

Abstract
Cytoplasmic S4–S5 loops have been shown to be involved in fast inactivation of voltage-gated ion channels. We studied mutations in these loops and their potential cooperative effects in domains D3 (N1151C, A1152C, I1160C/A) and D4 (F1473C, L1482C/A) of the human skeletal muscle Na+ channel α-subunit (hNav1.4) using expression in tsA201 cells and the whole cell patch-clamp technique. All cysteine mutations were accessible to intracellularly applied sulfhydryl reagents which considerably destabilized fast inactivation. For different combinations of corresponding D3/D4 double mutations, fast inactivation could be almost completely removed. Thermodynamic cycle analysis indicated an additive effect for N1151C/F1473C and a significant cooperative effect for I1160/L1482 double mutations. Application of oxidizing reagents such as Cu-phenanthroline to link two cysteines via a disulfide bridge did not reveal evidence for a direct physical interaction of cysteines in D3 and D4. In addition to the pronounced alterations of fast inactivation, mutations of I1160 shifted steady-state activation in the hyperpolarizing direction and slowed the kinetics of both activation and deactivation. Sulfhydryl reagents had charge-dependent effects on I1160C suggesting interaction with negative charges in another protein region. We conclude that fast inactivation of the Na+ channel involves both S4–S5 loops in D3 and D4 in a cooperative manner. D3/S4–S5 also plays an important role in activation and deactivation.
Voltage-gated Na+ and K+ channels provide the basis for the generation and conduction of action potentials in nerve and muscle cells. In response to membrane depolarization, they open from the resting, closed state, and then Na+ channels and some types of K+ channels inactivate. Voltage-gated ion channels have a common tetrameric structure of homologous domains (D1–D4) each with six transmembrane segments (S1–S6). Whereas K+ channels are constituted by four identical domains, the about fourfold longer Na+ channel α-subunits have four homologous but distinct domains. All four S4 segments contain positively charged residues conferring voltage dependence to the channel protein (Hille, 2001). As first proposed by Armstrong & Bezanilla (1977), fast inactivation of voltage-gated channels is supposed to function in a ball and chain or hinged-lid fashion with a tethered inactivation particle occluding the internal mouth of the pore from the cytoplasmic side (Hoshi et al. 1990; West et al. 1992).
S4–S5 loops are short sequences of 15–20 amino acids adjacent to the voltage sensors and exposed to the cytoplasm. They have been shown previously to play an important role in fast inactivation of both K+ and Na+ channels, and have been proposed to contribute to the formation of a receptor site for the inactivation particle (Isacoff et al. 1991; Holmgren et al. 1996; Smith & Goldin, 1997; Lerche et al. 1997; Filatov et al. 1998; McPhee et al. 1998; Tang et al. 1998; reviewed by Catterall, 2000). A direct interaction between an inactivation peptide and S4–S5 in Shaker K+ channels (Holmgren et al. 1996) as well as between the proposed Na+ channel inactivation particle IFM and an amino acid in D3/S4–S5 of the rat brain type IIa Na+ channel (rNav1.2) (Smith & Goldin, 1997) was suggested by experiments using substituted, complementary charges. Structure–function analysis using cysteine mutagenesis of the D4/S4–S5 loop of the rat or human skeletal muscle Na+ channel confirmed an important role in fast inactivation, but revealed that most parts of D4/S4–S5 are still accessible when the channel is inactivated (Lerche et al. 1997; Filatov et al. 1998), arguing against a direct function of the proximal part of D4/S4–S5 as a receptor site for the IFM. In a more recent study with K+ channels, the receptor site for the inactivation ball was located in the central cavity of the pore formed by the S6 segments (Zhou et al. 2001).
An important question for Na+ channels in contrast to K+ channels is whether the distinct domains play different roles in channel gating. Previous studies revealed that in particular D4 plays a prominent role in fast inactivation, whereas D1 to D3 are more important for channel activation (Stuhmer et al. 1989; Chahine et al. 1994; Lerche et al. 1996; Chen et al. 1996; Mitrovic et al. 1998; Cha et al. 1999). Second, the question for both Na+ and K+ channels arises as to whether the voltage sensors and other protein structures in different domains act independently or in a cooperative manner. Several studies revealed evidence for cooperative interactions of the voltage sensors during activation of K+ channels (Tytgat & Hess, 1992; Schoppa et al. 1992; Bezanilla et al. 1994; Zagotta et al. 1994; Schoppa & Sigworth, 1998; Smith-Maxwell et al. 1998a,b; Ledwell & Aldrich, 1999; Mannuzzu & Isacoff, 2000) and recent work also points to such cooperative effects in Na+ channels (Chanda et al. 2004).
Our paper is focused on cooperative effects of S4–S5 loops in domains D3 and D4 with regard to fast inactivation of the Na+ channel. We chose three sites in D3/S4–S5 (N1151, A1152, I1160) and two in D4/S4–S5 (F1473, L1482) which either harbour mutations causing ion channel disorders (Ptacek et al. 1994; Wang et al. 1995; Mitrovic et al. 1996; Richmond et al. 1997; Fleischhauer et al. 1998) or have otherwise been shown to be important for fast inactivation of the channel (Smith & Goldin, 1997; Lerche et al. 1997; Filatov et al. 1998; McPhee et al. 1998; Alekov et al. 2001). According to an alignment using positively charged residues in S4 segments and five conserved residues in S4–S5 loops, N1151/F1473 as well as I1160/L1482 should be corresponding residues in D3/S4–S5 and D4/S4–S5, respectively (Fig. 1). These two pairs were used for cooperativity studies.
Figure 1. Model of the adult human muscle Na+ channel α-subunit (Nav1.4).

All mutations within the intracellular S4–S5 loops of domains D3 and D4 are shown. The lower part of the figure shows a putative alignment of the amino acid sequences in S4 and S4–S5 segments of domains D1–D4 of Nav1.4, the Shaker K+ channel (Kv1.1). Positive charges of the voltage sensors (S4 segments) are shown in italic, conserved residues in S4–S5 are underlined, and the residues mutated in this study are shown in bold.
Methods
Mutagenesis
Site-directed mutagenesis to introduce the different alanine and cysteine mutations in S4–S5 loops of domains D3 and D4 of the α-subunit of the human skeletal muscle Na+ channel (Nav1.4, gene: SCN4A) was performed using the Altered Sites system employing the plasmid vector pSELECT (Promega Corporation; Lerche et al. 1997; Alekov et al. 2001) or PCR-based strategies. All mutations were verified by dideoxynucleotide sequencing. Full length wild-type (WT) and mutant constructs of single and double mutations were assembled in the expression vectors pRC/CMV for transfection into the mammalian cell line tsA201.
Expression in tsA201 cells
Transfection of plasmids containing the cDNA of WT or mutant Na+ channel α-subunits into tsA201 cells was performed using a standard calcium phosphate method. A CD8-cDNA containing plasmid was cotransfected to recognize transfected cells using anti-CD8 antibody-coated microbeads (Dynabeads M450, Dynal, Oslo, Norway) (Lerche et al. 1997).
Electrophysiology
Na+ currents from mammalian cells were recorded using an EPC-7 patch clamp amplifier (List Electronics, Darmstadt, Germany), a Digidata 1200 digitizer and pCLAMP 6 data acquisition software (Axon Instruments, Union City, CA, USA). Leakage and capacitive currents were automatically subtracted using a pre-pulse protocol (−P/4). Currents were filtered at 3 kHz and digitized at 20 kHz, except for tail currents which were filtered at 10 kHz and digitized at 50 kHz. All measurements were performed at room temperature of 21–23°C.
The experiments were conducted in tsA201 cells using the whole cell patch clamp technique. Na+ currents of 2–15 nA were recorded from transfected tsA201 cells, at least 10 min after establishing the whole cell configuration. Borosilicate glass pipettes were fire polished with a final tip resistance of 0.8–1.2 MΩ when filled with internal recording solution (see below). We carefully checked that the maximal voltage error due to residual series resistance after up to 90% compensation was always ≤ 5 mV.
Solutions
For whole-cell recordings, the pipette solution contained (mm): 105 CsF, 35 NaCl, 10 EGTA, 10 Hepes, pH 7.4. The bathing solution contained (mm): 150 NaCl, 2 KCl, 1.5 CaCl2, 1 MgCl2, 10 Hepes, pH 7.4.
The sulfhydryl reagents [2-(trimethylammonium) ethyl]methanethio-sulfonate bromide and sodium(2-sulfonatoethyl)methanethiosulfonate (MTSET and MTSES, Toronto Research Chemicals, North York, Ontario, Canada) were kept in a H2O-stock solution (500 mm) at −20°C and diluted 1: 50 (MTSES) or 1: 200 (MTSET) to the pipette solution immediately before the experiments. Reagents with lower water solubility (MTS-1-MTS) were prepared as stocks in dimethylsulfoxide (DMSO) and stored at −20°C. An aliquot of the stock solution was thawed each day and used as in the case of water soluble reagents.
Cu(II)1,10-phenanthroline3 stock solution was prepared by dissolving Cu(II)SO4 and 1,10-phenanthroline to reach final concentrations of 150 and 500 mm, respectively, in a 4: 1 water/ethanol solution (Careaga & Falke, 1992). This stock solution was diluted 1: 1000 to the pipette solution at the time of use.
Data analysis
All data were analysed using a combination of pCLAMP, Microsoft Excel and Origin (OriginLab Inc., Northampton, MA, USA) software. For statistical evaluation, Student's t test was applied. All data are shown as means ± s.e.m., unless otherwise indicated.
The membrane was depolarized to various test potentials from a holding potential of −140 mV. The time course of inactivation was best fitted to a double exponential function yielding two time constants of inactivation. The weight of the second slower time constant was relatively small (0–25%) except for mutations containing F1473C-ES for which we found an amplitude of 50% for the slow component. To describe the slowing of inactivation in the Results section, only the fast time constant was therefore used, which was termed τh. Persistent Na+ currents (Iss, for ‘steady-state’ current), were determined at the end of 70 ms-lasting depolarizing pulses to 0 mV and are given relative to the initial peak current (Ipeak). Recovery from inactivation was recorded from a holding potential of −100 mV. Cells were depolarized to −20 mV for 100 ms to inactivate all Na+ channels and then repolarized to various recovery potentials for increasing duration. The time course of recovery from inactivation was best fitted to a double exponential function with an initial delay. For comparison among WT, mutants and effects of MTSET/MTSES, only the fast time constant was used, since the slow one had a relatively small weight (< 25%) and did not vary much compared to the fast one, which was termed τrec.
Steady-state inactivation was determined using 300 ms conditioning pulses to various potentials followed by the test pulse to −20 mV at which the peak current reflected the percentage of non-inactivated channels. Inactivation curves were fitted to a standard Boltzmann function:
with Imax being the maximal current amplitude, V½ the voltage of half-maximal inactivation, and kV the slope factor. The activation curve (conductance–voltage relationship) was derived from the current–voltage relationship by measuring the peak current at various step depolarizations from the holding potential of −140 mV, and also fit to a Boltzmann function:
with g =I/(V–Vrev) being the conductance, gmax the maximal conductance, Vrev the Na+ reversal potential, V½ the voltage of half-maximal activation, and kV the slope factor.
Na+ current traces were fitted to the Hodgkin-Huxley equation (m3h, Hodgkin & Huxley, 1952) to quantify the time course of activation for the WT and some mutants:
where τm is the activation time constant, τh the inactivation time constant, A an amplitude factor, t the time after onset of the depolarization, and t0 the delay to activation of the channel.
To characterize deactivation of WT and mutant Na+ channels, a short depolarizing pulse (0.26 ms to 70 mV), which activated all channels without causing significant inactivation, was followed by the test pulse to the indicated potentials. The deactivation time constant, τd, was obtained by a single exponential fit to the tail current decay.
Entry into, recovery from and steady-state slow inactivation were characterized using cumulative protocols (Alekov et al. 2001). To measure the entry, cells were held at −100 mV, depolarized to 0 mV for increasing duration, repolarized for 100 ms to −100 mV to let the channels recover from fast inactivation, and then depolarized again shortly for 3 ms to determine the fraction of slow inactivated channels without inducing further slow inactivation. The time course of slow inactivation was well fitted to a single exponential function. Recovery from slow inactivation was measured for different time points at −120 mV after a 30 s conditioning pulse to 0 mV. Curves were fitted to a double exponential function. For steady-state slow inactivation 30 s conditioning pulses starting at −140 mV and stepping by 10 mV increasingly to 10 mV were used, with each followed by a 20 ms hyperpolarization to −140 mV (to let the channels recover from fast inactivation) and the 5 ms test pulse to −20 mV. Curves were fitted to a standard Boltzmann function as for fast inactivation (see above).
Double mutant cycle analysis
The mutational effects in Nav1.4 were analysed through a thermodynamic description of site-specific effects. We calculated the change in free energy due to inactivation of the WT and mutant proteins according to
where R is the gas constant, T is the absolute temperature in kelvins, and Keq is the equilibrium constant, kon/koff, with kon and koff being the on and off rate constants for fast inactivation. For strong depolarizations, the steady-state level of the current relative to the peak current can be approximated as Iss/Ipeak≈koff/(koff+kon). Thus, Keq was calculated as Keq = kon/koff≈(1 − Iss/Ipeak)/(Iss/Ipeak) (McPhee et al. 1995). We used the steady-state current determined at 0 mV, 70 ms after onset of the depolarization for these calculations (see above). At this voltage, WT and all mutant channels were fully activated, as can be seen in Fig. 3. As a measure of the effect on fast inactivation of the site-directed mutation, we calculated the difference ΔΔG(WT→MUT) =ΔGMUT−ΔGWT. In the absence of coupling between two mutations, the change in the free energy associated with that transition upon a double mutation equals the sum of changes in free energy due to the single mutations. The deviation from additivity gives a measure of the change in the coupling between the two mutations due to the specified transition (Horovitz & Fersht, 1990), in our case fast inactivation:
Figure 3. Activation and steady-state inactivation parameters of the studied mutants.

A, voltage dependence of steady-state activation (open symbols), and steady-state inactivation (filled symbols), for I1160C (upper diagram) and for I1160C/L1482A (lower diagram) before and after modification by MTSES/MTSET. Symbols as indicated in the legend. Lines represent fits to standard Boltzmann functions. Values for V½ are given in Table 1. B, V½ of activation curve for all mutants relative to the WT. C, V½ of steady-state inactivation curve for all mutants relative to the WT. Symbols of B and C as indicated in the legend. All values are shown as means ± s.e.m., n = 3–15.
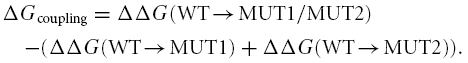 |
ΔG for WT and mutations was calculated for each cell separately. Standard deviations for ΔGcoupling were calculated by error propagation, i.e. the square root of the sum of the variances.
Results
To evaluate a potential cooperative effect of mutations in D3/S4–S5 and D4/S4–S5, we first studied the biophysical properties of the single mutations and second those of the different D3/D4 double mutants. We mainly introduced cysteines and modified those by intracellularly applied sulfhydryl reagents (Stauffer & Karlin, 1994), but we also introduced alanines at positions 1160 and 1482 to confirm the effects of cysteines in an independent experiment. Cooperative effects were analysed using thermodynamic mutant cycles. Third, we tested a direct physical interaction of D3/D4 cysteine double mutations using oxidizing reagents to induce disulfide bridges. In addition, we present interesting data on defects of channel activation and deactivation, and report results on slow inactivation.
Effects of single mutations in D3/S4–S5 and D4/S4–S5
Three cysteine mutations (N1151C, A1152C, and I1160C) and one alanine mutation (I1160A) were investigated in D3/S4–S5 using transient expression in tsA201 cells and the whole cell patch clamp technique. We aimed to characterize the accessibility of the introduced cysteines in the resting and inactivated states of the channel using MTSES. The negatively charged reagent has been shown to successfully modify cysteine mutants throughout D4/S4–S5; additional use of the positively charged MTSET did not yield further information (Lerche et al. 1997). Therefore, we here investigated charge-dependent effects, i.e. using both MTSES and MTSET, only at one site − I1160 – at which we observed large alterations in channel gating. Cysteines were modified by intracellularly applied MTSES (10 mm) or MTSET (2.5 mm) (denoted as C-ES or C-ET in the following). The reactions of sulfhydryl reagents with the different cysteine mutations were generally slow (Fig. 2, inset). Reaction rates were determined from the increase in persistent Na+ current or the slowing of inactivation. They ranged from 0.11 to 0.03 min−1. For L1482C the reaction was extremely slow with a rate of about 0.008 min−1.
Figure 2. Whole-cell currents of D3/S4–S5, D4/S4–S5 and D3/D4 double mutants.

Different pairs of mutations were investigated combining corresponding mutations at positions 1151 (D3/S4–S5) and 1473 (D4/S4–S5), as well as those at 1160 (D3/S4–S5) and 1482 (D4/S4–S5) (compare alignment in Fig. 1). Shown are representative raw current traces as recorded for each of the mutants expressed in tsA201 cells, in the absence and presence of MTSES (10 mm) or MTSET (2.5 mm), elicited by depolarizing the membrane between −105 mV and 67.5 mV in 7.5-mV steps from a holding potential of −140 mV. Note that – beside a distinct slowing of the inactivation time course and a persistent Na+ current at the end of the depolarization – the activation time course was slowed significantly for I1160C-ES, but not for I1160C-ET. The scale bars represent 1 nA (vertical) and 5 ms (horizontal), respectively. The inset shows the reaction of I1160C with MTSES monitored as the change with time of the persistent current relative to the peak current (Iss/Ipeak) 20 ms after onset of the depolarization, at two different holding potentials. At −20 mV (filled symbols), the reaction rate was 0.06 ± 0.03 min−1, whereas there was no detectable reaction at −140 mV (open symbols), n = 3–4.
The results in the absence and presence of sulfhydryl reagents revealed profound alterations of both activation and inactivation parameters, most pronounced for I1160C-ES or -ET. Families of raw current traces are shown in Fig. 2. It is noteworthy that I1160C could be modified by MTSES or MTSET only at a depolarized membrane but not when the cells were hyperpolarized (Fig. 2, inset), whereas modification of the other two cysteine mutations was independent of the membrane potential. This indicates a relative conformational change of D3/S4–S5 exposing C1160 to the cytoplasm upon membrane depolarization.
Steady-state activation and inactivation curves are presented in Fig. 3A. The activation curve was largely shifted in the hyperpolarizing direction, in particular for mutations at I1160, whereas steady-state inactivation was less affected (Fig. 3B and C, and Table 1).
Table 1.
Boltzmann parameters of activation and steady-state inactivation curves
| V½activ. (mV) | kactiv. (mV) | V½inactiv. (mV) | kinactiv. (mV) | |
|---|---|---|---|---|
| WT | −44.0 ± 1.2 | −5.8 ± 0.2 | −87.0 ± 1.3 | 5.3 ± 0.2 |
| N1151C | −48.5 ± 1.3* | −6.5 ± 0.1** | −89.9 ± 1.6 | 5.2 ± 0.1 |
| N1151C + MTSES | −53.7 ± 1.5*** | −6.0 ± 0.2 | −85.1 ± 1.2 | 5.1 ± 0.1 |
| A1152C | −44.6 ± 1.5 | −6.5 ± 0.3* | −83.8 ± 1.2 | 4.5 ± 0.2 |
| A1152C + MTSES | −50.4 ± 1.1** | −7.1 ± 0.1*** | −77.4 ± 0.6*** | 5.6 ± 0.2 |
| I1160C | −54.6 ± 1.5*** | −7.7 ± 0.3*** | −85.8 ± 1.1 | 5.2 ± 0.2 |
| I1160C + MTSES | −64.6 ± 0.7*** | −9.1 ± 0.7*** | −84.7 ± 1.3 | 5.9 ± 0.2** |
| I1160C + MTSET | −73.4 ± 3.3*** | −9.8 ± 0.7*** | −89.6 ± 2.6 | 8.3 ± 1.3 |
| I1160A | −57.1 ± 1.9*** | −6.3 ± 0.6 | −93.5 ± 0.9** | 5.2 ± 0.2 |
| F1473C | −39.3 ± 2.7 | −6.6 ± 0.4 | −74.0 ± 2.7*** | 7.5 ± 0.3*** |
| F1473C + MTSES | −52.9 ± 0.9** | −6.0 ± 0.4 | −74.1 ± 1.7*** | 7.9 ± 0.3*** |
| L1482C | −35.6 ± 1.3*** | −6.5 ± 0.2** | −77.4 ± 1.6*** | 6.3 ± 0.2** |
| L1482C + MTSES | −45.5 ± 1.3 | −6.2 ± 0.2 | −75.3 ± 0.8*** | 6.0 ± 0.1** |
| L1482A | −37.8 ± 2.9 | −7.4 ± 1.1* | −85.4 ± 1.2 | 7.4 ± 1.0** |
| N1151C/F1473C | −46.3 ± 1.4 | −6.7 ± 0.3* | −78.4 ± 1.1*** | 6.0 ± 0.3* |
| N51C/F73C + MTSES | −56.8 ± 0.4*** | −4.8 ± 0.2** | −68.6 ± 0.8*** | 5.9 ± 0.1 |
| I1160C/L1482C | −48.9 ± 1.6* | −6.8 ± 0.3* | −69.4 ± 1.6*** | 5.1 ± 0.3 |
| I60C/L82C + MTSES | −71.6 ± 2.0*** | −10.4 ± 0.5*** | −74.7 ± 2.2*** | 8.4 ± 1.1** |
| I1160C/L1482A | −56.4 ± 1.5*** | −5.6 ± 0.4 | −76.0 ± 1.6** | 6.3 ± 0.5* |
| I60C/L82A + MTSES | −60.5 ± 3.1*** | −8.0 ± 0.5*** | −74.2 ± 3.7** | 6.0 ± 0.7 |
| I60C/L82A + MTSET | −70.6 ± 0.7*** | −8.6 ± 1.5** | −86.1 ± 9.0 | 16.7 ± 6.8** |
| I1160A/L1482A | −54.8 ± 3.0** | −6.7 ± 0.4* | −90.9 ± 1.3 | 7.2 ± 0.6** |
Values for V½, the voltage of half-maximal activation and inactivation, and the slope factor kv were derived from Boltzmann fits to activation and inactivation curves (see Methods). n = 3–15. Significance levels are indicated as follows:
P < 0.05
P < 0.01
P < 0.001.
The time course of fast inactivation was slowed 3- to 4-fold for all mutations modified by the negatively charged MTSES without altering its voltage sensitivity, whereas kinetics were only slowed minimally for the positively charged I1160C-ET (Fig. 4A and B). The most prominent alteration of fast inactivation parameters was an increase of the persistent Na+ current. Seventy milliseconds after onset of the depolarization to 0 mV, we observed a persistent current of 20 ± 4% of the maximal peak current (Iss/Ipeak) for I1160C-ET and less pronounced increases for the other mutations compared to 0.4 ± 0.1% for wild-type (WT) channels (Fig. 4C). I1160A was the only mutation with a stabilizing effect on fast inactivation – left shift of the availability curve (Fig. 3C) and slowing of the recovery (Fig. 4D).
Figure 4. Kinetics and steady-state current of D3/S4–S5, D4/S4–S5 and D3/D4 double mutants.

A, fast inactivation time constant, τh, as a function of voltage for I1160C (upper diagram) and I1160C/L1482A (lower diagram) before and after modification by thiol reagents. Symbols as indicated in the legend. For comparison, τh at 0 mV (B), the sustained current relative to the peak current 70 ms after onset of the depolarization at 0 mV, Iss/Ipeak (C), and the time constant of recovery from inactivation at −100 mV, τrec (D), are shown. Symbols as indicated in the legend. All values are shown as means ± s.e.m., n = 3–13.
The mutations F1473C, L1482C and L1482A in D4/S4–S5 have been already investigated in previous studies of our group using different solutions (with 2 mm MTSES) and protocols at that time (Lerche et al. 1997; Alekov et al. 2001). Raw currents of these mutants as recorded in the study presented here are shown in Fig. 2, their gating parameters in Figs 3B and C and 4B–D, and Table 1. The results were very similar to those reported before. Additionally, a hyperpolarizing shift of steady state activation observed for F1473C-ES now reached significance. Using 10 mm MTSES, we now observed an increase in persistent sodium current for L1482C-ES compared to L1482C. The reaction of L1482C with MTSES was very slow (see above) and resulted in small effects (Figs 3B and C and 4B–D). Therefore, it could not be clearly determined if there was any voltage dependence of the availability of C1482. Also, a native cysteine might have become little accessible after mutating L1482 inducing these slowly progressing changes. However, as the observed effect was an increase in persistent Na+ current occurring as well for L1482C without reagents, L1482A and other mutations in that channel region (Lerche et al. 1997), we believe that it was specific for L1482C.
Effects on activation and deactivation mutating I1160
The left shift of the activation curve observed for many of the investigated mutants (Fig. 3B) might have arisen at least in part from the disrupted inactivation which can alter the apparent voltage dependence of activation. However, for mutants containing I1160C the activation, as well as deactivation, parameters were strongly affected – independent of the observed changes in inactivation – suggesting a direct effect on activation and deactivation. Beside the strong hyperpolarizing shift of the activation curve (up to −30 mV for I1160C-ET, Fig. 3A and B, and Table 1), we observed a distinct slowing of the activation time course for I1160C and I1160C-ES. Interestingly, this did not occur with introduction of a positive charge (I1160C-ET) (see raw currents in Fig. 2). The activation time constants, τm, for these mutations are shown in Fig. 5A over the whole voltage range investigated. For the more conservative mutant I1160A, a less pronounced but significant slowing of activation was also observed at −60 and −70 mV. The time course of activation was normal for all other single mutations in D3/S4–S5 or D4/S4–S5 (Fig. 2, time constants not shown). For the double mutations containing I1160C(-ES), we obtained similar results to those for I1160C(-ES) alone, but for those with I1160C-ET activation was again fast (Figs 2 and 5B).
Figure 5. Effects on the kinetics of activation and deactivation.

A, activation time constant, τm, as a function of voltage for I1160C(-ES/-ET), I1160A mutations. Activation was significantly slowed for I1160C in the absence and presence of MTSES, but not MTSET. B, voltage dependence of activation time constants for the double mutants I1160C(-ES/-ET)/L1482A and I1160C-ES/L1482C-ES. C, deactivation pulse protocol and representative recordings for WT and I1160C-ES. Scale bars represent 1 nA (vertical) and 1 ms (horizontal), respectively. D and E, time constant of deactivation, τd, for the single mutations (D) and the double mutations (E) as a function of voltage. Symbols as indicated in the legend (inset). Shown are means ± s.e.m., n = 3–6.
Representative examples of raw current traces of the deactivation time course are shown in Fig. 5C, and the deactivation time constants, τd, in Fig. 5D and E, for all examined single and double mutations, respectively. Deactivation was slowed up to 10-fold for all mutations containing I1160C(-ES/-ET) and for I1160A, regardless of whether the substituted modified or unmodified residue in position 1160 was neutral, or negatively or positively charged. In contrast to the differential effects of the introduced charges on the time course of activation, MTSET even had a slightly stronger effect on deactivation than MTSES. Thus, a positive charge in position 1160 prevents the slowing of activation kinetics observed for I1160C and I1160C-ES, but all other effects on gating were not qualitatively different for a positive charge in this position when compared to the polar cysteine residue or a negative charge.
Cooperativity of corresponding mutations in D3/S4–S5 and D4/S4–S5
Families of raw current traces for D3/D4 double mutations before and after modification by sulfhydryl reagents are shown in Fig. 2 in comparison to the respective single mutations. For N1151C/F1473C we observed similar effects as for F1473C, in particular a strong slowing of fast inactivation. None of the alterations in channel gating indicated a cooperative action of both mutations (Figs 3B and C and 4B–D).
In contrast, for the different combinations of double mutations in positions 1160/1482, the most obvious changes were much larger persistent Na+ currents than expected for a simple additive effect, suggesting a cooperative effect of mutations in those two positions. For the modified double mutations, fast inactivation could be almost completely removed (Figs 2 and 4C). The coupling energies for each pair of mutations were determined by calculating the change in the free energy of the channel state due to inactivation for all single mutations as well as for the double mutations and the differences between them in thermodynamic mutant cycles (see Methods). The coupling energies for all three pairs of mutations in the absence of sulfhydryl reagents were significantly different from zero (P < 0.05 or < 0.01, respectively, Fig. 6) indicating that these double mutations act interdependently with regard to their effects on fast inactivation.
Figure 6. Thermodynamic mutant cycles.

We used the persistent Na+ current, Iss/Ipeak (indicated for each mutation in brackets, means ± s.e.m.) to calculate the free energy ΔG =–RT ln(kon/koff). The coupling energy,
is shown in the middle of each cycle revealing an additive effect for N1151C/F1473C, and a significant cooperative effect for I1160C/L1482C (P < 0.01), I1160C/L1482A (P < 0.05) and I1160A/L1482A (P < 0.05). Shown are means ± s.d., n = 3–10.
The other gating parameters of 1160/1482 double mutations were altered in a similar way as already observed for the respective single mutations. Activation curves were largely shifted in the hyperpolarizing direction as for I1160C/A mutations, and the inactivation curves towards more depolarized membrane potentials as seen for L1482C/A mutations (Fig. 3A–C). The kinetics of fast inactivation were slowed and recovery from inactivation was accelerated (Fig. 4B and D). These changes point to a considerably destabilized fast inactivated state of the double mutant channel.
Exploring a direct interaction of D3/S4–S5 and D4/S4–S5 using cysteine double mutations, Cu-phenanthroline and MTS–linkers
The interaction of two protein regions can be examined by double cysteine mutagenesis, when experiments are designed to link the two cysteine side chains either directly via a disulfide bridge using oxidizing reagents such as Cu-phenanthroline, or with sulfhydryl reagents having a reactive MTS group at both ends (MTS-1-MTS). We performed such experiments with both pairs of combined cysteine mutations in D3/S4–S5 and D4/S4–S5 (N1151C/F1473C, I1160C/L1482C) using Cu-phenanthroline (0.15 mm) or H2O2 (up to 1%) as oxidizing reagents, and MTS-1-MTS as a sulfhydryl linker. All substances were applied intracellularly, i.e. added to the pipette solution in whole cell experiments.
For N1151C/F1473C, we did not observe significant effects on channel gating using either of the oxidizing substances. Applying the sulfhydryl linker, a slowing of the inactivation time course occurred, similar to that observed after modification by MTSES. There was no effect on channel gating pointing to a direct interaction of both cysteines via the MTS-linker, such as a channel block or an abolished inactivation that might have been expected for a severe conformational change of the protein implicated by the formation of a new disulfide bond (results not shown).
When MTS-1-MTS was applied to I1160C/L1482C there was also no substantial effect on channel gating other than those already known for MTSES or MTSET modification. In contrast, when Cu-phenanthroline (150 µm) was applied to this double mutation, we observed an inhibition of Na+ current at highly positive membrane potentials which developed in a use-dependent manner. The Cu-phenanthroline-induced inhibition was reversible without using reducing reagents, in the presence of Cu-phenanthroline, contrary to what would have been expected for the formation of a disulfide bond between the two introduced cysteines. Moreover, another oxidizing reagent, H2O2, was not able to induce current inhibition in contrast to Cu-phenanthroline. These results clearly indicate that there is no evidence for a direct interaction of both cysteine residues and that the observed current inhibition is due to a channel block by Cu-phenanthroline. However, when we examined the respective single mutations, I1160C and L1482C, we only observed a very weak block for both mutations − 14.3 ± 3.9% of blocked channels at +60 mV for I1160C, 18.2 ± 3.2% for L1482C compared with 67.9 ± 5.3% for the I1160C/L1482C (results for Cu-phenanthroline not shown). Thus, the double mutation strongly enhanced channel block which underlines the cooperative action of both residues already described for fast inactivation.
Slow inactivation
Since slow inactivation was affected by mutations L1482C/A in D4/S4–S5 as reported previously by our group (Alekov et al. 2001), we also looked for changes in slow inactivation of mutation I1160C at the corresponding site in D3/S4–S5 and the double mutation I1160C/L1482C. All mutations had small but significant effects on slow inactivation. I1160C shifted the steady-state availability curve by −7 mV (and by −10 mV after MTSES application) and accelerated the entry (at 0 mV) into slow inactivation by 1.8-fold. L1482C increased the steepness of the availability curve and slowed recovery from slow inactivation (at −120 mV) by 6.4-fold. Consequently, for the double mutation we observed a negatively shifted (−4 mV) and steeper availability curve, a 1.8-fold accelerated entry and a 2.5-fold slowed recovery, thus a consistent stabilization of slow inactivation. Application of MTSES did not substantially alter these results (results for slow inactivation are not shown).
Discussion
Although the four different domains of the Na+ channel certainly are specialized in their function, and in particular the voltage sensor D4/S4 is most important for fast inactivation in contrast to D1–D3/S4 (see Introduction), we here present evidence that S4–S5 loops of both D3 and D4 play crucial roles in fast inactivation of the channel and – more importantly – that the distal parts of these regions act in a cooperative manner. Mutations in both regions also affected voltage-dependent activation. However, most of the observed effects were relatively small compared to the alterations of fast inactivation, except for mutations at position 1160 affecting profoundly the voltage dependence and kinetics of activation and deactivation. In the following, we will discuss the potential molecular functions of these regions with regard to their relations to other parts of the channel based on our results and previous structure–function studies.
Potential role of D3/and D4/S4–S5 loops in fast inactivation and their cooperativity
In a previous study, Smith & Goldin (1997) examined the D3/S4–S5 loop and interaction with the IFM in rNav1.2 by introducing glutamines and charged residues. Major effects on inactivation were only observed for A1329 (corresponding to A1152 in hNav1.4) for which an interaction with the IFM was suggested by introduction of complementary charges. Our results for A1152C(-ES) largely agree with these previous investigations concerning the voltage dependence and kinetics of most gating parameters. With the other two mutations investigated, our results extend the previous work as we observed also for them significant alterations in channel gating, in particular for mutations of I1160. However, as will be discussed below, the voltage-independent accessibility of A1152C may argue against a direct contribution of A1152 to a receptor site of the IFM, as was suggested by the electrostatic interactions observed in the previous study.
We previously examined all amino acids of the D4/S4–S5 loop of hNav1.4 by systematic cysteine mutagenesis (Lerche et al. 1997). Since all proximal residues were accessible to sulfhydryl reagents with approximately the same kinetics at depolarized membrane potentials (i.e. inactivated channel) as at hyperpolarized potentials (i.e. resting closed channel), we concluded that they do not directly participate in a receptor site for the putative inactivation particle IFM. The results obtained in the study presented here confirm this impression also for D3/S4–S5, as all three residues in D3/S4–S5 were accessible in the inactivated state of the channel, two of them with essentially the same reaction kinetics as in the resting state, while I1160C was not accessible at a hyperpolarized membrane. However, as the reaction rates in the present study were very slow, this hypothesis has to be considered with caution. The examined cysteines could also be partially covered by different conformational changes in the resting and inactivated states yielding similarly slow reaction rates at both hyperpolarized and depolarized membrane potentials. Thus, the voltage-independent reaction rates do not exclude these residues as a receptor site for the IFM.
There also remains uncertainty for two distal residues in D4/S4–S5: L1482, since we could not examine voltage-dependent accessibility of C1482 properly – due to the very slow reaction rate and the small effect on inactivation – and N1484, since a cysteine mutation at this site led to a non-functional channel (Lerche et al. 1997; Filatov et al. 1998). N1484 is particularly important, as substitution of an alanine in the corresponding position of a rat brain Na+ channel removed fast inactivation almost completely (McPhee et al. 1998). Hence, this distal part of D4/S4–S5 might directly participate in a receptor site for the IFM.
Our data presented here demonstrate the importance of studying gating parameters in Na+ channels not only in isolated mutations of one domain but also their cooperativity in different domains. Persistent Na+ currents for the single mutations were relatively small and would not have indicated the crucial role of these loops for fast inactivation that was shown by the double mutations in positions 1160 and 1482. The persistent current increased from 1.2 ± 0.2% and 0.7 ± 0.1% for the single cysteine mutants to 23.7 ± 2% when these mutations were combined, strongly pointing to a cooperativity of the mutations concerning destabilization of the inactivated state. We attempted to quantify this cooperativity by applying a double mutant cycle analysis. Using this analysis we got a quantitative estimation of the interdependency of the I1160 and L1482 action with regard to inactivation, but no information if this is due to a direct or indirect interaction. Values of coupling energies reported in the literature for residue pairs which have been proven to be in direct contact by revealing their protein crystal structure vary between 1.3 and 7 kcal mol−1 (Schreiber & Fersht, 1995; Goldman et al. 1997; Dall'Acqua et al. 1998). For different paired substitutions of I1160 and L1482, we obtained coupling energies in the range 1.2–1.6 kcal mol−1, similar with what has been found for residues forming either a hydrogen bond or 2–3 van der Waals contacts (Goldman et al. 1997; Dall'Acqua et al. 1998).
Our experiments with Cu-phenanthroline also hint at a cooperative action of I1160C and L1482C, since channel block by Cu-phenanthroline was strongly enhanced in the double mutant. However, in multiple cross-linking experiments we could not find any conclusive evidence for a direct interaction of S4–S5 loops in D3 and D4. Therefore, the 1.2–1.6 kcal mol−1 coupling energy most probably does not result from a direct but rather an indirect interaction of I1160 with L1482 through other protein regions. How this interdependency of these two regions functions on a molecular level remains a matter of speculation. One possibility could be an interaction of S4–S5 with S6 segments in each domain and a cooperation of S6 segments in forming the receptor site for the inactivation particle.
Potential role of I1160 in channel activation and deactivation
A striking additional result of this study is the strong alteration of activation and deactivation gating by mutations in position 1160 at the C-terminal end of D3/S4–S5 indicating an important role of this residue for those processes. All mutations destabilized the resting closed relative to the open state by shifting the activation curve in the hyperpolarizing direction and I1160C (-ES/-ET) also reduced the apparent gating charge, since the slopes of the activation curves were significantly decreased (−1e, P < 0.0001, for cysteine alone, −1.6e, P < 0.00001, for I1160C-ES, and −1.8e, P < 0.0001, for I1160C-ET, respectively). These results indicate that a position of the voltage sensor or of the activation gate towards the activated state is favoured by the mutations and their modification by positively and negatively charged reagents. The effects on the voltage dependence of activation and also on deactivation kinetics were neither related to charge, as I1160C-ET exhibited the strongest negative shift in steady-state activation and slowed deactivation even slightly more than I1160C-ES, nor related to the size of the introduced residue, as also I1160A showed the same effects. Thus, the bulky hydrophobic isoleucine seems to exert a specific function important for these gating parameters.
In contrast, activation kinetics were slowed for all single and double mutants containing the polar cysteine or the negatively charged C-ES at position 1160, but not for the positive I1160C-ET for which activation kinetics were not significantly different from the WT. For the neutral alanine, kinetics were only a little slowed. The differential effects with introduced charges on the voltage dependence and the kinetics of activation indicate a dissociation of the two gating processes. The slowed activation kinetics could be caused either by slowed movement of the voltage sensors or by a slowed opening of the gate. In the latter case, this might suggest partial uncoupling of voltage sensor movement from the opening of the gate for I1160C(-ES).
The dissociation of effects between MTSES and MTSET furthermore hints to charge-dependent interactions of the introduced mutations with another protein region. For Shaker K+ channels and hyperpolarization-activated pacemaker channels recent studies revealed strong evidence for a direct interaction of S4–S5 with S6 segments thereby coupling voltage sensor movement to the opening of the pore, i.e. to activation of the channels (Chen et al. 2001; Lu et al. 2002; Decher et al. 2004). An alignment of these interacting regions from the Shaker K+ channel (Lu et al. 2002) and the corresponding regions of hNav1.4 reveal two negative charges in D3/S6 (D1296) and D4/S6 (E1601) that may potentially interact with I1160C-ES/-ET. In this regard we have to consider that interaction of I1160 with S6 in the native channel would have to be primarily hydrophobic, as isoleucine is a strongly hydrophobic residue. However, the polar cysteine and more particularly the negative charge of I1160C-ES could disrupt this hydrophobic interaction in the WT channel by a charge-dependent repulsion involving D1296 or E1601. This could be compensated for by a charge-dependent attraction of I1160C-ET with one of these residues and might therefore be responsible for the observed differences in kinetics by the introduction of distinct charges. The relatively small effects on activation kinetics upon introduction of the neutral alanine could be explained by less intense hydrophobic interactions compared to the native isoleucine.
In summary, our results reveal a crucial cooperative role of D3/S4–S5 and D4/S4–S5 for fast inactivation of the voltage-gated Na+ channel, even if these two loops may not directly contribute to a receptor site for the inactivation particle IFM and not directly interact with each other. I1160 is intimately involved in channel activation and deactivation and might interact with another protein region, which could be the S6 segment.
Acknowledgments
We thank A. Bellan-Koch for technical assistance and Drs R. Horn, N. Mitrovic and W. Peter for helpful discussion on the manuscript. This work was supported by the Deutsche Forschungsgemeinschaft (DFG, Le1030/5-1,/5-2). H.L. is a Heisenberg fellow of the DFG.
References
- Alekov AK, Peter W, Mitrovic N, Lehmann-Horn F, Lerche H. Two mutations in the IV/S4–S5 segment of the human skeletal muscle Na+ channel disrupt fast and enhance slow inactivation. Neurosci Lett. 2001;306:173–176. doi: 10.1016/s0304-3940(01)01895-x. 10.1016/S0304-3940(01)01895-X. [DOI] [PubMed] [Google Scholar]
- Armstrong CM, Bezanilla F. Inactivation of the sodium channel. II. Gating current experiments. J General Physiol. 1977;70:567–590. doi: 10.1085/jgp.70.5.567. 10.1085/jgp.70.5.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla F, Perozo E, Stefani E. Gating of Shaker K+ channels. II. The components of gating currents and a model of channel activation. Biophys J. 1994;66:1011–1021. doi: 10.1016/S0006-3495(94)80882-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Careaga CL, Falke JJ. Thermal motions of surface alpha-helices in the d-galactose chemosensory receptor. Detection by disulfide trapping. J Mol Biol. 1992;226:1219–1235. doi: 10.1016/0022-2836(92)91063-u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- Cha A, Ruben PC, George AL, Jr, Fujimoto E, Bezanilla F. Voltage sensors in domains III and IV, but not I and II, are immobilized by Na+ channel fast inactivation. Neuron. 1999;22:73–87. doi: 10.1016/s0896-6273(00)80680-7. [DOI] [PubMed] [Google Scholar]
- Chahine M, George AL, Jr, Zhou M, Ji S, Sun W, Barchi RL, Horn R. Sodium channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron. 1994;12:281–294. doi: 10.1016/0896-6273(94)90271-2. [DOI] [PubMed] [Google Scholar]
- Chanda B, Asamoah OK, Bezanilla F. Coupling interactions between voltage sensors of the sodium channel as revealed by site-specific measurements. J General Physiol. 2004;123:217–230. doi: 10.1085/jgp.200308971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Mitcheson J, Tristani-Firouzi M, Lin M, Sanguinetti M. The S4-S5 linker couples voltage sensing and activation of pacemaker channels. Proc Natl Acad Sci U S A. 2001;98:11277–11282. doi: 10.1073/pnas.201250598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LQ, Santarelli V, Horn R, Kallen RG. A unique role for the S4 segment of domain 4 in the inactivation of sodium channels. J General Physiol. 1996;108:549–556. doi: 10.1085/jgp.108.6.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dall'Acqua W, Goldman ER, Lin W, Teng C, Tsuchiya D, Li H, et al. A mutational analysis of binding interactions in an antigen-antibody protein-protein complex. Biochemistry. 1998;37:7981–7991. doi: 10.1021/bi980148j. [DOI] [PubMed] [Google Scholar]
- Decher N, Chen J, Sanguinetti MC. Voltage-dependent gating of hyperpolarization-activated, cyclic nucleotide-gated pacemaker channels: molecular coupling between the S4–S5 and C-linkers. J Biol Chem. 2004;279:13859–13865. doi: 10.1074/jbc.M313704200. 10.1074/jbc.M313704200. [DOI] [PubMed] [Google Scholar]
- Filatov GN, Nguyen TP, Kraner SD, Barchi RL. Inactivation and secondary structure in the D4/S4–5 region of the SkM1 sodium channel. J General Physiol. 1998;111:703–715. doi: 10.1085/jgp.111.6.703. 10.1085/jgp.111.6.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischhauer R, Mitrovic N, Deymeer F, Lehmann-Horn F, Lerche H. Effects of temperature and mexiletine on the F1473S Na+ channel mutation causing paramyotonia congenita. Pflugers Arch. 1998;436:757–765. doi: 10.1007/s004240050699. 10.1007/s004240050699. [DOI] [PubMed] [Google Scholar]
- Goldman ER, Dall'Acqua W, Braden BC, Mariuzza RA. Analysis of binding interactions in an idiotope-antiidiotope protein-protein complex by double mutant cycles. Biochemistry. 1997;36:49–56. doi: 10.1021/bi961769k. [DOI] [PubMed] [Google Scholar]
- Hille B. Ionic Channels in Excitable Membranes. 3. Sunderland MA USA: Sinauer Associates, Inc; 2001. [Google Scholar]
- Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren M, Jurman ME, Yellen GN. N-type inactivation and the S4–S5 region of the Shaker K+ channel. J General Physiol. 1996;108:195–206. doi: 10.1085/jgp.108.3.195. 10.1085/jgp.108.3.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horovitz A, Fersht AR. Strategy for analysing the co-operativity of intramolecular interactions in peptides and proteins. J Mol Biol. 1990;214:613–617. doi: 10.1016/0022-2836(90)90275-Q. 10.1016/0022-2836(90)90275-Q. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Isacoff EY, Jan YN, Jan LY. Putative receptor for the cytoplasmic inactivation gate in the Shaker K+ channel. Nature. 1991;353:86–90. doi: 10.1038/353086a0. 10.1038/353086a0. [DOI] [PubMed] [Google Scholar]
- Ledwell JL, Aldrich RW. Mutations in the S4 region isolate the final voltage-dependent cooperative step in potassium channel activation. J General Physiol. 1999;113:389–414. doi: 10.1085/jgp.113.3.389. 10.1085/jgp.113.3.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerche H, Mitrovic N, Dubowitz V, Lehmann-Horn F. Paramyotonia congenita: the R1448P Na+ channel mutation in adult human skeletal muscle. Ann Neurol. 1996;39:599–608. doi: 10.1002/ana.410390509. [DOI] [PubMed] [Google Scholar]
- Lerche H, Peter W, Fleischhauer R, Pika-Hartlaub U, Malina T, Mitrovic N, Lehmann-Horn F. Role in fast inactivation of the IV/S4–S5 loop of the human muscle Na+ channel probed by cysteine mutagenesis. J Physiol. 1997;505:345–352. doi: 10.1111/j.1469-7793.1997.345bb.x. 10.1111/j.1469-7793.1997.345bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Klem AM, Ramu YC. Coupling between voltage sensors and activation gate in voltage-gated K+ channels. J General Physiol. 2002;120:663–676. doi: 10.1085/jgp.20028696. 10.1085/jgp.20028696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhee JC, Ragsdale DS, Scheuer T, Catterall WA. A critical role for transmembrane segment IVS6 of the sodium channel alpha subunit in fast inactivation. J Biol Chem. 1995;270:12025–12034. doi: 10.1074/jbc.270.20.12025. 10.1074/jbc.270.20.12025. [DOI] [PubMed] [Google Scholar]
- McPhee JC, Ragsdale DS, Scheuer T, Catterall WA. A critical role for the S4–S5 intracellular loop in domain IV of the sodium channel alpha-subunit in fast inactivation. J Biol Chem. 1998;273:1121–1129. doi: 10.1074/jbc.273.2.1121. 10.1074/jbc.273.2.1121. [DOI] [PubMed] [Google Scholar]
- Mannuzzu LM, Isacoff EY. Independence and cooperativity in rearrangements of a potassium channel voltage sensor revealed by single subunit fluorescence. J General Physiol. 2000;115:257–268. doi: 10.1085/jgp.115.3.257. 10.1085/jgp.115.3.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrovic N, George AL, Jr, Horn R. Independent versus coupled inactivation in sodium channels. Role of the domain 2, S4 segment. J General Physiol. 1998;111:451–462. doi: 10.1085/jgp.111.3.451. 10.1085/jgp.111.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrovic N, Lerche H, Heine R, Fleischhauer R, Pika-Hartlaub U, Hartlaub U, et al. Role in fast inactivation of conserved amino acids in the IV/S4–S5 loop of the human muscle Na+ channel. Neurosci Lett. 1996;214:9–12. doi: 10.1016/0304-3940(96)12866-4. [DOI] [PubMed] [Google Scholar]
- Ptacek LJ, Tawil R, Griggs RC, Meola G, McManis P, Barohn RJ, et al. Sodium channel mutations in acetazolamide-responsive myotonia congenita, paramyotonia congenita, and hyperkalemic periodic paralysis. Neurology. 1994;44:1500–1503. doi: 10.1212/wnl.44.8.1500. [DOI] [PubMed] [Google Scholar]
- Richmond JE, VanDeCarr D, Featherstone DE, George AL, Jr, Ruben PC. Defective fast inactivation recovery and deactivation account for sodium channel myotonia in the I1160V mutant. Biophys J. 1997;73:1896–1903. doi: 10.1016/S0006-3495(97)78220-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoppa NE, McCormack K, Tanouye MA, Sigworth FJ. The size of gating charge in wild-type and mutant Shaker potassium channels. Science. 1992;255:1712–1715. doi: 10.1126/science.1553560. [DOI] [PubMed] [Google Scholar]
- Schoppa NE, Sigworth FJ. Activation of Shaker potassium channels. III. An activation gating model for wild-type and V2 mutant channels. J General Physiol. 1998;111:313–342. doi: 10.1085/jgp.111.2.313. 10.1085/jgp.111.2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber G, Fersht AR. Energetics of protein–protein interactions: analysis of the barnase–barstar interface by single mutations and double mutant cycles. J Mol Biol. 1995;248:478–486. doi: 10.1016/s0022-2836(95)80064-6. 10.1006/jmbi.1995.0235. [DOI] [PubMed] [Google Scholar]
- Smith MR, Goldin AL. Interaction between the sodium channel inactivation linker and domain III, S4–S5. Biophys J. 1997;73:1885–1895. doi: 10.1016/S0006-3495(97)78219-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Maxwell CJ, Ledwell JL, Aldrich RW. Role of the S4 in cooperativity of voltage-dependent potassium channel activation. J General Physiol. 1998a;111:399–420. doi: 10.1085/jgp.111.3.399. 10.1085/jgp.111.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Maxwell CJ, Ledwell JL, Aldrich RW. Uncharged S4 residues and cooperativity in voltage-dependent potassium channel activation. J General Physiol. 1998b;111:421–439. doi: 10.1085/jgp.111.3.421. 10.1085/jgp.111.3.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauffer DA, Karlin A. Electrostatic potential of the acetylcholine binding sites in the nicotinic receptor probed by reactions of binding-site cysteines with charged methanethiosulfonates. Biochemistry. 1994;33:6840–6849. doi: 10.1021/bi00188a013. [DOI] [PubMed] [Google Scholar]
- Stuhmer W, Conti F, Suzuki H, Wang XD, Noda M, Yahagi N, et al. Structural parts involved in activation and inactivation of the sodium channel. Nature. 1989;339:597–603. doi: 10.1038/339597a0. 10.1038/339597a0. [DOI] [PubMed] [Google Scholar]
- Tang L, Chehab N, Wieland SJ, Kallen RG. Glutamine substitution at alanine1649 in the S4–S5 cytoplasmic loop of domain 4 removes the voltage sensitivity of fast inactivation in the human heart sodium channel. J General Physiol. 1998;111:639–652. doi: 10.1085/jgp.111.5.639. 10.1085/jgp.111.5.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tytgat J, Hess P. Evidence for cooperative interactions in potassium channel gating. Nature. 1992;359:420–423. doi: 10.1038/359420a0. 10.1038/359420a0. [DOI] [PubMed] [Google Scholar]
- Wang Q, Shen J, Li Z, Timothy K, Vincent GM, Priori SG, et al. Cardiac sodium channel mutations in patients with long QT syndrome, an inherited cardiac arrhythmia. Hum Mol Genet. 1995;4:1603–1607. doi: 10.1093/hmg/4.9.1603. [DOI] [PubMed] [Google Scholar]
- West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na+-channel inactivation. Proc Natl Acad Sci U S A. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW. Shaker potassium channel gating. III: Evaluation of kinetic models for activation. J General Physiol. 1994;103:321–362. doi: 10.1085/jgp.103.2.321. 10.1085/jgp.103.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Morais-Cabral JH, Mann S, MacKinnon R. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 2001;411:657–661. doi: 10.1038/35079500. 10.1038/35079500. [DOI] [PubMed] [Google Scholar]