of this subunit in the absence of SUR (Tucker et al. 1997; Zerangue et al. 1999). Thus we compared the effect of nucleotides on recombinant channels formed from Kir6.2ΔC alone, with those of channels composed of Kir6.2 plus SUR1. Previous studies have shown that the ATP sensitivity of Kir6.2, when coexpressed with SUR1, is not altered by truncation of the C-terminal 26–36 amino acids (Tucker et al. 1997).
As shown in Fig. 1, addition of a ribose moiety to the phosphate tail of ATP or ADP markedly reduced the potency of both nucleotides on Kir6.2ΔC. The IC50 was 1.8 ± 0.2 mm (n = 5) for ADP-ribose compared with 260 ± 20 μm (n = 6) for ADP (Tucker et al. 1998), and it was 2.3 ± 0.1 mm (n = 5) for ATP-ribose compared with 115 μm for ATP (Gribble et al. 1998). Addition of both a ribose and an adenine moiety to the phosphate tail of ADP (AP2A) largely abolished nucleotide block. Remarkably, however, addition of an adenosine moiety to a three (AP3A), or four (AP4A), phosphate tail partially, or almost completely, restored nucleotide block: the IC50 was 538 ± 21 μm (n = 5) for AP3A, and 190 ± 18 μm (n = 6) for AP4A (Fig. 1D), compared with 115 μm for ATP. This suggests that the ATP-binding pocket widens along the length of the phosphate tail, and that the adenosine moiety of AP4A may even lie outside the main binding pocket.
Figure 1. Effects of modifying the phosphate tail on Kir6.2ΔC currents.

A, macroscopic current recorded from an inside-out patch in response to a series of voltage ramps from −110 to +100 mV from oocytes expressing Kir6.2ΔC. 1 mm ATP-ribose or 1 mm ATP was added as indicated by the horizontal bars. The dashed line indicates the zero current potential. B–D, concentration–inhibition curves for adenine nucleotide inhibition of Kir6.2ΔC currents. B, ADP (□, n = 5) (data from Tucker et al. 1998) and ADP-ribose (•, n = 6); C, ATP (□, n = 11) (data from Gribble et al. 1998) and ATP-ribose (•, n = 5); D, A[P]nA. (AP2A: ▪, n = 5; AP3A: ○, n = 6; AP4A: ▴, n = 6) compared to ATP (□, n = 11). The macroscopic conductance in the presence of nucleotide (G) is expressed as a fraction of that measured in nucleotide-free solution (GC). The lines are the best fits to eqn (1). Mean nucleotide concentrations at which inhibition is half-maximal (IC50) are given in the text. Chemical structures of the nucleotides are shown to the right of the individual concentration–inhibition curves.
Because SUR1 enhances the potency of ATP ∼20-fold (IC50= 6 μm for Kir6.2–SUR1 and 115 μm for Kir6.2ΔC: Gribble et al. 1998), we examined whether the sulphonylurea receptor modifies the ATP-binding pocket of Kir6.2. Addition of a ribose to the phosphate tail of ATP or ADP decreased the potency of both nucleotides for Kir6.2–SUR1, but the effect was smaller than was found for Kir6.2ΔC. The IC50 was 44 ± 5 μm (n = 5) for ATP-ribose compared to 6 μm for ATP (Gribble et al. 1998), and 103 ± 4 μm (n = 5) for ADP-ribose compared with 64 ± 1 μm (n = 6) for ADP (Fig. 2). Furthermore, in contrast to what was observed for Kir6.2ΔC, where AP2A did not produce measurable block, there was only a small difference in the potencies of ADP and AP2A as Kir6.2–SUR1 blockers, the IC50 values being 64 μm and 164 ± 9 μm (n = 6), respectively (Fig. 2). These data therefore suggest that SUR may widen the ATP-binding pocket of Kir6.2, enabling it to accommodate the ribose or adenosine moiety more easily. As was the case for Kir6.2ΔC, however, greater inhibition of Kir6.2–SUR1 was produced by AP4A and AP3A than AP2A (Fig. 2D). A similar inhibitory potency for AP4A and ATP has also been observed for native KATP channels (Martin et al. 1998).
Figure 2. Effects of modifying the phosphate tail on Kir6.2–SUR1, Kir6.2▵N-SUR1 and Kir6.2▵N▵C currents.

A, macroscopic current recorded from an inside-out patch in response to a series of voltage ramps from −110 to +100 mV from oocytes expressing Kir6.2–SUR1. 0.1 mm ATP or 0.1 mm ATP-ribose was added as indicated by the horizontal bars. The dashed line indicates the zero current potential. The solution did not contain Mg2+. B–D, concentration–inhibition curves for adenine nucleotide inhibition of Kir6.2–SUR1 currents. B, ADP (•, n = 6) and ADP-ribose (□, n = 5); C, ATP (dashed line, n = 15) and ATP-ribose (▪, n =6); D, A[P]nA (AP2A: ▪, n = 6; AP3A: ○, n = 6; AP4A: ▴, n = 6). E and F, concentration–inhibition curves for adenine nucleotide inhibition of Kir6.2ΔN–SUR1 (E) and Kir6.2ΔNΔC (F) currents as indicated: ADP (□, n = 5) and ADP-ribose (▪, n = 5). The lines are the best fits to eqn (1). IC50 values are given in the text. The data for the ATP concentration–response curve are taken from our previous publication (Gribble et al. 1998).
There is evidence that the N-terminus is involved in coupling SUR1 to Kir6.2 (Babenko et al. 1999; Koster et al. 1999; Reimann et al. 1999; Babenko & Bryan, 2002). In particular, truncation of the N-terminus of Kir6.2 by as few as 14 amino acids (Kir6.2ΔN) results in an enhanced open probability and a lower block by tolbutamide when coexpressed with SUR1. We therefore measured concentration–inhibition curves for ADP and ADP-ribose for channels containing Kir6.2ΔN. As observed for ATP (Babenko et al. 1999; Koster et al. 1999; Reimann et al. 1999), truncation of the N-terminus reduces the inhibitory potency of ADP (IC50= 206 ± 18 μm, n = 5 for Kir6.2ΔN–SUR1 channels compared with 64 μm for Kir6.2–SUR1 channels), an effect that can be attributed to the increased open probability (Reimann et al. 1999). Importantly, however, there was no clear difference in the ability of ADP and ADP-ribose to block Kir6.2ΔN–SUR1 channels (Fig. 2E; IC50= 251 ± 27 μm, n = 5, n.s.). In contrast, ADP-ribose was significantly less potent at blocking Kir6.2ΔNΔC channels than ADP (Fig. 2F): IC50 values were 219 ± 19 μm (n = 5) and 1.92 ± 0.52 mm (n = 5; P < 0.05), respectively. Thus the data qualitatively resemble those observed for channels lacking an N-terminal truncation, and suggest that the effect of SUR1 on the nucleotide-binding pocket of Kir6.2 is not mediated (solely) via the first 14 residues of the protein.
We next examined the effect of changing the structure of the adenine ring. It has previously been shown that KATP channel inhibition is unique to ATP and that GTP, ITP, CTP and UTP are ineffective (Spruce et al. 1987; Ashcroft 1988; Tucker et al. 1998; Babenko & Bryan, 2001). Inosine differs from adenine at two positions: the presence of -NH rather than nitrogen at the 1′ position, and of oxygen rather than -NH2 at the 6′ position, of the purine ring (Fig. 3A). We therefore tested purine triphosphate (PuTP) and MeATP, which differ from adenine only at the 6′ position (Fig. 3A). PuTP (1 mm) was without effect on either Kir6.2ΔC or Kir6.2–SUR. In contrast, while MeATP had little effect on Kir6.2ΔC, it produced a significant block of Kir6.2–SUR1 (Fig. 3B and C). These data indicate that the 6′ position (rather than the 1′ position) of ATP is critical for ATP binding and further suggest that SUR may also modify the ATP-binding pocket of Kir6.2 at the level of the adenine moiety.
Figure 3. Effects of modifying the adenine ring structure.

A, chemical structures of adenine, inosine, methyladenine and purine. B, macroscopic currents recorded from inside-out patches in response to a series of voltage ramps from −110 to +100 mV from oocytes expressing either Kir6.2ΔC alone (above) or Kir6.2 plus SUR1 (below). 1 mm purine triphosphate (PuTP), methyladenine triphosphate (MeATP) or 0.1 mm ATP was added as indicated by the horizontal bars. The dashed line indicates the zero current potential. C, mean macroscopic slope conductance (G) in the presence of MeATP and PuTP as indicated, expressed as a fraction of the mean of that measured in nucleotide-free solution before and after nucleotide application (GC), for Kir6.2ΔC or Kir6.2–SUR1 as indicated.
Discussion
Addition of a ribose moiety to the end of the phosphate chain reduced the sensitivity of Kir6.2ΔC to ADP and ATP by 7-fold and 16-fold, respectively. This suggests the binding pocket of Kir6.2 is narrower than a ribose moiety at the level of both the β- and γ-phosphates of ATP, with the constriction being greater at the γ-phosphate. In contrast, addition of a ribose moiety to the terminal phosphate barely affected the sensitivity of Kir6.2–SUR1 to ADP (1.6-fold reduction) and produced a smaller (7.5-fold) decrease in the ATP sensitivity of Kir6.2–SUR1 than Kir6.2ΔC. These results are consistent with the idea that the presence of SUR1 results in an increase in the width of the phosphate-binding groove of Kir6.2 at the level of the both the β- and γ-phosphates. Interestingly, even in the presence of SUR1 the groove appears to be narrower at the level of the γ-phosphate than the β-phosphate.
It is striking that the length of the intervening phosphate chain had a marked effect on the inhibitory potency of A[P]nA, the IC50 for block of Kir6.2ΔC currents being 6 mm for AP2A, 540 μm for AP3A and 190 μm for AP4A. These results suggest that the phosphate chain of the nucleotide lies in a narrow groove that is not wide enough to accommodate the second adenosine group (∼8 Å) at the level of the β- or γ-phosphates. Since it can do so when four intervening phosphates are present (the inhibitory potencies of AP4A and ATP are similar), it is likely that the terminal phosphate of AP4A lies outside the binding pocket, in free solution. This would make the phosphate-binding groove between three and four phosphates long, or ∼11 Å (Fig. 4). The relative potencies of A[P]nA were similar for Kir6.2–SUR1 channels, being 164 μm for AP2A, 32 μm for AP3A and 13 μm for AP4A, compared with 6 μm for ATP. Thus SUR1 does not alter the length of the phosphate-binding groove, despite increasing its width.
Figure 4. Planar model of the ATP-binding groove.

A, planar model of the ATP-binding site of Kir6.2, with the phosphate chain of ATP lying in a long narrow groove. C, in the absence of SUR1, this groove is too narrow to accommodate a ribose moiety. B and D, coexpression with SUR1 produces a conformational change in Kir6.2 that increases the width of the groove (B), enabling nucleotides with bulky moieties attached to the γ-phosphate to be accommodated more easily (D). E and F, histogram of IC50 values for inhibition of Kir6.2ΔC (E) and Kir6.2—SUR1 (F) by the nucleotides indicated. The length of the phosphate chain is indicated.
Our data indicate that MeATP, but not PuTP, blocks Kir6.2–SUR1 currents. The nature of the group at the 6′ position of ATP therefore plays a key role in nucleotide binding: an -NH2 (as in ATP) or -CH3 (as in MeATP) group confers the ability to bind, whereas an -O (as in ITP) or -H (PuTP) does not. A recent molecular model for ATP binding proposes that the amine group of the purine ring makes a hydrogen bond with the backbone carbonyl of residue R301 (Trapp et al. 2003). This interaction will be abolished in PuTP and ITP, and could therefore account for their inability to bind. The differential sensitivities of Kir6.2ΔC and Kir6.2–SUR1 to MeATP is interesting. It is possible that SUR enhances ATP sensitivity by facilitating access of MeATP to R301 by widening the phosphate-binding groove that guards the entrance to the purine-binding pocket, as described above. Alternatively, SUR1 may influence the structure of the binding site for the purine ring directly, enabling MeATP to be accommodated more easily.
In conclusion, our data suggest that SUR1 can modify the structure of the ATP-binding site of Kir6.2, enabling ATP to bind more tightly. This effect of SUR1 is consistent with the fact that the ATP-binding site(s) lies on the outer side of the tetramer, where it can be embraced easily by SUR1. The ability of SUR1 to modify the nucleotide-binding pocket does not appear to involve the first 14 amino acids of Kir6.2, but a role for the remainder of the N terminus is not excluded.
Acknowledgments
We thank the Wellcome Trust, the Royal Society and the European Union (GrowBeta) for support. M.D. was a Robert Turner Visiting Scholar. F.M.A. is the Royal Society GlaxoSmithKline Research Professor.
References
- Aguilar-Bryan L, Nichols CG, Wechsler SW, Clement JP, Boyd AE, Gonzalez G, et al. Cloning of the β-cell high-affinity sulphonylurea receptor: a regulator of insulin secretion. Science. 1995;268:423–425. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- Ämmälä C, Bokvist K, Galt S, Rorsman P. Inhibition of ATP-regulated K+ channels by a photoactivatable ATP-analogue in mouse pancreatic β-cells. Biochim Biophys Acta. 1991;1092:347–349. doi: 10.1016/s0167-4889(97)90011-2. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM. Adenosine 5′-triphosphate-sensitive potassium channels. Annu Rev Neurosci. 1988;11:97–118. doi: 10.1146/annurev.ne.11.030188.000525. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM, Kakei M. ATP-sensitive K-channels: modulation by ATP and Mg2+ ions. J Physiol. 1989;416:349–367. doi: 10.1113/jphysiol.1989.sp017765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babenko AP, Bryan J. A conserved inhibitory and differential stimulatory action of nucleotides on K(IR)6.0/SUR complexes is essential for excitation-metabolism coupling by K(ATP) channels. J Biol Chem. 2001;276:49083–49092.. doi: 10.1074/jbc.M108763200. [DOI] [PubMed] [Google Scholar]
- Babenko AP, Bryan J. SUR-dependent modulation of KATP channels by an N-terminal KIR6.2 peptide. Defining intersubunit gating interactions. J Biol Chem. 2002;277:43997–44004. doi: 10.1074/jbc.M208085200. [DOI] [PubMed] [Google Scholar]
- Babenko AP, Gonzalez G, Bryan J. The N-terminus of Kir6.2 limits spontaneous bursting and modulates the ATP-inhibition of KATP channels. Biochem Biophys Res Commun. 1999;255:231–238. doi: 10.1006/bbrc.1999.0172. [DOI] [PubMed] [Google Scholar]
- Gribble FM, Ashfield R, Ämmäla C, Ashcroft F. Properties of cloned ATP-sensitive K+ currents expressed in Xenopus oocytes. J Physiol. 1997;498:87–98. doi: 10.1113/jphysiol.1997.sp021843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble FM, Tucker SJ, Haug T, Ashcroft FM. MgATP activates the β-cell KATP channel by interaction with its SUR1 subunit. Proc Natl Acad Sci U S A. 1998;95:7185–7190. doi: 10.1073/pnas.95.12.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement JP, Namba N, Inazawa J, Gonzalez G, et al. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 1995;270:1166–1170. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- Koster JC, Sha Q, Shyng S-L, Nichols CG. ATP inhibition of KATP channels: control of nucleotide sensitivity by the N-terminal domain of the Kir6.2 subunit. J Physiol. 1999;515:19–30. doi: 10.1111/j.1469-7793.1999.019ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin F, Pintor J, Rovira JM, Ripoll C, Miras-Portugal MT, Soria B. Intracellular diadenosine polyphosphates: a novel second messenger in stimulus-secretion coupling. FASEB J. 1998;12:1499–1506. doi: 10.1096/fasebj.12.14.1499. [DOI] [PubMed] [Google Scholar]
- Matsuo M, Tanabe K, Kioka N, Amachi T, Ueda K. Different binding properties and affinities for ATP and ADP among sulfonylurea receptor subtypes, SUR1, SUR2A, and SUR2B. J Biol Chem. 2000;275:28757–28763. doi: 10.1074/jbc.M004818200. [DOI] [PubMed] [Google Scholar]
- Reimann F, Tucker SJ, Proks P, Ashcroft FM. Involvement of the N-terminus of Kir6.2 in coupling to the sulphonylurea receptor. J Physiol. 1999;518:325–336. doi: 10.1111/j.1469-7793.1999.0325p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakura H, Ämmälä C, Smith PA, Gribble FM, Ashcroft FM. Cloning and functional expression of the cDNA encoding a novel ATP-sensitive potassium channel expressed in pancreatic β-cells, brain, heart and skeletal muscle. FEBS Lett. 1995;377:338–344. doi: 10.1016/0014-5793(95)01369-5. [DOI] [PubMed] [Google Scholar]
- Seino S. ATP-sensitive potassium channels: a model of heteromultimeric potassium channel/receptor assemblies. Annu Rev Physiol. 1999;61:337–362. doi: 10.1146/annurev.physiol.61.1.337. [DOI] [PubMed] [Google Scholar]
- Seino S, Mikit T. Physiological and pathophysiological roles of ATP-sensitive K+ channels. Prog Biophys Mol Biol. 2003;81:133–176. doi: 10.1016/s0079-6107(02)00053-6. [DOI] [PubMed] [Google Scholar]
- Shyng S, Ferrigni T, Nichols CG. Regulation of KATP channel activity by diazoxide and MgADP. Distinct functions of the two nucleotide binding folds of the sulfonylurea receptor. J General Physiol. 1997;110:643–654. doi: 10.1085/jgp.110.6.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruce AE, Standen NB, Stanfield PR. Studies of the unitary properties of adenosine-5'-triphosphate-regulated potassium channels of frog skeletal muscle. J Physiol. 1987;382:213–236. doi: 10.1113/jphysiol.1987.sp016364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe K, Tucker SJ, Matsuo M, Proks P, Ashcroft FM, Seino S, et al. Direct photoaffinity labeling of the Kir6.2 subunit of the ATP-sensitive K+ channel by 8-azido-ATP. J Biol Chem. 1999;274:3931–3933. doi: 10.1074/jbc.274.7.3931. [DOI] [PubMed] [Google Scholar]
- Tanabe K, Tucker SJ, Ashcroft FM, Proks P, Kioka N, Amachi T, Ueda K. Direct photoaffinity labeling of Kir6.2 by [gamma-(32)P]ATP-[gamma]4-azidoanilide. Biochem Biophys Res Commun. 2000;272:316–319. doi: 10.1006/bbrc.2000.2780. [DOI] [PubMed] [Google Scholar]
- Trapp S, Haider S, Jones P, Sansom MSP, Ashcroft FM. Identification of residues contributing to the ATP-binding site of Kir6.2. EMBO J. 2003;22:2093–2912. doi: 10.1093/emboj/cdg282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker SJ, Gribble FM, Proks P, Trapp S, Ryder TJ, Haug T, et al. Molecular determinants of KATP channel inhibition by ATP. EMBO J. 1998;17:3290–3296. doi: 10.1093/emboj/17.12.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM. Truncation of Kir6.2 produces ATP-sensitive K-channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–183. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- Wang C, Wang K, Wang W, Cui Y, Fan Z. Compromised ATP binding as a mechanism of phosphoinositide modulation of ATP-sensitive K+ channels. FEBS Lett. 2002;532:177–182. doi: 10.1016/s0014-5793(02)03671-2. [DOI] [PubMed] [Google Scholar]
- Zerangue N, Schwappach B, Jan YN, Jan LY. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K (ATP) channels. Neuron. 1999;22:537–548. doi: 10.1016/s0896-6273(00)80708-4. [DOI] [PubMed] [Google Scholar]