

Abstract
This article analyzes mammalian genome rearrangements at higher resolution than has been published to date. We identify 3171 intervals, covering ∼92% of the human genome, within which we find no rearrangements larger than 50 kilobases (kb) in the lineages leading to human, mouse, rat, and dog from their most recent common ancestor. Combining intervals that are adjacent in all contemporary species produces 1338 segments that may contain large insertions or deletions but that are free of chromosome fissions or fusions as well as inversions or translocations >50 kb in length. We describe a new method for predicting the ancestral order and orientation of those intervals from their observed adjacencies in modern species. We combine the results from this method with data from chromosome painting experiments to produce a map of an early mammalian genome that accounts for 96.8% of the available human genome sequence data. The precision is further increased by mapping inversions as small as 31 bp. Analysis of the predicted evolutionary breakpoints in the human lineage confirms certain published observations but disagrees with others. Although only a few mammalian genomes are currently sequenced to high precision, our theoretical analyses and computer simulations indicate that our results are reasonably accurate and that they will become highly accurate in the foreseeable future. Our methods were developed as part of a project to reconstruct the genome sequence of the last ancestor of human, dogs, and most other placental mammals.
Using computer simulations, we have shown (Blanchette et al. 2004) that the genome sequence of the so-called Boreoeutherian ancestor (Fig. 1) can be computationally predicted at high accuracy within most euchromatic intervals that are free of large-scale rearrangements, given adequate data from living mammals. For instance, when sequences from 20 appropriately chosen mammalian species are available, we expect that >98% of the reconstructed nucleotides will be identical to the corresponding ancestral base. Because all mammals have experienced large-scale genomic rearrangements since their last common ancestor, in order to determine regional correspondence we analyze these rearrangements to infer a partition of each genome into intervals where nucleotide-level reconstruction methods can be applied.
Figure 1.

Position of the Boreoeutherian ancestor. Branch labels give the estimated number of chromosomal breaks from our study, also categorized as (interchromosomal, intrachromosomal). If conserved segments i and j are adjacent in the ancestral genome but not in the descendant genome, then we call the break interchromosomal if i and j are on different chromosomes in the descendant, and intrachromosomal otherwise. We suspect that many of the predicted intrachromosomal breaks in rat are assembly artifacts.
The regional correspondence between modern and ancestral chromosomes has been predicted with increasing accuracy by a number of groups using a variety of methods. Currently, the main experimental technique is chromosomal painting (for surveys, see Wienberg 2004 and Froenicke et al. 2006), in which fluorescently labeled chromosomes from one species are hybridized to chromosomes from another species. Although the requirement of optical visibility means that the cytogenetic approach can recognize only rearrangements with conserved segments longer than 4 Mb (Froenicke et al. 2006) and cannot identify intrachromosomal rearrangements (Wienberg 2004), the chromosomal painting approach has the advantage that data are available for over 80 mammals (50 primates). Alternatively, computational methods that attempt to identify orthologous genomic intervals have much higher resolution, potentially down to under a kilobase. However, only a handful of vertebrate genomes are currently sequenced with sufficient precision and completeness to be informative for such an analysis. Using a combination of these two approaches, Murphy et al. (2005) estimated the rearrangement rates in the lineages leading to human, mouse, rat, cat, cattle, dog, pig, and horse, and predicted that the Boreoeutherian ancestor had 24 chromosomes.
To predict large-scale relationships among modern and ancestral genomes with sufficient accuracy for our needs, we have devised new methods. Conserved genomic segments are identified directly from freely available data and analyzed by a new computer program, as described below. We also estimate the accuracy of our results, compare them with published analyses, and explore the biological properties of rearrangement sites. The computer software described herein and details of our predictions for human, mouse, rat, and dog are freely available at http://www.bx.psu.edu/miller_lab/.
Results
Segmenting the genomes based on pair-wise alignments
To predict segments of the ancestral genome, we start with “nets” (Kent et al. 2003), downloaded from the UCSC Human Genome Browser (http://genome.ucsc.edu/) (Kent et al. 2002). A net is an alignment between putatively orthologous regions in two genomes, where it is predicted that no large-scale rearrangements occurred since the last common ancestor. We split nets if necessary to guarantee that they never contain an indel (insertion or deletion) of length exceeding a chosen threshold, e.g., 50 kb. Based on nets, we progressively construct sets of genomic intervals called, respectively, “orthology blocks,” “conserved segments,” and “contiguous ancestral regions” (abbreviated CARs). Each set contains pair-wise orthologous genomic intervals, one from each species under consideration, which for the current study means human (build hg18, March 2006) (Human Genome Sequencing Consortium 2001), mouse (build mm8, Feb. 2006) (Mouse Genome Sequencing Consortium 2002), rat (build rn3, June 2003) (Rat Genome Sequencing Consortium 2004), and dog (build canFam2, May 2005) (Lindblad-Toh et al. 2005). Since the intervals in a given set are orthologous, the set corresponds to a genomic interval in the last common ancestor of those species. We categorized the set according to restrictions on the kinds of large-scale evolutionary operations predicted to have happened in the lineages leading to the modern species, as summarized in Table 1.
Table 1.
Types of orthologous-interval sets discussed in this article

By “large rearrangement,” we mean inversions and translocations involving genome segments exceeding some appropriate threshold in length (50 kb for the current study), or chromosome fissions or fusions.
Our methods for constructing orthology blocks and conserved segments are illustrated in Figure 2, while later sections and the Supplemental material explain the construction of CARs. Figure 2A shows human–mouse nets. Four mouse intervals are depicted, as ordered and oriented by the orthologous human segments. The second and third mouse intervals are actually adjacent (and appropriately orientated) on a mouse chromosome, and the intervening bases, if any, do not align to human; this is depicted by a thin line connecting the representations of those intervals.
Figure 2.

(A) Nets. Human is the reference species. The line between intervals indicates that a genomic interval of zero or more unaligned bases exists in the nonreference species between the adjacent intervals (see text). (B) Orthology blocks. (C) Conserved segments, including outgroup nets. The order and orientation of OB2 and OB3 are conserved in all four species, so we merge them into a conserved segment.
Figure 2B shows the human–mouse, human–rat, and human–dog nets for a segment of the human sequence and illustrates the creation of orthology blocks. A dashed line between orthology blocks lies halfway between two intervals that are adjacent relative to human. For instance, the line at human position 2 in Figure 2B is midway between two mouse intervals. When the gap between two adjacent intervals in one species overlaps a gap relative to another species, as at position 1, we use the point halfway between the larger of the interval endpoints to the left and the smaller of the endpoints on the right. We discard all orthology blocks that cover <50 kb of human, because experiments showed that they tend to be unreliable (e.g., aligned segments are not always clearly orthologous). For this reason we describe our orthology blocks as having “50-kb resolution.” Application of this process created 3171 genomic intervals, which include 92.36% of the available human genome sequence.
As illustrated in Figure 2C, we fuse runs of consecutive orthology blocks whenever the order and orientation of these blocks are conserved in each of the contemporary genomes. In terms of the convention used in Figure 2A, this means that for all nonhuman species, the boundary between the blocks is crossed either by a net or by a thin line. The results of the fusion process are independent of the order that fusions are performed. We call each resulting union of blocks a conserved segment. We found 1338 conserved segments, each containing an average of ∼2.4 orthology blocks, which include 94.81% of the available human genome sequence. To help the process of inferring adjacencies between conserved segments in the ancestral sequence, we add nets from outgroup species to the conserved segments (Fig. 2C). The intervals in a conserved segment from an outgroup species are not required to be consecutive on the same chromosome.
Figure 3 shows the length distributions of orthology blocks and conserved segments across the whole genome.
Figure 3.

Length distribution of orthology blocks and conserved segments. Both orthology blocks and conserved segments are grouped into bins of 500 kb. Counts scaled by natural logarithm are plotted against lengths (in Mb).
Predicting contiguous ancestral regions from modern adjacencies
Adjacencies of genomic content (e.g., genes) have been used as a binary character to infer phylogeny in a parsimony framework (for a survey, see Savva et al. 2003). However, in a different context, where the phylogeny is known, our objective is to predict the ancestral order and orientation based on adjacencies in modern genomes. Consider an end of a conserved segment that does not correspond to a human telomere or centromere. How can we identify the conserved segment that was adjacent in the ancestral genome? If the segment that is currently adjacent in human is identical to the one that is adjacent in dog (but a different segment is adjacent in mouse and rat), the most parsimonious assumption is that the first and second segments were adjacent in the ancestral genome (and that a disruption occurred in the rodent lineage at this genomic position).
If the same segment is adjacent to the chosen segment in human, mouse, and rat but not in dog, we need more information to confidently predict the ancestral configuration, since there is a chance that the dog adjacency is ancestral and that the breakage occurred on the short branch from the human–dog ancestor to the human–rodent ancestor (see Fig. 1). To help resolve these cases, we add outgroup information in the form of matches to opossum (build monDom4, Jan 2006, http://www.ncbi.nlm.nih.gov/; K. Linblad-Toh, pers. comm.) and chicken sequence (build galGal2, Feb 2004) (Hillier et al. 2004) to the conserved segments (Fig. 2C). If the outgroup information does not resolve the issue (by agreeing with either the human adjacency or the dog adjacency), we assume the more likely scenario, i.e., that the break occurred in the lineage leading to dog.
We have generalized these observations to develop a computational procedure for predicting the order and orientation of conserved segments (and hence of orthology blocks) in the ancestor, based on observed adjacency relationships in the modern genomes. In broadest outline, the method is analogous to Fitch’s parsimony method (Fitch 1971) for phylogenetic reconstruction, but adjacencies replace nucleotides as phylogenetic characters. Details of the method can be formulated precisely using concepts from graph theory (see Methods and a detailed example in the Supplemental material). We call each predicted ancestral run of segments a “contiguous ancestral region,” abbreviated CAR. We found 29 CARs in total from the data we used (see Fig. 4 and Table 2). If we add the human sequence between conserved segments that are adjacent in both human and the ancestor (since a nucleotide-level reconstruction can include those intervals), 96.8% of the available human genome sequence is included. In Figure 4 we also use some chromosome painting results to combine CARs (leaving gaps in the figure) into our prediction of the genome structure of the Boreoeutherian ancestor. In Figure 4, black tick marks indicate joins with relatively weak support. For example, the leftmost tick mark on CAR 1 (which corresponds to human chr21 and chr3) shows a predicted ancestral adjacency between two conserved segments (conserved segments 238 and 239 in the Supplemental material) that are adjacent in human, mouse, and rat but not in dog and the outgroups.
Figure 4.

Map of the Boreoeutherian ancestral genome. For lengths of each CAR and corresponding parts in mouse, rat, and dog, see Table 2. Numbers above bars indicate the corresponding human chromosomes. Black tick marks below the bars indicate ambiguous joins (Fig. 7 in Methods; for details, see Supplemental material). Our predicted CARs are colored and ordered to facilitate comparison with Froenicke et al. (2006). Gaps between CARs are joins suggested by Froenicke et al. (2006). Diagonal lines within each block show the orientation and position in the human chromosome (Bourque et al. 2006).
Table 2.
Number of conserved segments involved in each of 29 CARs

This table also shows how each contiguous ancestral region has been scattered among chromosomes in human, mouse, rat, and dog.
In order to estimate the breakages on each lineage, we also reconstructed the intermediate ancestral genomes, i.e., the rodent ancestor and human–rodent ancestor. Boreoeutherian adjacencies were propagated to the intermediate ancestors. See the Methods section for the inference algorithm.
Identification of small inversions
Within each conserved segment, we identified in-place inversions that are too small to create a new orthology block. Because we currently lack a good outgroup species (such as elephant or armadillo), it was frequently difficult to confidently predict ancestral orientation. In ambiguous cases, we assumed that human is in the ancestral orientation relative to the immediately flanking regions, in part because the human assembly is more accurate than the others. However, this means that inversions in the human lineage are currently underestimated.
This method assigns 856 inversions to human, 3210 to mouse, 3067 to rat, and 4924 to dog. Among the 4924 inversions assigned to the dog lineage, only 703 were confirmed by an outgroup (e.g., opossum agreed with human); many of the remaining 4221 will be resolved by better outgroup data, e.g., from elephant. Figure 5 shows the length distributions of observed inversions assigned to each species. Among human inversions, 29 were assigned to the short branch leading from the Boreoeutherian ancestor to the human–rodent ancestor. The shortest inversions we found are 87 bp (in human), 31 bp (in mouse), 36 bp (in rat), and 34 bp (in dog). Figure 6 gives a detailed map of CAR 16, including small inversions. The detailed coordinates of these inversions and the tools for identifying them are freely available from http://www.bx.psu.edu/miller_lab/.
Figure 5.

Length distribution of predicted inversions in human (A), mouse (B), rat (C), and dog (D). Inversions of lengths >10 kb are not represented in the plots. Inversions lengths are grouped into bins of 250 bp.
Figure 6.

Detailed map of human chromosome 13q onto CAR 16. There are16 large-scale rearrangement-free pieces. Vertical lines above each piece indicate positions of small in-place inversions. The star indicates an inversion (around hg18.chr13:57,380,591–57,383,765) that happened on the branch from the Boreoeutherian ancestor to the human–rodent ancestor.
Properties of the breakpoints
Of 1309 pairs of conserved intervals that were predicted to be adjacent in the Boreoeutherian ancestor, 149 (11%) were separated by events in at least two independent lineages (12 were separated in three lineages). When we omit rat (because of the potential assembly problems indicated in Fig. 1), we find breakpoint reuse for 57 of 742 (8%). The ratio of breakpoint reuse we found is lower than what was reported in Murphy et al. (2005). One reason is that we use higher resolution to partition the genomes. Another reason is that we only use four Boreoeutherian descendant species in our study. Some of the breakpoints identified using dog and rodent might be reused in other Boreoeutherian descendants. Simulations, described below, suggest that this frequency of breakpoint reuse is approximately what one would expect if breakage was equally likely for every genomic position, but a careful analysis is beyond the scope of this study. See Peng et al. (2006), Sankoff (2006), and references therein for an introduction to the long-standing debate about validity of the uniform breakage model.
We inspected intervals around breakpoints in the human sequence, looking for properties that might help explain why breaks occur at some positions but not others. We used 50-kb intervals centered on the end of a conserved segment where the adjacent segments in the ancestor and human differ. Breaks that occurred only in the human lineage were treated separately from those that were reused in another species, giving 16.97 Mb of human-specific breakpoint regions and 11.96 Mb around reused breakpoints. For small inversions in human, we used 1-kb intervals centered on each endpoint, covering 1.60 Mb.
Our observations are summarized in Table 3. GC content around breakpoints is slightly higher than the genome average, but not as elevated as reported for dog chromosomal breaks by Webber and Ponting (2005). The breakpoint regions are substantially enriched for RefSeq genes, consistent with what Murphy et al. (2005) observed in larger (∼1 Mb) regions around breakpoints. The density of SINEs is also much higher than average. Finally, we observed that a large amount of DNA (41.72%) in human-specific breakpoint regions is in human segmental duplications (Bailey et al. 2001).
Table 3.
Genomic content of breakpoint regions

Gene density is not given for inversions because we used regions too short to give meaningful results. Repeats were identified with RepeatMasker, and segmental duplications were obtained from UCSC Genome Browser segmental duplication track.
Theoretical analysis
We wanted to assess what proportion of the ancestral adjacencies inferred by our method might be correct. We started with a theoretical analysis, which necessitated some rather severe assumptions. Following the method of Sankoff and Blanchette (1999), we imagine a genome π with n elements that evolves through a series of rearrangements. An entry of π can be thought of as one of the 2n numbers 1, −1, 2, −2, .., n, −n, where the sign designates the element’s orientation. No assumptions are made about the relative frequencies of inversions, translocations, fusions, and fissions. Instead, if f precedes g in the genome π = ..fg.. at a particular time, then g may be changed to h (other than f, −f or g), with each of the 2n − 3 possibilities equally likely. The probabilities that the successor of f is changed in time t along a specific branch are pivotal parameters of the model. We estimated those parameters using data observed in real genomes. A surprisingly complex computation (see the Supplemental material) then estimated that ∼90.18% of the adjacencies we predict in the Boreoeutherian ancestor are correct.
Mathematical tractability of this model is purchased at a high cost in realism. For instance, the assumption that all replacements are equally likely contradicts the observations that inversions are more frequent than are other rearrangements and that short inversions are more common than are long ones.
Simulations
We used computer simulations to inject more realism into the analysis, employing a realistic evolutionary tree with branch lengths based on substitution frequencies. Starting with a hypothetical human–chicken ancestor having 6000 orthology blocks and 25 chromosomes, we simulated inversions, translocations, fusions, and fissions along each branch. Rearrangements were distributed as 90% inversions, 5% translocations, 3.75% fusions, and 1.25% fissions. We modeled lengths of inverted blocks with a γ distribution, with shape and scale parameters α = 0.7 and scale θ = 500, respectively. Since we required inversion lengths not to exceed 50 blocks, we truncated the γ distribution at 50 (probabilities for shorter lengths are renormalized). Simulation parameters, especially the rates of rearrangements, were further tuned according to our observed data. For example, we also allowed each branch to have its own adjustment parameter for each operation, to account for the differences among branches. The simulated genomes produced by this approach are consistent with actual mammalian genomes in terms of number of conserved segments, number of breakpoints, chromosome count, etc. Some important features of the simulated data are compared with what were seen in real data in Table 4.
Table 4.
Comparison between our simulated data and real data

Simulated statistics are the average from 50 simulated data sets. The standard deviations of numbers in simulated data are in parentheses.
For the species shown in Figure 1, we repeated the simulation 50 times, in each case running our program for inferring CARs on the resulting data set and comparing the predicted adjacencies with the known (simulated) ones. For determining the success rate, we considered only the ancestral joins that were broken in at least one lineage, since the unbroken joins will be found by essentially any procedure.
When using human, mouse, rat, and dog, with opossum and chicken as outgroups (Fig. 1), the frequency of correctly predicted adjacencies was 98.96% (SD = 0.39) for the Boreoeutherian ancestor, 98.37% (SD = 0.55) for the human–rodent ancestor, and 97.07% (SD = 1.01) for the mouse–rat ancestor. Note that as for inference of nucleotides (Blanchette et al. 2004), the prediction accuracy is also higher for the Boreoeutherian ancestor than for some younger ancient genomes.
We also reconstructed the Boreoeutherian ancestor without using opossum and chicken; the accuracy decreased to 97.40% (SD = 0.69). If we retain the outgroups but leave out rat, the accuracy drops to 98.29% (SD = 0.67). However, if chimp, cow, and macaque are included in the reconstruction, the simulation indicates that joins in the Boreoeutherian ancestor are computed with 99.34% (SD = 0.29) accuracy.
Comparison with other reconstructions
A comparison with other reconstructions identifies which part of the ancestral genome can be confidently reconstructed and highlights regions where further investigation is needed. Our predicted CARs agree well with predictions from chromosome painting with respect to interchromosomal operations (cf. Fig. 1 in Froenicke et al. 2006 and Fig. 4). Of eight strongly supported interchromosomal breaks in human predicted by Froenicke et al. our reconstruction agrees with five (see Table 5). In the prediction of Murphy et al. (2005), joins of Hsa16q/Hsa19q and Hsa16p/Hsa7 were reconstructed with weak support, using data from cat and pig, and cat and cow, respectively, all of which were unavailable for computing CARs. Also, in both cases, the species (cat and pig or cat and cow) come from the same superorder, so they did not provide strong evidence as to the Boreoeutherian ancestral state. Therefore, more species are needed. Moreover, the endpoints of the relevant conserved segments for these two joins are all connected with ambiguity in our prediction (see Fig. 4), suggesting other possible scenarios which are very likely to be joins of Hsa16q/Hsa19q and Hsa16p/Hsa7. The join of Hsa16p/Hsa7 corresponds to the centromere of Hsa16 (the first weakly supported join on CAR17) and the left side of CAR22 in the picture. We think the small sample size of Boreoeutherian descendant genomes in the current study is likely responsible for the failure to recover these adjacencies.
Table 5.
Comparison with Froenicke et al. (2006) and Murphy et al. (2005)

The naming convention of human chromosomes’ regions follows Figure 1 in Froenicke et al. 2006. “+” indicates the method that made that join, −, otherwise. If the join was made by our method, we also list the corresponding conserved segment numbers and show which species support that join.
The predictions of Murphy et al. (2005) which were mainly based on the MGR algorithm (Bourque and Pevzner 2002), are more or less similar to Froenicke et al.’s (2006) results. However, five of Murphy’s putative weakly supported interchromosomal breaks (Hsa1/Hsa22, Hsa5/Hsa19, Hsa2/Hsa18, Hsa1/Hsa10, Hsa2/Hsa20) are not supported by chromosome painting data. Our program made none of these joins, which is in agreement with Froenicke et al. To further examine the differences between reconstruction algorithms, we ran our CAR-building program on Murphy et al.’s data set with 307 conserved segments from their Supplemental materials. Table 6 compares our predicted ancestral joins with theirs.
Table 6.
Results of running our CAR-building program on data from Murphy et al. (2005)

Endpoint joins have two cases: (1) the join between the first conserved segment in a chromosome and the beginning of the chromosome; (2) the join between the last conserved segment in a chromosome and the end of the chromosome. Non-human-consecutive joins refer to two consecutive elements in human that are not consecutive in the ancestor, indicating a breakpoint in human.
Discussion
A number of additional mammals are already being sequenced “at low redundancy” for the purpose of identifying human regions under negative selection for substitutions (Margulies et al. 2005). The resulting sequence data are extremely useful for predicting ancestral nucleotides. However, they lack the long-range contiguity needed for accurate identification of large-scale evolutionary events. We look forward to the day when a high-accuracy assembly of, say, elephant or armadillo provides us with an ideal outgroup for large-scale reconstruction of the Boreoeutherian ancestral sequence.
Our computational methods need further refinement. In particular, we anticipate improvements in the handling of large duplications and deletions. Additional progress may be possible through the modeling of other evolutionary events, such as gene conversion or expansion/contraction of short tandem repeats caused by strand slippage. Moreover, advances are needed to facilitate simultaneous reconstruction of ancestral genomes at all internal nodes of the phylogenetic tree.
An accurate conceptual model of large-scale evolutionary events will be critical for successful reconstruction of ancestral genomes. The discrepancy between our theoretical analysis of reconstruction accuracy and the results from simulation underline the need for more work in this area.
A number of challenges remain before the genome sequences of mammalian ancestors can be accurately predicted at nucleotide resolution. However, we believe that this goal is an appropriate focus for our twin aims of understanding the evolutionary history of every position in the human genome and of providing an Internet resource that optimally organizes and presents the ever-expanding wealth of mammalian and vertebrate sequence data in the context of the ancestral sequence they have in common.
Methods
Inferring CARs
Given information about adjacencies between conserved segments in each modern species, our goal is to infer segment order in the ancestral genome. To get a clean and precise statement of the problem, we formalize it using graph theory. We develop an algorithm that identifies a most parsimonious scenario for the history of each individual adjacency, although the whole-genome prediction is not guaranteed to optimize traditional measures like the number of breakpoints. We introduce weights to the graph edges to model the reliability of each adjacency.
By encoding each genomic element as a number, we view a chromosome as a signed permutation where the sign corresponds to the orientation (strand) of the encoded element. For example, π = 1–4 −3 5 2 denotes a chromosome of five genomic elements, in which elements 1, 5, 2 are on the positive strand and 3, 4 are on the negative strand. Currently, we do not allow duplications, i.e., two elements with the same absolute value. Each element πi in a chromosome π = π1 π2 . . . πi-1 πi πi+1 . . . πn has a unique predecessor πi−1 and successor πi+1,; we define that π1 has predecessor 0 and πn has successor 0, where 0 is a special symbol for the left and right end of each chromosome. Inferring CARs in the ancestral genome is equivalent to finding the unambiguous predecessor and successor of each element in the ancestral genome.
Recall that the Fitch algorithm infers the ancestral DNA sequence at the root of a phylogenetic tree from the DNA sequences at the leaves, site-by-site in two stages (Felsenstein 2004). For each site, in a bottom-up fashion, it first determines a set Cv of candidate nucleotides at each internal node v according to the following rule: If v is a leaf, Cv just contains its nucleotide label; otherwise, if v has children u, w, then Cv equals Cu ∪ Cw or Cu ∩ Cw depending on whether Cu and Cw are disjoint or not. Then, in a top-down fashion, it assigns a nucleotide bv from Cv to v according the following rule: Let v′ be the parent of v; if the nucleotide bv′ assigned to v′ belongs to Cv, then, bv = bv′. Otherwise, set bv to be any nucleotide in Cv. Although nucleotide assignment in the second stage is not unique, any assignment gives an evolutionary history with the minimum number of substitution events.
To infer the ancestral predecessor of an element x, our algorithm does exactly the same thing as described above. In a bottom-up fashion, for each node u, we compute a set Pu(i) of the “predecessors” for each element i according to the following rule:
If u is a leaf, then Pu(i) consists of the unique predecessor in the genome associated with u. Otherwise, assume u has children v and w; then, Pu(i) is equal to Pv(i) ∪ Pw(i) or Pv(i) ∩ Pw(i) depending on whether Pv(i) and Pw(i) are disjoint or not.
The above inference method outputs a “predecessor graph” at each internal node. In this graph there is an arc from each element in Pu(i) to element i. In general, such a predecessor graph may not be a union of vertex-disjoint paths. This is because two different series of evolutionary events may transform different genomes into the same one, and hence, we often do not have enough information to determine the true predecessor and successor relationships in the ancestor. However, the vast majority of the ancestral adjacencies are likely to be present as edges in the inferred graph.
We treat outgroups in a consistent manner. We first infer the predecessor set PR(i) in the common ancestor R of all the species, including outgroups. Then we propagate PR(i) down the tree until we reach the target ancestor T. During the propagation process, if O and A are ancestor and descendant on one branch, respectively, for each element i, we adjust the inferred predecessor set PA(i) of i at the mammalian ancestor A as follows: If PO(i) and PA(i) share common elements, we just take them as the predecessor set of i at A; otherwise, PA(i) is unchanged. In our reconstruction, we first infer the predecessor set of human–chicken common ancestor. Then we used it to adjust the human–opossum common ancestor. And finally, we used human-opossum ancestor to adjust the human–dog common ancestor.
Similarly, we infer a “successor graph” at each internal node in the given phylogeny. In a successor graph, there is an arc from element i to each element in its inferred successor set S(i) at each internal node.
The predecessor and successor graphs are the same at each leaf, but those at an internal node are generally different, although they typically have many common parts. We find the consistent parts by taking the intersection graph G of the predecessor and successor graphs. In the intersection graph, an element could still be involved in three kinds of ambiguities, as depicted in Figure 7. If none of these ambiguous cases is present, the graph itself forms the set of paths that covers all the elements and provides the reconstructed ancestral genome structure.
Figure 7.

Three potential ambiguous cases. (A) i has several possible predecessors; (B) i has several possible successors; (C) i forms a cycle with j.
When ambiguity exists, we need to resolve it and choose appropriate directed edges to form CARs. We assign a weight to each of the directed edges in the intersection graph using the following approach. If the edge (i, j) is neither one of the incoming edges of case (a) nor one of the outgoing edges of case (b), we set wα(i, j) = 1. Otherwise, the weight wα(i, j) is recursively determined from the leaves by
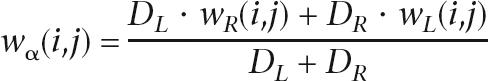 |
where DL and DR are the branch lengths to the left child L and right child R branching from ancestral node α. Here, wL(i, j) and wR(i, j) are the edge weights to left child and right child. Therefore, in the graph, all the edge weights are between 0 and 1.
On a leaf genome, if (i, j) is present in the predecessor graph, we set w(i, j) = 1; otherwise w(i, j) = 1. This weight can be determined by a post-order traversal. Note that if an edge (i, j) is involved in ambiguous case (a) or (b), then w(i, j) < 1. The greater the weight of an edge (i, j), the more likely it is that i and j should be joined. The underlying assumption is that rearrangement is more likely to happen on longer branches.
Our purpose is to have a set of paths that cover all the nodes in the directed graph and at the same time maximize the total edge weights in the paths (we allow degenerate paths where there is only one node in the path). We propose a greedy heuristic approach. We first sort all the edges by weight and start to add edges to the vertex-disjoint paths representing the CARs. When an edge (i, j) is being added, we make sure that it will not cause ambiguous cases. If it will, we discard that edge and choose the next available one. The process is performed until no edge can be added. In the graph resulting from this step, all the edges that are not involved in ambiguous cases (a) and (b) will be retained. For ambiguous case (c) where a cycle is formed, we can discard any one edge to break the cycle. In fact, we can easily prove that if there is a cycle, the weight of each edge in that cycle is 1.
When we are adding elements into an existing path, particular care is needed to avoid putting j and −j in the same CAR. In addition, we add both (i, j) and its symmetric version, (−j, −i). For each path found by this approach, a symmetric path in the opposite orientation is also found, since we have nodes for both i and −i. The two paths correspond to the same CAR, and we choose one of them.
A detailed example of the algorithm can be found in the Supplemental materials.
Acknowledgments
We thank the Broad Institute for making the opossum assembly publicly available, Francesca Chiaromonte for several helpful suggestions, Elliott H. Margulies for providing the phylogenetic tree used in ENCODE project, and Guillaume Bourque for the MGR program. We especially would like to thank the anonymous reviewers for their comments on the initial draft of this paper. J.M., R.C.B., and W.M. were supported by NIH grant HG02238. L.X.Z. was supported by ARF grant 146-000-068-112. W.J.K, B.J.R, and D.H. were supported by NHGRI grant 1P41HG02371 and NCI contract 22XS013A, and D.H. was supported additionally by the Howard Hughes Medical Institute.
Footnotes
[Supplemental material is available online at www.genome.org and http://www.bx.psu.edu/miller_lab/.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.5383506
References
- Bailey J.A., Yavor A.M., Massa H.F., Trask B.J., Eichler E.E., Yavor A.M., Massa H.F., Trask B.J., Eichler E.E., Massa H.F., Trask B.J., Eichler E.E., Trask B.J., Eichler E.E., Eichler E.E. Segmental duplications: Organization and impact within the current human genome project assembly. Genome Res. 2001;11:1005–1017. doi: 10.1101/gr.187101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette M., Green E.D., Miller W., Haussler D., Green E.D., Miller W., Haussler D., Miller W., Haussler D., Haussler D. Reconstructing large regions of an ancestral mammalian genome in silico. Genome Res. 2004;14:2412–2423. doi: 10.1101/gr.2800104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourque G., Pevzner P.A., Pevzner P.A. Genome-scale evolution: Reconstructing gene orders in the ancestral species. Genome Res. 2002;12:26–36. [PMC free article] [PubMed] [Google Scholar]
- Bourque G., Tesler G., Pevzner P.A., Tesler G., Pevzner P.A., Pevzner P.A. The convergence of cytogenetics and rearrangement-based models for ancestral genome reconstruction. Genome Res. 2006;16:311–313. doi: 10.1101/gr.4631806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Inferring phylogenies. Sinauer; Sunderland, MA: 2004. [Google Scholar]
- Fitch W.M. Toward defining the course of evolution: Minimum change for a specific tree topology. Syst. Zool. 1971;20:406–416. [Google Scholar]
- Froenicke L., Caldes M.G., Graphodatsky A., Muller S., Lyons L.A., Robinson T.J., Volleth M., Yang F., Wienberg J., Caldes M.G., Graphodatsky A., Muller S., Lyons L.A., Robinson T.J., Volleth M., Yang F., Wienberg J., Graphodatsky A., Muller S., Lyons L.A., Robinson T.J., Volleth M., Yang F., Wienberg J., Muller S., Lyons L.A., Robinson T.J., Volleth M., Yang F., Wienberg J., Lyons L.A., Robinson T.J., Volleth M., Yang F., Wienberg J., Robinson T.J., Volleth M., Yang F., Wienberg J., Volleth M., Yang F., Wienberg J., Yang F., Wienberg J., Wienberg J. Are molecular cytogenetics and bioinformatics suggesting diverging models of ancestral mammalian genomes? Genome Res. 2006;16:306–310. doi: 10.1101/gr.3955206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier L.W., Miller W., Birney E., Warren W., Hardison R.C., Ponting C.P., Bork P., Burt D.W., Groenen M.A., Delany M.E., Miller W., Birney E., Warren W., Hardison R.C., Ponting C.P., Bork P., Burt D.W., Groenen M.A., Delany M.E., Birney E., Warren W., Hardison R.C., Ponting C.P., Bork P., Burt D.W., Groenen M.A., Delany M.E., Warren W., Hardison R.C., Ponting C.P., Bork P., Burt D.W., Groenen M.A., Delany M.E., Hardison R.C., Ponting C.P., Bork P., Burt D.W., Groenen M.A., Delany M.E., Ponting C.P., Bork P., Burt D.W., Groenen M.A., Delany M.E., Bork P., Burt D.W., Groenen M.A., Delany M.E., Burt D.W., Groenen M.A., Delany M.E., Groenen M.A., Delany M.E., Delany M.E., et al. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004;432:695–716. doi: 10.1038/nature03154. [DOI] [PubMed] [Google Scholar]
- Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D., Pringle T.H., Zahler A.M., Haussler D., Zahler A.M., Haussler D., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W.J., Baertsch R., Hinrichs A., Miller W., Haussler D., Baertsch R., Hinrichs A., Miller W., Haussler D., Hinrichs A., Miller W., Haussler D., Miller W., Haussler D., Haussler D. Evolution’s cauldron: Duplication, deletion, and rearrangement in the mouse and human genomes. Proc. Natl. Acad. Sci. 2003;100:11484–11489. doi: 10.1073/pnas.1932072100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindblad-Toh K., Wade C.M., Mikkelsen T.S., Karlsson E.K., Jaffe D.B., Kamal M., Clamp M., Chang J.L., Kulbokas E.J., III, Zody M.C., Wade C.M., Mikkelsen T.S., Karlsson E.K., Jaffe D.B., Kamal M., Clamp M., Chang J.L., Kulbokas E.J., III, Zody M.C., Mikkelsen T.S., Karlsson E.K., Jaffe D.B., Kamal M., Clamp M., Chang J.L., Kulbokas E.J., III, Zody M.C., Karlsson E.K., Jaffe D.B., Kamal M., Clamp M., Chang J.L., Kulbokas E.J., III, Zody M.C., Jaffe D.B., Kamal M., Clamp M., Chang J.L., Kulbokas E.J., III, Zody M.C., Kamal M., Clamp M., Chang J.L., Kulbokas E.J., III, Zody M.C., Clamp M., Chang J.L., Kulbokas E.J., III, Zody M.C., Chang J.L., Kulbokas E.J., III, Zody M.C., Kulbokas E.J., III, Zody M.C., Zody M.C., et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005;438:803–819. doi: 10.1038/nature04338. [DOI] [PubMed] [Google Scholar]
- Margulies E.H., Vinson J.P., Miller W., Jaffe D.B., Lindblad-Toh K., Chang J.L., Green E.D., Lander E.S., Mullikin J.C., Clamp M., Vinson J.P., Miller W., Jaffe D.B., Lindblad-Toh K., Chang J.L., Green E.D., Lander E.S., Mullikin J.C., Clamp M., Miller W., Jaffe D.B., Lindblad-Toh K., Chang J.L., Green E.D., Lander E.S., Mullikin J.C., Clamp M., Jaffe D.B., Lindblad-Toh K., Chang J.L., Green E.D., Lander E.S., Mullikin J.C., Clamp M., Lindblad-Toh K., Chang J.L., Green E.D., Lander E.S., Mullikin J.C., Clamp M., Chang J.L., Green E.D., Lander E.S., Mullikin J.C., Clamp M., Green E.D., Lander E.S., Mullikin J.C., Clamp M., Lander E.S., Mullikin J.C., Clamp M., Mullikin J.C., Clamp M., Clamp M. An initial strategy for the systematic identification of functional elements in the human genome by low-redundancy comparative sequencing. Proc. Natl. Acad. Sci. 2005;102:4795–4800. doi: 10.1073/pnas.0409882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouse Genome Sequencing Consortium Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Murphy W.J., Larkin D.M., Everts-van der Wind A., Bourque G., Tesler G., Auvil L., Beever J.E., Chowdhary B.P., Galibert F., Gatzke L., Larkin D.M., Everts-van der Wind A., Bourque G., Tesler G., Auvil L., Beever J.E., Chowdhary B.P., Galibert F., Gatzke L., Everts-van der Wind A., Bourque G., Tesler G., Auvil L., Beever J.E., Chowdhary B.P., Galibert F., Gatzke L., Bourque G., Tesler G., Auvil L., Beever J.E., Chowdhary B.P., Galibert F., Gatzke L., Tesler G., Auvil L., Beever J.E., Chowdhary B.P., Galibert F., Gatzke L., Auvil L., Beever J.E., Chowdhary B.P., Galibert F., Gatzke L., Beever J.E., Chowdhary B.P., Galibert F., Gatzke L., Chowdhary B.P., Galibert F., Gatzke L., Galibert F., Gatzke L., Gatzke L., et al. Dynamics of mammalian chromosome evolution inferred from multispecies comparative maps. Science. 2005;309:613–617. doi: 10.1126/science.1111387. [DOI] [PubMed] [Google Scholar]
- Peng Q., Pevzner P.A., Tesler G., Pevzner P.A., Tesler G., Tesler G. The fragile breakage versus random breakage models of chromosome evolution. PLoS Comp. Biol. 2006;2:e14. doi: 10.1371/journal.pcbi.0020014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rat Genome Sequencing Consortium Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004;428:493–521. doi: 10.1038/nature02426. [DOI] [PubMed] [Google Scholar]
- Sankoff D. The signal in the genomes. PLoS Comput. Biol. 2006;2:e35. doi: 10.1371/journal.pcbi.0020035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankoff D., Blanchette M., Blanchette M. RECOMB ’99: Proceedings of the third annual international conference on computational molecular biology. ACM Press; New York: 1999. Probability models for genome rearrangement and linear invariants for phylogenetic inference; pp. 302–309. [Google Scholar]
- Savva G., Dicks J., Roberts I.N., Dicks J., Roberts I.N., Roberts I.N. Current approaches to whole genome phylogenetic analysis. Brief. Bioinform. 2003;4:63–74. doi: 10.1093/bib/4.1.63. [DOI] [PubMed] [Google Scholar]
- Webber C., Ponting C.P., Ponting C.P. Hotspots of mutation and breakage in dog and human chromosomes. Genome Res. 2005;15:1787–1797. doi: 10.1101/gr.3896805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienberg J. The evolution of eutherian chromosomes. Curr. Opin. Genet. Dev. 2004;14:657–666. doi: 10.1016/j.gde.2004.10.001. [DOI] [PubMed] [Google Scholar]