

Abstract
Background
Cytochrome P450 monooxygenases (CYP) play an important role in the defense against inhaled toxicants, and expression of CYP enzymes may differ among various lung cells and tissue compartments.
Methods
We studied the effects of tobacco smoke in volunteers and investigated gene expression of 19 CYPs and 3 flavin-containing monooxygenases, as well as isoforms of gluthathione S-transferases (GST) and uridine diphosphate glucuronosyltransferases (UGT) and the microsomal epoxide hydrolase (EPHX1) in bronchoalveolar lavage cells and bronchial biopsies derived from smokers (n = 8) and nonsmokers (n = 10). We also investigated gene expression of nuclear transcription factors known to be involved in the regulation of xenobiotic metabolism enzymes.
Results
Gene expression of CYP1A1, CYP1B1, CYP2S1, GSTP1, and EPHX1 was induced in bronchoalveolar lavage cells of smokers, whereas expression of CYP2B6/7, CYP3A5, and UGT2A1 was repressed. In bronchial biopsies of smokers, CYP1A1, CYP1B1, CYP2C9, GSTP1, and GSTA2 were induced, but CYP2J2 and EPHX1 were repressed. Induction of CYP1A1 and CYP1B1 transcript abundance resulted in increased activity of the coded enzyme. Finally, expression of the liver X receptor and the glucocorticoid receptor was significantly up-regulated in bronchoalveolar lavage cells of smokers.
Conclusions
We found gene expression of pulmonary xenobiotic metabolizing enzymes and certain key transcription factors to be regulated in bronchoalveolar lavage cells and bronchial biopsies of smokers. The observed changes demonstrate tissue specificity in xenobiotic metabolism, with likely implications for the metabolic activation of procarcinogens to ultimate carcinogens of tobacco smoke.
Keywords: cytochrome P450 monooxygenases, metabolism, smoking, transcription factors, xenobiotic metabolizing enzymes
The lung serves as a primary site for xenobiotic metabolism, and many xenobiotics are substrates for cytochrome P450 catalyzed oxidations. Notably, the lung is composed of > 40 different cell types with different levels of metabolic competence (Ding and Kaminsky 2003). Alveolar macrophages and bronchial epithelial cells are part of > 40 different cell types in lung tissue and differ in their biological functions (Hocking and Golde 1979; Hukkanen et al. 2002). Indeed alveolar macrophages clear the airways of tobacco smoke particles and therefore constitute an important defense mechanism (Drath et al. 1979). Furthermore, the lung epithelium takes part in the detoxification of tobacco smoke through metabolic activation by phase I and phase II enzymes (Crawford et al. 1998; Han et al. 2005). Although a number of CYP isoforms have been identified in human lung tissue, a comprehensive survey of most human pulmonary xenobiotic metabolizing enzymes in different human lung tissue compartments has not been performed.
Tobacco smoke constitutes a complex mixture of thousands of toxicants, some of which require tissue-specific activation to become genotoxic carcinogens and others become metabolically activated poisons (Smith et al. 2003; Yamazaki et al. 1992). For smokers, the risk of developing lung cancer has been shown to be, at least in part, dependent on pulmonary metabolism of smoke constituents (Rubin 2001). Particular evidence stems from studies with smokers in which genetic polymorphisms of certain xenobiotic metabolizing enzymes were linked to the development of lung cancer (Bartsch et al. 2000; London et al. 1999). Currently available data suggest that genetic variability in coding sequences of P450 enzymes, antioxidants, or DNA repair genes contribute to the risk of developing bronchogenic carcinomas (Caporaso 2002).
Cigarette smoke may affect the capacity of lung tissue to dispose of foreign chemicals by changing expression and coded activity of xenobiotic metabolizing enzymes. Additionally, cigarette smoke may affect the pharmacokinetics of inhaled drugs by modulating activity of specific drug metabolizing enzymes (Durak et al. 1996).
Therefore, knowledge of expression and activity of xenobiotic and drug metabolizing enzymes in lung tissue of smokers gives us a better understanding of metabolically induced toxicity of tobacco smoke constituents and allows us to assess the capacity for tissue specific detoxification.
We investigated the gene expression of 19 cytochrome P450 monooxygenases (CYPs) and 3 flavin-containing monooxygenases (FMOs), as well as uridine diphosphate glucuronosyltransferase (UGT) 2A1 (UGT2A1), epoxide hydrolase (EPHX1), glutathione-S-transferase (GST) A2 (GSTA2), GSTP1, and GSTM1 in lung cells obtained from bronchoalveolar lavage (BAL) fluid and in bronchial biopsies (BBs) of smokers (n = 8) and nonsmokers (n = 10).
Additionally, transcriptional regulation of CYPs depends, at least in part, on promoter activation through binding of transcription factors and nuclear receptors to cognate DNA recognition sites (Borlak and Thum 2001; (Pascussi et al. 2003). We therefore studied expression of the aryl hydrocarbon receptor (AHR), constitutive androstane receptor (CAR), pregnane X receptor (PXR), liver X receptor (LXR), and glucocorticoid receptor (GR) (Gibson et al. 2002; Tirona et al. 2003) for their established role in CYP gene regulation in BAL cells and BBs derived from smokers and nonsmokers.
Methods
Study subjects
All subjects were volunteers and gave written consent after being fully informed about the purpose and nature of the study. The study was approved by the Ethical Committee of Hannover Medical School.
Ten nonsmokers and eight smokers were enrolled. All subjects had no history of allergic or other diseases. Only subjects with forced expiratory volume in 1 sec (FEV1) > 75% (predicted); normal electrocardiogram, differential blood cell count, blood coagulation, serum parameters (gamma-glutamyl-transferase, aspartate aminotransferase, alanine aminotransferase, urea, creatinine, sodium, potassium, IgE); and negative skin-prick test (ALK-SCHERAX Arzneimittel GmbH, Hamburg, Germany) were included. The characteristics of the study subjects are summarized in Table 1.
Table 1.
Subject characteristics.
| Subjects | Age | Sex | FEV1 (% predicted) | FEV1/FVC (%) | Cigarettes/day | Pack-years | Cotinine (ng/mL) | Interval (min)a |
|---|---|---|---|---|---|---|---|---|
| Nonsmokers (n = 10) | 23 | F | 108.5 | 73.7 | 0 | 0 | 8.5 | — |
| 36 | F | 113.7 | 85.8 | 0 | 0 | 13.5 | — | |
| 21 | F | 92.2 | 77.1 | 0 | 0 | 0 | — | |
| 24 | F | 101.2 | 78.8 | 0 | 0 | 0 | — | |
| 28 | M | 106.0 | 76.3 | 0 | 3.75 | 20.4 | — | |
| 24 | F | 123.5 | 86.7 | 0 | 0 | 2.3 | — | |
| 25 | F | 105.5 | 83.8 | 0 | 0 | 0 | — | |
| 26 | M | 98.6 | 81.7 | 0 | 0 | 0 | — | |
| 26 | F | 85.5 | 79.7 | 0 | 0 | 0 | — | |
| 23 | F | 106.0 | 83.7 | 0 | 0 | 0 | — | |
| Median (range) | 24.5 (21–36) | — | 105.8 (85.5–123.5) | 80.7 (73.7–86.7) | 0 (0) | 0 (0–3.75) | 0 (0–20.4) | — |
| Smokers (n = 8) | 28 | M | 107.6 | 85.2 | 20 | 13.0 | 194.8 | 87 |
| 30 | M | 98.1 | 75.8 | 20 | 3.0 | 175.8 | 137 | |
| 22 | F | 113.2 | 83.2 | 20 | 7.0 | 400.0 | 157 | |
| 30 | M | 95.4 | 73.5 | 20 | 13.0 | 241.4 | 88 | |
| 29 | M | 89.2 | 78.6 | 20 | 2.5 | 156.8 | 93 | |
| 27 | M | 108.2 | 74.6 | 25 | 15.0 | 310.0 | 136 | |
| 26 | M | 99.0 | 83.7 | 20 | 12.0 | 369.0 | 88 | |
| 45 | M | 77.2 | 75.3 | 20 | 31.0 | 102.8 | 70 | |
| Median (range) | 28.5 (22–45) | — | 98.6 (77.2–113.2) | 77.2 (73.5–85.2) | 20 (20–25) | 12.5 (2.5–31.0) | 218.1 (102–400) | 90.5 (70–157) |
Abbreviations: F, female; M, male.
Time interval between smoking the last cigarette to bronchoscopy.
None of the subjects had records of drug abuse; for females, pregnancy was excluded before study participation. None of the subjects suffered acute bronchitis 4 weeks before bronchoscopy. Nonsmokers could not have smoked a cigarette for at least 5 years. Inclusion criteria for smokers were a minimum consumption of 15 cigarettes/day for at least 2 years. Levels of cotinine, a stable metabolite of nicotine, were measured in urine to estimate current nicotine exposure. Only smokers with cotinine levels ≥ 100 ng/mL and nonsmokers with cotinine levels ≤ 25 ng/mL were included. In addition, the interval between bronchoscopy and smoking of the last cigarette prior to bronchoscopy was 57–157 min.
Bronchoscopic procedure
Prior to bronchoscopy, all subjects received atropine (0.5 mg subcutaneously) and midazolam (0.05–0.1 mg/kg). Lidocaine (maximum: 6 mg/kg) was given as a local anesthetic of the upper and lower airways. Drug administration and timing was strictly controlled. The bronchoscope (BF 160 P, Olympus Optical Co. Europe GmbH, Hamburg, Germany) was wedged into the medial segment of the middle lobe, and BAL was performed with 6 × 20 mL sterile saline solution. Lavage fluid from the first 20 mL was discarded. After lavage, the bronchoscope was passed to the anterior segment of the left upper lobe and three bronchial biopsies were taken distal from the carina. The biopsy samples were immediately placed in RNA isolation buffer (Macherey-Nagel GmbH & Co. KG, Düren, Germany), snap-frozen in liquid nitrogen, and stored at −80°C to await further analysis.
Processing of BAL cells
BAL fluid samples were processed as described previously (Krug et al. 2001). Briefly, cells were filtered through a 100-μm filter. An aliquot of the BAL cells was used to determine the total count of nucleated cells using a Neubauer hemocytometer. After counting, two aliquots containing 1 × 106 cells were separated, pelleted, snap-frozen in liquid nitrogen, and stored at −80°C until RNA isolation. Differential cell counts were obtained from cytospin slides, with 300 cells/slide being counted (Table 2).
Table 2.
Basic BAL fluid data given as median (range).
| Macrophages
|
Neutrophils
|
Lymphocytes
|
Eosinophils
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Subjects (n) | Recovery (mL) | Total cells (× 106) | Percent of cells | No. (× 106) | Percent of cells | No. (× 106) | Percent of cells | No. (× 106) | Percent of cells | No. (× 106) |
| Nonsmokers (10) | 77.5 (60–88) | 5.9 (2.5–7.0) | 90 (84–95) | 5.2 (2.2–6.7) | 2 (1–3) | 0.1 (0.1–0.2) | 7 (3–13) | 0.3 (0.1–0.8) | 0 (0–1) | 0 (0–0.1) |
| Smokers (8) | 76.5 (62–86) | 9.3 (5.3–23.6)** | 90 (85–92) | 8.1 (4.9–21.2)** | 4 (1–8)** | 0.5 (0.1–1.4)** | 4.5 (3–5) | 0.4 (0.2–1.2) | 0 (0–4) | 0 (0–0.3) |
p < 0.01 compared with nonsmokers.
RNA isolation and reverse transcription
RNA was isolated from BAL pellets (1 × 106 cells/pellet) and biopsy materials using the Total RNA Isolation System (Macherey-Nagel GmbH & Co. KG) according to the manufacturer’s recommendation. Quality and quantity of isolated RNA was assayed by capillary electrophoresis (Bioanalyzer 2100; Agilent Technologies Deutschland GmbH, Böblingen, Germany) following the manufacturers instructions. We used 1 μg total RNA from each sample for reverse transcription, as described previously (Thum and Borlak 2002). The resulting cDNA was frozen at −20°C until further experimentation.
Thermocycler reverse transcriptase-polymerase chain reaction (RT-PCR)
PCR was carried out using a thermal cycler (T3; Biometra GmbH, Goettingen, Germany) as described previously (Thum and Borlak 2001). In brief, the following PCR conditions were used: denaturation, 94°C (45 sec); primer annealing, 54–58°C (60 sec); and extension, 72°C (60 sec). Detailed oligonucleotide sequence information is given in Table 3. We checked for DNA contamination by direct amplification of RNA extracts before conversion to cDNA. Any contamination of RNA extracts with genomic DNA could therefore be excluded. The optimal PCR cycles were derived by studying PCR products at different numbers of PCR cycles. As a consequence, PCR-reactions were performed within the linear range of amplification, and transcript expression levels were calculated as the ratio of the gene of interest (numerator) versus an established housekeeping gene (cyclophilin A, denumerator). We observed no significant differences in the gene expression results when experiments were repeated with the same cDNA, indicating good reproducibility of the method. Amplification products were separated on a 1.5% agarose gel, stained with ethidium bromide, photographed on a transilluminator (Kodak Image Station 440; Kodak GmbH, Stuttgart, Germany), and quantified using the Kodak 1D 3.5 network software.
Table 3.
Oligonucleotide primers and gene regulation in BAL cells and BB samples.
| Accession no. | Gene | Forward primer (5′–3′) | Reverse primer (5′–3′) | Product length (bp) | BAL | BB |
|---|---|---|---|---|---|---|
| CYPs | ||||||
| D01150 | CYP1A1 | TCACAGACACAGCCTGATTGAG | GATGGGTTGACCCATAGCTT | 432 | ↑ | ↑ |
| NM_000104.2 | CYP1B1 | GCAGAATTGGATCAGGTCGT | TGGTCAGGTCCTTGTTGATG | 301 | ↑ | ↑ |
| M38504 | CYP1A2 | TGGCTTCTACATCCCCAAGAAAT | TTCATGGTCAGCCCGTAGAT | 308 | ||
| U22027 | CYP2A6/7 | GTGTGGACATGATGCCGT | AGGACTTGAGGCGGAAGT | 1,151 | ||
| X13494 | CYP2B6/7 | CCATACACAGAGGCAGTCAT | GGTGTCAGATCGATGTCTTC | 357 | ↓ | |
| XM_050922 | CYP2C8 | GATCATGTAATTGGCAGACACA | CCTGCTGAGAAAGGCATGAAG | 311 | ||
| XM_050918 | CYP2C9 | AGCTTGGAAAACACTGCAGT | CCTGCTGAGAAAGGCATGAAG | 437 | ↑ | |
| NM_000772 | CYP2C18 | CTGTAACTGATATGTTTGGG | CCTGCTGAGAAAGGCATGAAG | 431 | ||
| NM_000769 | CYP2C19 | GTAATCACTGCAGCTGACTTAC | CCTGCTGAGAAAGGCATGAAG | 431 | ||
| M33189 | CYP2D6 | TGATGAGAACCTGCGCATAG | ACCGATGACAGGTTGGTGAT | 332 | ||
| AF084225 | CYP2E1 | AGCACAACTCTGAGATATGG | ATAGTCACTGTACTTGAACT | 365 | ||
| J02906 | CYP2F1 | ATGAACTTGCCGCACCGCGT | ACAGGCTCCACTTACGGTGC | 283 | ||
| AF272142 | CYP2J2 | CCCACCAAACTCTCTTCAGC | CATTCTCTGCACCTCATGGA | 389 | ↓ | |
| AF335278 | CYP2S1 | ACCCCAACATCTTCAAGCAC | TTCATCTGGTCTGCGTGGT | 312 | ↑ | |
| X12387 | CYP3A4 | CCAAGCTATGCTCTTCACCG | TCAGGCTCCACTTACGGTGC | 323 | ||
| L26985 | CYP3A5 | TGTCCAGCAGAAACTGCAAA | TTGAAGAAGTCCTTGCGTGTC | 472 | ↓ | |
| NM 000765.1 | CYP3A7 | CTATGATACTGTGCTACAGT | TCAGGCTCCACTTACGGTCT | 474 | ||
| AF208532 | CYP4A11 | CAAGTGACCTCCCTGCTCAT | CTGATCTCCCCAGAATCACC | 280 | ||
| NM 000779.1 | CYP4B1 | TGACCATGTGCATCAAAGGAG | AAAGCCATTCTTGGAGCGCA | 397 | ||
| Other drug-metabolizing enzymes | ||||||
| NM_000120 | EPHX1 | TGATGAGGGAGAGCGGGCTAC | TCAGCAGGTCGTCCAGGGAG | 227 | ↑ | ↓ |
| NM_002021 | FMO1 | GCTGTTCGAGTCCTGAAAGG | GCCAAAGAAGACGGTCAGAG | 234 | ||
| NM_006894 | FMO3 | GGCAGGGCTAGCATTTACAA | GATGTCCGGAACAAACCATT | 301 | ||
| NM_001461 | FMO5 | CTCTCAGTTTCATATTGCCCAG | ACATTATTTCTCTTATCTCTCAGG | 400 | ||
| NM_000846 | GSTA2 | GCCCAAGCTCCACTACTTCA | GCAAGCTTGGCATCTTTTTC | 354 | ↑ | |
| NM_000561 | GSTM1 | TTCCCAATCTGCCCTACTTG | GGGCTCAAATATACGGTGGA | 347 | ↑ | ↑ |
| XM_040116 | GSTP1 | ACCTCCGCTGCAAATACATC | GGGAGGTTCACGTACTCAGG | 313 | ↓ | |
| XM_003547 | UGT2A1 | TTGGCCAATGGAAGGTAGTC | TCCTGAGACACCATGTGGAA | 297 | ||
| Nuclear transcription factors | ||||||
| NM_001621 | AHR | CTGCCTTTCCCACAAGATGT | GAAATTCAGCTCGGTCTTCG | 352 | ||
| NM_005122 | NR1I3 | GTCATGGCCAGTAGGGAAGA | GTCCGGATCAGCTCTTCTTG | 350 | ↑ | |
| M10901 | NR3C1 | GCTCTGGGGTGGAGATCATA | TCCTTCCCTCTTGACAATGG | 300 | ↑ | |
| U22662 | NR1H3 | TCAACCCCATCTTCGAGTTC | GGGGACAGAACAGTCATTCG | 351 | ||
| AF061056 | NR1I2 | TTGTTCGGCATCACAGGTAG | GGGATCTGAGGGATTTCTCC | 351 | ||
| Housekeeping gene | ||||||
| NM_021130 | Cyclophilin | CTTGCCATTCCTGGACCCAA | TTTCGTGCTCTGAGCACTGG | 347 | ||
Accession numbers from GenBank (2005). Arrows indicate up- or down-regulation of significantly affected genes.
Ethoxyresorufin-O-demethylase (EROD) assay
Immediately after bronchoscopy, approximately 0.1 × 106 cells were separated from BAL fluid and put into 500 μL Dulbecco’s modified Eagle’s medium supplemented with 5% fetal calf serum and 2 mM glutamine. Then, 7-ethoxyresorufin (2 μM, Sigma-Aldrich Chemie GmbH, Deisenhofen, Germany; dissolved in DMSO) and dicumarol (10 μM; Sigma-Aldrich Chemie GmbH) were added, and cells were incubated at 37°C for 4 hr and centrifuged for 5 min (1,200 × g, 4°C). The resulting supernatant was snap-frozen in liquid nitrogen and stored at −80°C to await fluorescence detection essentially as described by Grant et al. (1987). Samples (250 μL) were treated with 250 μL ammonium acetate (pH 4.5), and aliquots were treated with 100 U/mL β-glucuronidase (Sigma-Aldrich Chemie GmbH) overnight at 37°C to assay product release of β-glucuronide conjugates. After addition of 500 μL glycine buffer (pH 10.3), fluorometric analysis was carried out on a spectrofluorophotometer (Bio-Rad Laboratories GmbH, München, Germany). The fluorometric analysis of the resultant product resorufin was done with an excitation at 530 nm and an emission wave length of 585 nm. Calibration of the system was performed with resorufin and an appropriate standard curve with a concentration range of up to 100 nM.
Statistical analysis
We used the Mann-Whitney U test for intergroup comparisons. A p-value < 0.05 was considered statistically significant. Unless otherwise stated, the data are expressed as median and range or as individual results.
Results
Subject characteristics and BAL data
The clinical characteristics of the study subjects are given in Table 1. There was no significant difference in FEV1 (% predicted) and FEV1/forced vital capacity (FVC) between smokers and nonsmokers.
Recovery of BAL fluid did not differ between smokers and nonsmokers. The total cell count in BAL fluid from smokers was nearly double the cell count in nonsmokers (p < 0.01). This was mainly due to higher numbers of macrophages and neutrophils. We found no significant differences in the absolute counts of lymphocytes and eosinophils in BAL samples between smokers and nonsmokers (Table 2).
Gene expression of CYPs and FMOs
Expression of CYP1A2, CYP2C8, CYP2C18, CYP2C19, CYP2D6, CYP3A7, CYP4A11, FMO1, and FMO3 was below the limit of detection (LOD) in all investigated samples (data not shown).
Transcript profiling in BAL cells
Expression of CYP1A1 (p < 0.01), CYP1B1 (p < 0.001), and CYP2S1 (p < 0.001) was higher in BAL cells from smokers compared with those of nonsmokers (Figure 1), whereas expression of CYP2B6/7 (p < 0.01) and CYP3A5 (p < 0.001) was lower; expression of CYP2A6/7, CYP2E1, CYP2C9, CYP2J2, and FMO5 was not altered. CYP2F1 transcripts were detected in 3 of 10 nonsmokers and in 3 of 8 smokers, but the amount was too small for semiquantitative analysis (data not shown). Similarly, CYP4B1 was detected in 5 smokers and 1 nonsmoker, and CYP3A4 was detected in 2 nonsmokers but was < LOD in smokers (data not shown).
Figure 1.

Gene expression of various CYPs in BAL cells and BB material obtained from nonsmokers (n = 10) and smokers (n = 8). NS, not significant. Data are represented as plots from individual volunteers, with solid lines indicating medians. Results are presented as the ratio of the gene of interest/cyclophilin; only signficantly altered genes are shown.
*p < 0.05. **p < 0.01. ***p < 0.001.
Transcript profiling in BBs
In smokers, expression of CYP1A1 (p < 0.05), CYP1B1 (p < 0.001), and CYP2C9 (p < 0.05) was increased (p < 0.05) compared with non-smokers, and there was a trend toward increased expression of CYP3A5 (p = 0.0545; Figure 1); however, the expression of CYP2J2 was significantly (p < 0.05) repressed. Expression of CYP2B6/7, CYP2E1, CYP2F1, CYP2S1, CYP4B1, and FMO5 was similar to that of nonsmokers. CYP2A6/7 transcripts were detected in 2 of 10 nonsmokers and 3 of 8 smokers (data not shown). CYP3A4 was < LOD in all BBs investigated.
Gene expression of EPHX1, GSTs, and UGTs
In BAL cells of smokers, gene expression of EPHX1 and GSTP1 was significantly higher that that of nonsmokers, whereas expression of the UGT2A1 gene was significantly lower (p < 0.001) (Figure 2). Expression of GSTA2 and GSTM1 was not different between the study groups (Figure 2).
Figure 2.

Gene expression of FMO5, various phase II enzymes, and key regulatory transcription factors in BAL cells and BB material obtained from nonsmokers (n = 10) and smokers (n = 8). NS, not significant. Data are represented as plots from individual volunteers, with solid lines indicating medians. Results are presented as the ratio of the gene of interest/cyclophilin; only significantly altered genes are shown.
*p < 0.05. **p < 0.01. ***p = < 0.001.
Further, in BBs taken from smokers, expression of EPHX1 was significantly lower (p < 0.001), whereas the expression of GSTA2 and GSTP1 were up to triple that in non-smokers. Expression of GSTM1 and UGT2A1 was not significantly different in BBs of the two groups (Figure 2).
Gene expression of nuclear receptors
Gene expression of LXR (p < 0.001) and GR (p < 0.01) in BAL cells of smokers (Figure 2) was up to three times that of nonsmokers, whereas expression of the cytosolic AHR and PXR was similar. The expression of AHR, LXR, PXR and GR was not different in BBs of both study groups. Expression of CAR was < LOD in all samples studied.
Enzyme activity of CYP1A1 and CYP1B1
7-Ethoxyresorufin served as substrate for CYP1A1 and CYP1B1. In BAL cells derived from smokers, EROD activity was up to 3-fold that of nonsmokers (p < 0.05; Figure 3), but glucuronides were < LOD, as determined by β-glucuronidase treatment.
Figure 3.
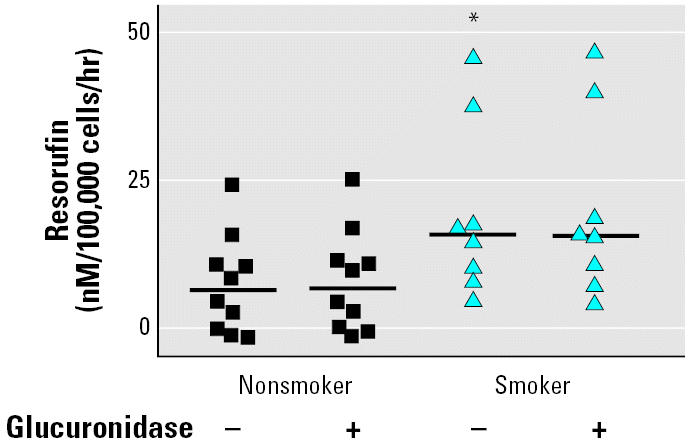
Metabolism of 7-ethoxyresorufin in BAL cells obtained from nonsmokers (n = 10) and smokers (n = 8) with and without glucuronidase treatment. Data represent mean ± SE of the individual incubation experiments, with approximately 100,000 cells per experiment.
*p < 0.05.
Discussion
In this study we aimed to investigate the regulation of gene expression of major human pulmonary xenobiotic metabolizing enzymes as well as regulatory nuclear transcription factors in smoking and nonsmoking healthy volunteers. Differences were observed when we compared BAL cells and BBs of smokers and nonsmokers. Our findings provide new insight into lung tissue–specific responses to tobacco smoke as detailed below.
Effects of tobacco smoke on pulmonary xenobiotic metabolizing enzymes
Lung tissue and cell types within the lung differ in their capacity to oxidize xenobiotics (Hecht 1999) because CYPs are not uniformly expressed (Ding and Karminsky 2003). As the majority of airborne toxicants enter the body through the respiratory tract, the pulmonary epithelium is exposed to high concentrations before systemic circulation, which, in turn, requires an effective local defense system. Our findings of a simultaneous induction of CYP1A1 and CYP1B1 in different lung compartments after exposure to tobacco smoke demonstrate up-regulation of the pulmonary enzyme system and agree well with other reports (McLemore et al. 1990). Regulation of CYP1A1 and CYP1B1 is not fully understood, but transcriptional activation by the AHR constitutes a major mechanism. Upon translocation into the nucleus, the AHR forms a heterodimer with its nuclear counterpart ARNT (AHR nuclear translocator) and drives gene expression of the AHR-responsible gene family, including CYP1A1 and CYP1B1 (Borlak and Thum 2001). Thus, the strong induction of CYP1A1 and CYP1B1 can be explained, at least in part, in terms of AHR-mediated transcriptional activation. Although we did not observe changes of the AHR at the gene expression level, Martey et al. (2005) recently reported that cigarette smoke extract led to an activation of the AHR in cultured human lung fibroblasts based on induced nuclear translocation of the AHR (Martey et al. 2005). The observed up-regulation of AHR-responsive genes in lung tissue of smokers is likely mediated by the activation of AHR. The present study does, however, contradict the findings of Anttila et al. (1991), who did not find CYP1A1 expression in alveolar macrophages, regardless of smoking status. This may be due to different experimental approaches because Anttila et al. employed immunohistochemistry, whereas we used a sensitive RT-PCR method determine CYP1A1 and CYP1B1 expression and a fluorometric assay to demonstrate increased activity of the coded enzymes. Indeed, Willey et al. (1996) were initially unable to detect CYP1A1 expression in alveolar macrophages, but upon application of more sensitive techniques these investigator were able to detect CYP1A1 gene expression (Willey et al. 1997).
CYP2A enzymes metabolize a variety of carcinogens including 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), which can lead to the development of lung cancer (Koskela et al. 1999). In the present study, we detected expression of CYP2A6/7 in both alveolar macrophages and bronchial biopsy material, but we did not find any difference in regard to smoking status. This contrasts the finding of Crawford et al. (1998), who demonstrated repression of CYP2A6/7 in human bronchial epithelial cells of smokers. The somewhat high interindividual variation of CYP2A6/7 gene expression observed in the present study and in the study of Crawford et al. (1998) make comparison of the data difficult.
We also observed significant induction of CYP2S1 in BAL cells of smokers. Expression of CYP2S1 in lung tissue has been previously reported (Rylander et al. 2001), but regulation by cigarette smoke has not been determined so far. One study (Rivera et al. 2002) demonstrated inducibility of CYP2S1 in mouse lungs after systemic administration of dioxins. The observed induction of CYP1A1 and CYP2S1 in bronchoalveolar macrophages and bronchial biopsies of smokers is of importance because of their contribution to the metabolic activation of tobacco smoke components, namely polycyclic aromatic hydrocarbons, which can lead to the development of lung cancer (Georgiadis et al. 2005).
CYP2B6/7 and CYP3A5 play an important role in the metabolic activation of NNK and of benzo[a]pyrene (Code et al. 1997; Hecht 1999), but transcript levels and enzyme activity were repressed in BAL cells derived from smokers. Previously, Hukkanen et al. (2003) demonstrated decreased CYP3A5 expression in alveolar macrophages of smokers. We now extend the findings of Hukkanen et al. (2003) to BBs and report a trend toward increased CYP3A5 expression in smokers (p < 0.0545), which could result in local metabolic activation of carcinogens. Notably, the CYP3A family of monooxygenases plays a key role in pulmonary drug metabolism. Inhaled drugs, such as salmeterol, tiotropium, theophylline, or glucocorticoids (e.g., budesonide, prednisone), with an established role in chronic obstructive pulmonary disease or asthma treatment (Global Initiative for Asthma 2005; Global Initiative for Chronic Obstructive Lung Disease 2005) are substrates for CYP3A isoforms (Jonsson et al. 1995; Zevin and Benowitz 1999). Therefore, modulation of CYP3A enzyme activity in smokers may require dose adjustment for some inhaled drugs.
Furthermore, the CYP epoxygenases CYP2C and CYP2J2 are key players in the metabolism of arachidonic acid, resulting in the production of epoxyeicosatrienoic acids, some of which affect vascular and bronchial smooth muscle tone (Fisslthaler et al. 1999; Thum and Borlak 2004; Zeldin et al. 1995). In the present study, we observed reduced expression of CYP2J2 in BBs of smokers, whereas CYP2C9 was significantly induced in this lung compartment. This shift in the expression of the CYP epoxygenase gene might alter production of epoxy fatty acids, some of which are considered to be signalling molecules affecting vascular- and smooth muscle cell tone. Undoubtedly, future studies are needed to determine the regulation and the role of epoxyeicosatrienoic acids in smokers.
In addition, genetic variability in the coding of various CYP genes may affect enzyme activity (reviewed by Daly et al. 1994; Ingelman-Sundberg 2001). Likewise, changes in the expression of CYP transcripts may influence an individual’s capacity to convert different precarcinogenic compounds into their ultimate carcinogens; therefore, these CYP transcripts are of major importance for an individual’s susceptibility of developing chemically induced cancer. Certain CYP mono-oxygenases have an established role in an activation of precarcinogens to ultimate carcinogens of inhaled tobacco smoke toxicants (e.g., CYP1A1, CYP1A2, CYP1B1, CYP2E1, CYP3A4). The simultaneous induction of CYP1A1 and CYP1B1 in both types of lung cells obtained from BAL and pulmonary epithelial cells after smoking cigarettes, as found in the present study, may therefore enhance an individual susceptibility for chemically induced lung cancer.
In addition, expression of EPHX1 was increased in BAL cells of smokers, but decreased in BBs. This is important because in the absence of EPHX1 the tobacco smoke carcinogen benzo[a]pyrene is primarily metabolized to the noncarcinogenic 7,8-benzo-phenolic product (Shou et al. 1994). Bartsch et al. (1992) observed a significant increase in the activity of pulmonary EPHX1 within smokers, but EPHX1 activity was determined in preparations of parenchymal lung tissue, which consists of many different cell types. Besides, the lung tissue specimens were taken from patients with either lung cancer or non-neoplastic lung diseases. Thus, a direct comparison between the present study and that of Bartsch et al. (1992) cannot be made. Our results demonstrate that enzyme activity of EPHX1 can be expressed differently in two lung tissue compartments.
We also observed induction of the carcinogen-metabolizing enzymes GSTP1 in BAL cells and BBs and GSTA2 in BBs from smokers. This is likely an adaptive response to chronic exposure to tobacco smoke components. GSTP1 overexpression inhibited cytotoxic effects of cigarette smoke extract on human fibroblast-derived cells and depletion of GSTP1-induced apoptosis in lung fibroblasts (Ishii et al. 2001, 2003). Moreover, Crawford et al. (2000) showed that mRNA levels of GSTs expressed by bronchial epithelial cells from patients with bronchogenic carcinoma are significantly lower compared with subjects without carcinoma. Thus, increased expression of GSTP1 in healthy smokers might protect against accumulation of carcinogens.
Effects on nuclear receptors
GR and LXR were significantly up-regulated in BAL cells, but no clear correlation between CYP mono-oxygenases and nuclear receptor gene expression was observed. However, the up-regulation of GR fits well to the increased expression of CYP2C9 because GR plays an important role in transcriptional regulation of the CYP2C9 gene (Gerbal-Chaloin et al. 2002). CYP regulation is, in most cases, not dependent only on a specific nuclear factor, but requires a complex network of interacting factors (Waxman 1999).
Study limitations
We did not recruit for a sex-balanced study group; for example, 8 of 10 were females in the nonsmoking group, whereas 7 of 8 were males in the smoking group. Nonetheless, when gene expression was compared between men and women, we found no significant differences. Furthermore, within different lung tissue compartments, we observed opposite effects in expression of certain genes (i.e., CYP3A5, EPHX1) for smokers, but these were independent of the sex. Although drug administration before the bronchoscopic procedures was strictly controlled, we cannot rule out minor effects of the drugs used (e.g., midazolam, lidocain, atropine) on gene expression within lung tissue. Notably, the same drugs were given to both smokers and nonsmokers, usually ≤15 min before the bronchoscopic procedures. This short interval makes major effects on gene expression unlikely.
Conclusions
We observed profound effects of tobacco smoke exposure in BBs and BAL cells of volunteers and found CYP1A1, CYP1B1, and GSTP1 to be up-regulated in BBs and alveolar macrophages of smokers, whereas others transcripts were differentially expressed and, in some cases, even oppositely regulated (i.e., CYP3A5, EPHX1). Differences in the expression of xenobiotic metabolizing enzymes may suggest local and tissue-specific susceptibility in metabolically activated toxicity.
Footnotes
We thank C.R. Lai (Drug Research and Medical Biotechnology, Fraunhofer ITEM) and B. Volkmann (Immunology/Allergology and Clinical Inhalation, Fraunhofer ITEM) for technical assistance.
This work was supported by a grant from the Lower Saxony Ministry of Culture and Science to J.B.
References
- Anttila S, Hietanen E, Vainio H, Camus AM, Gelboin HV, Park SS, et al. Smoking and peripheral type of cancer are related to high levels of pulmonary cytochrome P450IA in lung cancer patients. Int J Cancer. 1991;47:681–685. doi: 10.1002/ijc.2910470509. [DOI] [PubMed] [Google Scholar]
- Bartsch H, Nair U, Risch A, Rojas M, Wikman H, Alexandrov K. Genetic polymorphism of CYP genes, alone or in combination, as a risk modifier of tobacco-related cancers. Cancer Epidemiol Biomarkers Prev. 2000;9:3–28. [PubMed] [Google Scholar]
- Bartsch H, Petruzzelli S, De Flora S, Hietanen E, Camus AM, Castegnaro M, et al. Carcinogen metabolism in human lung tissues and the effect of tobacco smoking: results from a case–control multicenter study on lung cancer patients. Environ Health Perspect. 1992;98:119–124. doi: 10.1289/ehp.9298119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borlak J, Thum T. Induction of nuclear transcription factors, cytochrome P450 monooxygenases, and glutathione S-transferase alpha gene expression in Aroclor 1254-treated rat hepatocyte cultures. Biochem Pharmacol. 2001;61:145–153. doi: 10.1016/s0006-2952(00)00537-2. [DOI] [PubMed] [Google Scholar]
- Caporaso NE. Why have we failed to find the low penetrance genetic constituents of common cancers? Cancer Epidemiol Biomarkers Prev. 2002;11:1544–1549. [PubMed] [Google Scholar]
- Code EL, Crespi CL, Penman BW, Gonzalez FJ, Chang TK, Waxman DJ. Human cytochrome P4502B6: inter-individual hepatic expression, substrate specificity, and role in procarcinogen activation. Drug Metab Dispos. 1997;25:985–993. [PubMed] [Google Scholar]
- Crawford EL, Khuder SA, Durham SJ, Frampton M, Utell M, Thilly WG, et al. Normal bronchial epithelial cell expression of glutathione transferase P1, glutathione transferase M3, and glutathione peroxidase is low in subjects with bronchogenic carcinoma. Cancer Res. 2000;60:1609–1618. [PubMed] [Google Scholar]
- Crawford EL, Weaver DA, DeMuth JP, Jackson CM, Khuder SA, Frampton MW, et al. Measurement of cytochrome P450 2A6 and 2E1 gene expression in primary human bronchial epithelial cells. Carcinogenesis. 1998;19:1867–1871. doi: 10.1093/carcin/19.10.1867. [DOI] [PubMed] [Google Scholar]
- Daly AK, Cholerton S, Armstrong M, Idle JR. Genotyping for polymorphisms in xenobiotic metabolism as a predictor of disease susceptibility. Environ Health Perspect. 1994;102(suppl 9):55–61. doi: 10.1289/ehp.94102s955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X, Kaminsky LS. Human extrahepatic cytochromes P450: function in xenobiotic metabolism and tissue-selective chemical toxicity in the respiratory and gastrointestinal tracts. Annu Rev Pharmacol Toxicol. 2003;43:149–173. doi: 10.1146/annurev.pharmtox.43.100901.140251. [DOI] [PubMed] [Google Scholar]
- Drath DB, Karnovsky ML, Huber GL. Tobacco smoke. Effects on pulmonary host defense. Inflammation. 1979;3:281–288. doi: 10.1007/BF00914185. [DOI] [PubMed] [Google Scholar]
- Durak H, Sayit E, Aktogu S, Degirmenci B, Yurekli Y, Ertay T, et al. Lung clearance of inhaled 99Tcm-erythromycin lacto-bionate in non-smokers and smokers. Nucl Med Commun. 1996;17:895–901. doi: 10.1097/00006231-199610000-00012. [DOI] [PubMed] [Google Scholar]
- Fisslthaler B, Popp R, Kiss L, Potente M, Harder DR, Fleming I, et al. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–497. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- Georgiadis P, Topinka J, Vlachodimitropoulos D, Stoikidou M, Gioka M, Stephanou G, et al. Interactions between CYP1A1 polymorphisms and exposure to environmental tobacco smoke in the modulation of lymphocyte bulky DNA adducts and chromosomal aberrations. Carcinogenesis. 2005;26:93–101. doi: 10.1093/carcin/bgh294. [DOI] [PubMed] [Google Scholar]
- GenBank 2005. GenBank Overview. Available: http://www.ncbi.nlm.nih.gov/Genbank/index.html [accessed 5 November 2005].
- Gerbal-Chaloin S, Daujat M, Pascussi JM, Pichard-Garcia L, Vilarem MJ, Maurel P. Transcriptional regulation of CYP2C9 gene. Role of glucocorticoid receptor and constitutive androstane receptor. J Biol Chem. 2002;277:209–217. doi: 10.1074/jbc.M107228200. [DOI] [PubMed] [Google Scholar]
- Gibson GG, Plant NJ, Swales KE, Ayrton A, Sankary W. Receptor-dependent transcriptional activation of cytochrome P4503A genes: induction mechanisms, species differences and interindividual variation in man. Xenobiotica. 2002;32:165–206. doi: 10.1080/00498250110102674. [DOI] [PubMed] [Google Scholar]
- Global Initiative for Asthma 2005. Global Strategy for Asthma Management and Prevention. Available: http://www.ginasthma.org/Guidelineitem.asp??l1=2&l2=1&intId=60 [accessed 5 November 2005].
- Global Initiative for Chronic Obstructive Lung Disease 2005. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease. Available: http://www.goldcopd.org/Guidelineitem.asp?l1=2&l2=1&intId=989 [accessed 5 November 2005].
- Grant MH, Burke MD, Hawksworth GM, Duthie SJ, Engeset J, Petrie JC. Human adult hepatocytes in primary monolayer culture. Maintenance of mixed function oxidase and conjugation pathways of drug metabolism. Biochem Pharmacol. 1987;36:2311–2316. doi: 10.1016/0006-2952(87)90596-x. [DOI] [PubMed] [Google Scholar]
- Han W, Pentecost BT, Pietropaolo RL, Fasco MJ, Spivack SD. Estrogen receptor alpha increases basal and cigarette smoke extract-induced expression of CYP1A1 and CYP1B1, but not GSTP1, in normal human bronchial epithelial cells. Mol Carcinog. 2005;44:202–211. doi: 10.1002/mc.20128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst. 1999;91:1194–1210. doi: 10.1093/jnci/91.14.1194. [DOI] [PubMed] [Google Scholar]
- Hocking WG, Golde DW. The pulmonary-alveolar macrophage (first of two parts) N Engl J Med. 1979;301:580–587. doi: 10.1056/NEJM197909133011104. [DOI] [PubMed] [Google Scholar]
- Hukkanen J, Pelkonen O, Hakkola J, Raunio H. Expression and regulation of xenobiotic-metabolizing cytochrome P450 (CYP) enzymes in human lung. Crit Rev Toxicol. 2002;32:391–411. doi: 10.1080/20024091064273. [DOI] [PubMed] [Google Scholar]
- Hukkanen J, Vaisanen T, Lassila A, Piipari R, Anttila S, Pelkonen O, et al. Regulation of CYP3A5 by glucocorticoids and cigarette smoke in human lung-derived cells. J Pharmacol Exp Ther. 2003;304:745–752. doi: 10.1124/jpet.102.038208. [DOI] [PubMed] [Google Scholar]
- Ingelman-Sundberg M. Genetic susceptibility to adverse effects of drugs and environmental toxicants. The role of the CYP family of enzymes. Mutat Res. 2001;482:11–19. doi: 10.1016/s0027-5107(01)00205-6. [DOI] [PubMed] [Google Scholar]
- Ishii T, Fujishiro M, Masuda M, Nakajima J, Teramoto S, Ouchi Y, et al. Depletion of glutathione S-transferase P1 induces apoptosis in human lung fibroblasts. Exp Lung Res. 2003;29:523–536. doi: 10.1080/01902140303777. [DOI] [PubMed] [Google Scholar]
- Ishii T, Matsuse T, Igarashi H, Masuda M, Teramoto S, Ouchi Y. Tobacco smoke reduces viability in human lung fibroblasts: protective effect of glutathione S-transferase P1. Am J Physiol Lung Cell Mol Physiol. 2001;280:1189–1195. doi: 10.1152/ajplung.2001.280.6.L1189. [DOI] [PubMed] [Google Scholar]
- Jonsson G, Astrom A, Andersson P. Budesonide is metabolized by cytochrome P450 3A (CYP3A) enzymes in human liver. Drug Metab Dispos. 1995;23:137–142. [PubMed] [Google Scholar]
- Koskela S, Hakkola J, Hukkanen J, Pelkonen O, Sorri M, Saranen A, et al. Expression of CYP2A genes in human liver and extrahepatic tissues. Biochem Pharmacol. 1999;57:1407–1413. doi: 10.1016/s0006-2952(99)00015-5. [DOI] [PubMed] [Google Scholar]
- Krug N, Erpenbeck VJ, Balke K, Petschallies J, Tschernig T, Hohlfeld JM, et al. Cytokine profile of bronchoalveolar lavage-derived CD4+, CD8+, and γδT cells in people with asthma after segmental allergen challenge. Am J Respir Cell Mol Biol. 2001;25:125–131. doi: 10.1165/ajrcmb.25.1.4194. [DOI] [PubMed] [Google Scholar]
- London SJ, Idle JR, Daly AK, Coetzee GA. Genetic variation of CYP2A6, smoking, and risk of cancer. Lancet. 1999;353:898–899. doi: 10.1016/S0140-6736(98)04984-8. [DOI] [PubMed] [Google Scholar]
- Martey CA, Baglole CJ, Gasiewicz TA, Sime PJ, Phipps RP. The aryl hydrocarbon receptor is a regulator of cigarette smoke induction of the cyclooxygenase and prostaglandin pathways in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2005;289:391–399. doi: 10.1152/ajplung.00062.2005. [DOI] [PubMed] [Google Scholar]
- McLemore TL, Adelberg S, Liu MC, McMahon NA, Yu SJ, Hubbard WC, et al. Expression of CYP1A1 gene in patients with lung cancer: evidence for cigarette smoke-induced gene expression in normal lung tissue and for altered gene regulation in primary pulmonary carcinomas. J Natl Cancer Inst. 1990;82:1333–1339. doi: 10.1093/jnci/82.16.1333. [DOI] [PubMed] [Google Scholar]
- Pascussi JM, Gerbal-Chaloin S, Drocourt L, Maurel P, Vilarem MJ. The expression of CYP2B6, CYP2C9 and CYP3A4 genes: a tangle of networks of nuclear and steroid receptors. Biochim Biophys Acta. 2003;1619:243–253. doi: 10.1016/s0304-4165(02)00483-x. [DOI] [PubMed] [Google Scholar]
- Rivera SP, Saarikoski ST, Hankinson O. Identification of a novel dioxin-inducible cytochrome P450. Mol Pharmacol. 2002;61:255–259. doi: 10.1124/mol.61.2.255. [DOI] [PubMed] [Google Scholar]
- Rubin H. Synergistic mechanisms in carcinogenesis by polycyclic aromatic hydrocarbons and by tobacco smoke: a bio-historical perspective with updates. Carcinogenesis. 2001;22:1903–1930. doi: 10.1093/carcin/22.12.1903. [DOI] [PubMed] [Google Scholar]
- Rylander T, Neve EP, Ingelman-Sundberg M, Oscarson M. Identification and tissue distribution of the novel human cytochrome P450 2S1 (CYP2S1) Biochem Biophys Res Commun. 2001;281:529–535. doi: 10.1006/bbrc.2001.4390. [DOI] [PubMed] [Google Scholar]
- Shou M, Korzekwa KR, Crespi CL, Gonzalez FJ, Gelboin HV. The role of 12 cDNA-expressed human, rodent, and rabbit cytochromes P450 in the metabolism of benzo[a]pyrene and benzo[a]pyrene trans-7,8-dihydrodiol. Mol Carcinog. 1994;10:159–168. doi: 10.1002/mc.2940100307. [DOI] [PubMed] [Google Scholar]
- Smith GBJ, Bend JR, Bedard LL, Reid KR, Petsikas D, Massey T. Biotransformation of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) in peripheral human lung microsomes. Drug Metab Dispos. 2003;31:1134–1141. doi: 10.1124/dmd.31.9.1134. [DOI] [PubMed] [Google Scholar]
- Thum T, Borlak J. Reprogramming of gene expression in cultured cardiomyocytes and in explanted hearts by the myosin ATPase inhibitor butanedione monoxime. Transplantation. 2001;71:543–552. doi: 10.1097/00007890-200102270-00010. [DOI] [PubMed] [Google Scholar]
- Thum T, Borlak J. Testosterone, cytochrome P450, and cardiac hypertrophy. FASEB J. 2002;16:1537–1549. doi: 10.1096/fj.02-0138com. [DOI] [PubMed] [Google Scholar]
- Thum T, Borlak J. Mechanistic role of cytochrome P450 monooxygenases in oxidized low-density lipoprotein-induced vascular injury. Therapy through LOX-1 receptor antagonism? Circ Res. 2004;94:1–13. doi: 10.1161/01.RES.0000110081.03480.E9. [DOI] [PubMed] [Google Scholar]
- Tirona RG, Lee W, Leake BF, Lan LB, Cline CB, Lamba V, et al. The orphan nuclear receptor HNF4alpha determines PXR- and CAR-mediated xenobiotic induction of CYP3A4. Nat Med. 2003;9:220–224. doi: 10.1038/nm815. [DOI] [PubMed] [Google Scholar]
- Waxman DJ. P450 gene induction by structurally diverse xenochemicals: central role of nuclear receptors CAR, PXR, and PPAR. Arch Biochem Biophys. 1999;369:11–23. doi: 10.1006/abbi.1999.1351. [DOI] [PubMed] [Google Scholar]
- Willey JC, Coy E, Brolly C, Utell MJ, Frampton MW, Hammersley J, et al. Xenobiotic metabolism enzyme gene expression in human bronchial epithelial and alveolar macrophage cells. Am J Respir Cell Mol Biol. 1996;14:262–271. doi: 10.1165/ajrcmb.14.3.8845177. [DOI] [PubMed] [Google Scholar]
- Willey JC, Coy EL, Frampton MW, Torres A, Apostolakos MJ, Hoehn G, et al. Quantitative RT-PCR measurement of cytochromes p450 1A1, 1B1, and 2B7, microsomal epoxide hydrolase, and NADPH oxidoreductase expression in lung cells of smokers and nonsmokers. Am J Respir Cell Mol Biol. 1997;17:114–124. doi: 10.1165/ajrcmb.17.1.2783. [DOI] [PubMed] [Google Scholar]
- Yamazaki H, Inui Y, Yun CH, Guengerich FP, Shimada T. Cytochrome P450 2E1 and 2A6 enzymes as major catalysts for metabolic activation of N-nitrosodialkylamines and tobacco-related nitrosamines in human liver microsomes. Carcinogenesis. 1992;13:1789–1794. doi: 10.1093/carcin/13.10.1789. [DOI] [PubMed] [Google Scholar]
- Zeldin DC, Plitman JD, Kobayashi J, Miller RF, Snapper JR, Falck JR, et al. The rabbit pulmonary cytochrome P450 arachidonic acid metabolic pathway: characterization and significance. J Clin Invest. 1995;95:2150–2160. doi: 10.1172/JCI117904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zevin S, Benowitz NL. Drug interactions with tobacco smoking. An update. Clin Pharmacokinet. 1999;36:425–438. doi: 10.2165/00003088-199936060-00004. [DOI] [PubMed] [Google Scholar]