

Abstract
A major obstacle to successful islet transplantation for both type 1 and 2 diabetes is an inadequate supply of insulin-producing tissue. This need for transplantable human islets has stimulated efforts to expand existing pancreatic islets and/or grow new ones. To test the hypothesis that human adult duct tissue could be expanded and differentiated in vitro to form islet cells, digested pancreatic tissue that is normally discarded from eight human islet isolations was cultured under conditions that allowed expansion of the ductal cells as a monolayer whereupon the cells were overlaid with a thin layer of Matrigel. With this manipulation, the monolayer of epithelial cells formed three-dimensional structures of ductal cysts from which 50-to 150-μm diameter islet-like clusters of pancreatic endocrine cells budded. Over 3–4 weeks culture the insulin content per flask increased 10- to 15-fold as the DNA content increased up to 7-fold. The cultivated human islet buds were shown by immunofluorescence to consist of cytokeratin 19-positive duct cells and hormone-positive islet cells. Double staining of insulin and non-β cell hormones in occasional cells indicated immature cells still in the process of differentiation. Insulin secretion studies were done over 24 h in culture. Compared with their basal secretion at 5 mM glucose, cysts/cultivated human islet buds exposed to stimulatory 20 mM glucose had a 2.3-fold increase in secreted insulin. Thus, duct tissue from human pancreas can be expanded in culture and then be directed to differentiate into glucose responsive islet tissue in vitro. This approach may provide a potential new source of pancreatic islet cells for transplantation.
One of the main obstacles to successful islet transplantation for both type 1 and 2 diabetes is the limitation of available insulin-producing tissue (1). Only about 3,000 cadaver pancreases become available in the U.S. each year while about 35,000 new cases of type 1 diabetes are diagnosed each year (2) This lack of tissue has given a high priority to efforts to stimulate the growth of new pancreatic islet tissue. Most studies have shown there is limited in vitro growth of adult islet cells of any species (3), but several recent reports have found cell proliferation using cultures of adult human islet preparations with extracellular matrix and growth factors (4–8) but these have been associated with loss of insulin production.
From studies on rat pancreatic regeneration (9, 10) we were impressed with the capacity of adult pancreatic duct cells to both expand and differentiate. These data led to the hypothesis (10, 11) that adult duct cells have the potential to lose their specific duct phenotype with rapid proliferation, reverting to multiipotent cells that then can differentiate into islet cells with the appropriate external stimuli. The potential of extracellular matrix as such an external stimulus has been suggested for other cell types (12). Extracellular matrix, in particular laminin, was shown to induce β-casein expression in cultured mammary duct cells (13). Additionally, an overlay of Matrigel, an extracellular matrix preparation, induced the expression of liver-specific genes in clonally expanded hepatocytes (14). Herein we show expansion of human ductal tissue in vitro and its subsequent differentiation to islet cells after being overlaid with Matrigel. Over 3–4 weeks culture there was a significant increase in insulin as well as formation of islet-like structures that we have called cultivated human islet buds (CHIBs).
Materials and Methods
Initial Tissue and Culture Conditions.
Human islet isolations were performed in the Islet Core Laboratory of the Juvenile Diabetes Foundation Center for Islet Transplantation at Harvard Medical School using the method of Ricordi and coworkers (15). After purification on a Ficoll gradient, the top interface (1.062/1.096 densities) was 50–95% islet with varying amounts of duct and degranulated acinar tissue, the middle interface (1.096/1.11 densities) contained 1–15% islets, duct, and degranulated acini, and the pellet was mostly well granulated acinar tissue with less than 1% islets. In the top and middle layers there were sheets of ductal epithelium from larger ducts whereas the clumps of exocrine cells found in all layers consisted of small intercalated ducts continuing into the acini. From eight collagenase (Liberase, Roche) digested pancreases (donor age 27–59 years), tissue from these layers was cultured in 50 ml of CMRL 1066 (5.6 mM glucose) media plus 10% FBS in Falcon nontreated T-75 flasks (#3012 Becton Dickinson) at 37°C, 5% CO2. At 1–4 days the nonadherent tissue (both viable and dead) was removed with a media change, and the adherent, or residual, cells were expanded for up to 1 week with additional media changes every 2–3 days. At about 1 week, when most, if not all, adherent cells were in monolayer, the media was changed to 20 ml of serum-free DMEM/F12 (8 mM glucose) medium with 1 g/liter ITS supplement (5 mg/liter insulin + 5 mg/liter transferrin + 5 mg/liter selenium, Sigma), 100 units/ml penicillin, 100 μg/ml streptomycin, 2 g/liter BSA, 10 mM nicotinamide, and keratinocyte growth factor (KGF)(10 ng/ml, Roche). We previously had found that DMEM/F12 (8 mM glucose, plus nicotinamide) facilitated growth of rat and pig duct cells in vitro. KGF has been reported to be a duct mitogen (16), and we previously had found it to stimulate ductal proliferation in vitro without evident changes in cell phenotype. These cells then were grown for about 1–2 weeks until reaching near confluence or forming substantial plaques of epithelial cells. The cells then were layered with Matrigel, a commercial preparation of murine basement membrane (Collaborative Research-Becton Dickinson) as per instructions of supplier for thin layer gel with the exception of an increased gelling time at 37°C. Briefly, the cells were coated with 50 μl Matrigel per cm2 and allowed to gel overnight before additional media was added. Cell samples were taken at different time points over the course of 6 weeks. Dithizone (diphenylthiocarbozone), which stains insulin-containing cells bright red, was used to assess quickly the presence of insulin-producing cells.
Tissue Fixation and Immunochemistry.
Monolayer cultures were fixed for 30 min in either 4% (para) formaldehyde (PFA) in 0.1 M phosphate buffer or in Bouin's solution, and then rinsed in the phosphate buffer. Three-dimensional structures (cysts) that formed from these monolayers were harvested by mechanical shearing with a stream of media. Harvested cysts were fixed in PFA for 60 min, enrobed in 2% agar to keep the pellet together through processing and embedding, immersed in the same fixative for another 90 min, washed, and stored in 0.1 M phosphate buffer until routine embedding in paraffin; sections of these were used for immunostaining. Other cysts were fixed in 2.5% glutaraldehyde/0.1 M phosphate buffer for 2 h, washed, and stored in phosphate buffer until being embedded in plastic resin (Araldite, E. F. Fullam, Lanthan, NY) for semithin (1 μm) sections or ultrathin sections for ultrastructural analysis.
Double immunofluorescent staining were done sequentially by using primary antibodies made in different species: Guinea pig anti-human insulin (1:200, Linco Research Immunoassay, St. Charles, MO), rabbit anti-bovine glucagon (1:2,000, kindly donated by M. Appel, University of Massachusetts Medical School, Worcester) and rabbit anti-bovine pancreatic polypeptide (1:3,000, gift of R. E. Chance, Eli Lilly, Indianapolis, IN), rabbit antisynthetic somatostatin (1:300, made in our own laboratory), a mixture of the latter three antibodies (anti-glucagon, -somatostatin, and -pancreatic polypeptide) for identifying the non-β islet cells (17); monoclonal mouse anti-human cytokeratin 19 (CK 19) antibody (1: 100, Dako) (18)or rabbit pancytokeratin (1: 100, Dako); IDX-1 antibody (Hm-253, dilution 1:500 from J. Habener, Massachusetts General Hospital, Boston) (19). The conjugated secondary antibodies used for immunofluorescence were Texas red-conjugated donkey anti-guinea pig IgG, FITC-conjugated donkey anti-rabbit IgG and streptavidin-conjugated FITC (1:100 dilution for all, Jackson ImmunoResearch). Biotinylated horse anti-mouse IgG and normal horse serum were purchased from Vector Laboratories. For cytokeratin and IDX-1 staining of sectioned tissue, antigens were retrieved by microwaving in citrate buffer (three times of 4 min each with the maximum strength of a domestic microwave) (20). Monolayer cultures were incubated for 10–20 min in 0.3% Triton X-100 (Fisher) with 1% lamb serum (GIBCO/BRL) before primary antibody incubation.
Insulin and DNA Content.
Harvested cysts or cells removed from flasks by treatment trypsin/EDTA (1× trypsin-EDTA solution, Cellgro, Mediatech Laboratories, Cody, NY; 10–15 min at 37°C) were brought up to 1 ml high-salt buffer (2.15 M NaCl/0.1 M NaH2PO4/0.04 M NaHPO4/EDTA, pH 7.4) and then were sonicated three times, 10 sec each at 4–6 W and then stored at −20°C until assayed. Insulin was measured by using a RIA kit for human insulin from Linco. DNA content was measured fluorometerically by using Hoechst 33258 dye as described by dyna quant (Hoefer).
RNA Extraction and Analysis.
Total RNA from samples was extracted following manufacturer-suggested protocols using Ultraspec (Biotecx Laboratories, Houston). cDNA synthesis was performed as described (19). PCR was carried out in 50-μl reactions using 3 μl of the diluted cDNA reaction product (corresponding to 20 ng RNA equivalent) as template mixed with 47 μl of PCR mix [1× Taq buffer (Promega), 1.5 mM MgCl2 (Promega), 10 pm of each insulin primers (forwards and backwards) (Genosys, The Woodlands, TX), 4 μl of 4:6 ratio of 18S primers/competimers (Classic 18S Internal Standards, Ambion, Austin TX), 80 μM cold dNTPs (GIBCO/BRL), 5 units AmpliTaq Gold DNA polymerase (Perkin–Elmer), and 2.5 μCi [α-32P]dCTP (New England Nuclear)]. Reverse transcription–PCR (RT-PCR) for insulin with 18S ribosomal subunit as internal control was run on the samples. Primers were as follows: human insulin 5′-TCA CAC CTG GTG GAA GCTC; human insulin 3′-ACA ATG CCA CGC TTC TGC (which yield a 179-bp PCR product); and for internal control 18S primers: competimers [Classic 18S Internal Standards (which yield a 488-bp PCR product)]. The thermal cycling protocol began with a denaturing step of 97°C for 10 min, then 19 cycles of 94°C 1 min, 55°C 1 min, 72°C 1 min, and finished with 72°C for 10 min. For human glucagon 5′-ATG AAC GAG GAC AAG CGC; 3′-TTC ACC AGC CAA GCA ATG (which yields a 236-bp product) and human cyclophilin 5′-CCC ACC GTG TTC TTC GAC, 3′-ATC TTC TGC TGG TCT TGCC were used. The reaction volume differed from that above in that 7.5 pmol of each glucagon primer and 25 pmol of each cyclophilin primer were used; the thermal cycle profile was the same except that 23 cycles were used and the annealing temperature was 59°C. Screens were scanned by using a Molecular Dynamics Storm PhosphorImager and reaction products were quantitated with imagequant software (Molecular Dynamics). Results are calculated as a percentage of internal standard and presented as mean ± SEM. Reaction conditions were standardized so as to observe linear amplification of PCR products (for both insulin and ribosomal 18S, glucagon and cyclophilin) for different amounts of cDNA (10–50 ng RNA equivalent) and cycle numbers (18–32 cycles). Graded dilutions (1–20%) of a human islet preparation (H99–22, 90% islet purity, 676 ng insulin/μg DNA) were run to establish a standard curve of insulin mRNA to 18 S mRNA and of glucagon mRNA to cyclophilin mRNA. By including two samples from this curve as standards in any other RT-PCR experiment, an estimate of the % islet for a sample could be made.
Insulin Secretion.
Three-dimensional structures (cysts/CHIBs) from 1–2 flasks of tissue from pancreas 19, 24, and 25 were harvested at 3–5 weeks culture and washed three times in RPMI (5 mM glucose, 10 mM Hepes, pencillin/streptomycin, 5% FBS). From each flask, 12 aliquots of 40 cysts/CHIBs were incubated in 1.5 ml of the same media in 12-well plates for 4 h at 37°C, the media were removed for measurement of preincubation insulin levels, and fresh media were added for a 24-h incubation. After this 24 h period, media were again removed and measured for basal insulin secretion, and fresh media with either 5 mM or 20 mM glucose were added. At the end of this second 24-h incubation, the final media were removed for measurement with a human insulin RIA kit (Linco).
Results
To promote the attachment of duct cells rather than islet cells, nonsticky culture flasks were used; these flasks have been used to maintain islets in suspension. With pure islet preparations obtained from the top layer of the density gradient, little tissue became adherent even with 7 days culture. It was noted, however, that clumps of nonislet tissue obtained from the top, middle, or pellet layers can adhere to this nonsticky surface starting at about 24 h. It was mainly in the less pure islet preparations that there were adherent cell clumps within 2–4 days. Although there was considerable loss of floating tissue as has been reported for pancreatic acinar tissue in culture (21, 22), the quantity of cell clumps that adhered increased with time. If the nonadherent clumps were removed when the adherent density reached an empirically determined level (covering about 10% of surface), the adherent cells had little to no dithizone staining and included few fibroblasts. Initial samples for insulin and DNA contents were taken at the removal of nonadherent tissue and before the clumps flattened into monolayers. The adherent tissue was only 2.5–24% of the original DNA and 2.5–11% of the original insulin content (Table 1). However, if the nonadherent tissue remained longer in the cultures, both the amount of adherent islet tissue (dithizone positive) and fibroblasts increased (data not shown). With additional time, cells grew from the adherent clumps and formed monolayer plaques of cells with clear epithelial morphology (Fig. 1).
Table 1.
Insulin and DNA content of 75-cm2 flask containing cultured human ductal cells
| Original aliquot, 50 ml | Initial adherent | 2 wk | 3 wk | 4 wk | |
|---|---|---|---|---|---|
| H99-13 (top + middle, 58% islet) | |||||
| Insulin, ng | 3,200 | 78.4 | — | 888 | — |
| DNA, μg | 160 | 3.8 | — | 22.1 | — |
| I/DNA, ng/μg | 20 | 20.5 | — | 40.2 | — |
| H99-19 (middle, 5% islet) | |||||
| Insulin, ng | nd | 70.8 | 123 | 344 | 863 |
| DNA, μg | nd | 30.7 | 39.8 | 29.8 | 41 |
| I/DNA, ng/μg | 2.3 | 3.1 | 11.5 | 21.1 | |
| H99-20 (middle, <5% islet) | |||||
| Insulin, ng | 1,600 | 174 | — | 1,788 | 2,564 |
| DNA, μg | 250 | 60 | — | 42.9 | 46.8 |
| I/DNA, ng/μg | 6.4 | 2.9 | — | 41.2 | 54.8 |
At 2–4 days after islet isolation the majority of the tissue aliquot originally placed into the culture flask was removed, leaving only the tissue adhering to the nontreated surface. Much of the original tissue died as would be expected for acinar tissue. The cell clumps spread to form monolayers; at 2–3 weeks, these monolayers were coated with a thin Matrigel layer. nd, Not determined.
Figure 1.

(A) The adherent cells are primarily epithelial cells, immunostained for pan-cytokeratin (FITC green); staining for pan-cytokeratin and cytokeratin 19 were similar. Insulin-positive cells (Texas red) are scattered and infrequent. (B and C) Double staining of insulin (red) and transcription factor IPF-1 (FITC, green). Besides insulin-producing β cells, many duct cells express this transcription factor, both in the nucleus and in the cytoplasm. In B a number of cells express IPF-1 in the nucleus and/or cytoplasm without insulin staining; the field has the same density of cells as A. In addition, as in C, scattered clumps of cells had cytoplasmic IPF-1 staining with little nuclear staining and no insulin staining. Both A and B are 7-day cultured tissue of pancreas H99–12 pellet, whereas C is 7-day cultured tissue of pancreas H99–10 middle layer. (Magnification bars = 50 μm.)
Once the clumps had attached and formed monolayers, the media were changed to serum-free media with added keratinocyte growth factor to favor stimulation of ductal epithelial growth over that of fibroblasts. Over the next 5–10 days the plaques of epithelial cells became nearly confluent. Most of these cells were immunopositive for cytokeratin (results using anti-cytokeratin 19 and anti-pan-cytokeratin were identical), and varying numbers were also islet promoter factor-1 (IPF-1) (PDX-1/IDX-1/STF-1) positive (Fig. 1). The occasional insulin-positive β cells had strong IPF-1 nuclear staining. In addition, many duct cells expressed this transcription factor, both in the nucleus and in the cytoplasm. Scattered cells, both singly and in patches, had cytoplasmic IPF-1 staining with little nuclear staining and again no insulin staining. The large, cytokeratin positive cells in cobblestone patterns are characteristic of pancreatic ductal epithelium. Islets that were included flattened into clusters of small epithelial cells without cytokeratin 19 staining. At the stage of 75–90% confluency, the cultured cells were overlaid with the matrix.
During the first 1–2 weeks with Matrigel, there was movement of the epithelial cells into three-dimensional cystic structures, ranging from 50 to 400 μm in diameter, which often had multiple buds of dithizone-positive cells (Fig. 2). These structures, termed CHIBs, were observed in cultures from all layers and all eight pancreases. The frequency of cysts/CHIBs appeared to depend more on the extent of epithelial confluency than on the layer or pancreas of origin. Control flasks without the matrix overlay produced few, if any, cystic structures but in some preparations some solid spheres formed from the monolayer.
Figure 2.
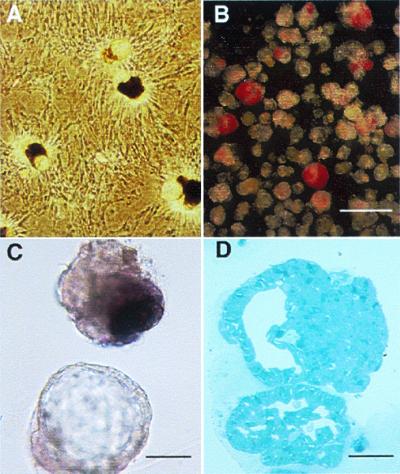
(A) After ducts were overlaid with matrix, three-dimensional structures of ductal cysts with protruding buds of islet tissue (CHIBs) were observed rising from the monolayer lawn of cells. (B and C) There are variable numbers of dithizone-stained β cells in these harvested cycts/CHIBs; many of the structures are solely cysts whereas other have 50- to 150-μm islet buds. (D) The structure of budding islet cells from a cyst is seen in this toluidine blue 1-μm section. (Magnification bar = 500 μm in B, 100 μm in C, and 50 μm in D.)
There was significant increase of both the cultured tissue and its insulin content during the 3–6 weeks culture (last 2–3 weeks with Matrigel). Data from the three pancreas from which samples of the full content of 75-cm2 flasks were taken initially and at several intermediate time points are shown in Table 1. The insulin/DNA ratio of the starting adherent material (8.2 ± 4.2 ng insulin/μg DNA) was 1–2% that of the islet preparations whether using the mean values from the four purest human islet preparations (top layers) to date (90 ± 2% islet purity, 920 ± 170 ng insulin/μg DNA) or of the purified islets (top layers) from four pancreases of Table 2 (75 ± 4% islet purity, 380 ± 130 ng insulin/μg DNA). Over the 3–4 weeks culture period the insulin/DNA ratio per flask increased, but more importantly the insulin content per flask increased 10- to 15-fold whereas the DNA content increased 0.8- to 7-fold. In contrast, the cultured tissue from the pellet layers showed increases in insulin/DNA ratios but had no increase in insulin and considerable loss of DNA (starting: 63 ± 52 ng insulin, 64.7 ± 13.6 mg DNA n = 3, 3–4 wk: 50 ± 10 ng insulin, 24.4 ± 4.2 mg DNA, n = 3). However, dithizone-positive CHIBs were formed from these cultures of pellet tissue.
Table 2.
Enrichment of insulin content in cysts/CHIBs
| Tissue origin | Insulin/DNA (ng/μg), individual samples | Insulin/DNA (ng/μg), mean ± SEM | |
|---|---|---|---|
| H99-08 | Top | 852, 333, 249, 327 | 440 ± 139 |
| H99-10 | Top | 48, 66 | 57 |
| H99-12 | Top | 149, 61, 149 | 120 ± 29 |
| H99-13 | Top + middle | 178, 218, 68 | 154 ± 45 |
| H99-19 | Middle | 46, 110 | 78 |
| H99-20 | Middle | 110, 152, 94, 27, 74 | 91 ± 21 |
Starting at 10 days after Matrigel, some cysts/CHIBs could be collected after becoming dislodged during media changes. Other samples were harvested at the end of an experiment by mechanical shearing. The time of culture for the cysts/CHIB samples was between 5 and 6 weeks (range 27 to 65 days) after isolation, 2–3 weeks after Matrigel (range 10 to 41 days).
After 2 weeks of matrix overlay, cysts/CHIBs would lift off with the mild agitation of media changes. Others were harvested at the end of the experiment by mechanical shearing with a forceful stream of media. However, this harvesting was imprecise, leaving some CHIBs still attached and lifting off some of the simple ductal cysts as well as some of the remaining monolayer or “lawn.” As shown in Table 2, the cysts/CHIBs were enriched in insulin. There was considerable variation in this enrichment with various batches of cysts/CHIBs even from the same pancreas and the same time period, partly because of the imprecision of shearing.
Demonstration that the insulin was produced by the tissue and not just adsorbed from the media was confirmed by three approaches: dithizone staining, immunofluorescent staining of the cysts/CHIBs, and semiquantitative RT-PCR for insulin. The CHIBs were composed of cytokeratin 19-positive duct cells and hormone-positive islet cells (Fig. 3). As even suggested by the dithizone-stained samples (as shown already in Fig. 2), the proportion of endocrine tissue in the cysts/CHIBs varied among the different pancreas; many were simple ductal cysts whereas others were cysts with multiple islet buds. The non-β endocrine cells were often equal in proportion to the β cells (Fig. 3 B–D). A few cells with double staining for insulin and the non-β cell hormones suggested that some endocrine cells were immature and still in the process of differentiation. Many of the cells within CHIBs had clearly differentiated phenotypes by ultrastructural analysis; both endocrine and mature duct cells were identifiable; however, some cells that had left the ductal epithelium were not granulated (Fig. 4).
Figure 3.

Double immunostained sections of CHIBs. (A) Cytokeratin 19 (FITC, green)-positive duct cells make up most of this CHIB with insulin-positive cells (Texas red) in several islet buds. Another CHIB shown with cell positive for insulin (red in B and D) and for the non-β cell hormones (glucagon, somatostatin, and pancreatic polypeptide) (green in C and D); D is the overlay of these red and green images. There are a few cells that coexpress both β and non-β cell hormones (yellow in D), indicating that some of the cells are immature and still in differentiation. (Magnification bar = 50 μm.)
Figure 4.

By ultrastructural analysis of CHIBs, mature and immature phenotypes could be seen. The duct cells (D), with characteristic short stubby microville and apical junctional complexes, line the lumen of a cyst. Adjacent to the ductal epithelium is a row of unidentifiable cells (U) that do not have characteristic granules of islet endocrine cells. β, α, and δ cells are identified by their granules. (Magnification bar = 2 μm.)
The analysis of mRNA by RT-PCR initially showed very low levels of insulin mRNA in the starting material but increases were found as CHIBs developed. Using the standard curve of insulin mRNA/18S mRNA for different dilutions of purified islets, the initial insulin mRNA levels were the equivalent of 0.9 ± 0.4% islet (n = 6, all middle layers) whereas, from pancreas H99–20, 5.9% islet at 4 weeks and 5.0% islet in cysts/CHIBs. Similarly, glucagon mRNA levels increased from the initial adherent tissue being equivalent of 1.3 ± 0.7% islet and harvested CHIBs being 14.1 ± 7.2% islet and remaining lawn 4.8 ± 0.8% islet (n = 3).
Studies were performed to determine whether the new β cells in these CHIBs could secrete insulin in response to glucose. To address this question, insulin secretion was studied over sequential and parallel 24-h time periods (Fig. 5) in tissue from three pancreases. The data from each pancreas were qualitatively the same. There were no differences in insulin concentration in the basal samples at 5 mM glucose for either the first or second 24-h period (pancreas 19: first, 1.8 ± 0.1 ng/ml, n = 24 replicates; second, 1.8 ± 0.1 ng/ml, n = 12 replicates). However, in those samples exposed to a stimulatory 20 mM glucose during the second 24-h period, there was a 2.4-fold increase in insulin (pancreas 19: 4.3 ± 0.5 ng/ml, n = 12 replicates), demonstrating the glucose responsiveness of the CHIBs.
Figure 5.

The cysts/CHIBs are responsive to glucose in vitro secretion studies. Forty cysts/CHIBs were incubated in six replicates from each of two groups for three pancreases. After an initial 4-h preincubation, each sample was incubated for 24 h in RPMI with 5 mM glucose for basal secretion determination. The media were replaced with either fresh media with 5 mM (hatched bars) or 20 mM (solid bars) glucose for an additional 24 h. Insulin secretion over the 24-h period was expressed as percent of the same tube at basal. Parallel experiments with middle layer tissue from two flasks of pancreas H99–19 and of H99–25 and one flask from H99–24 are shown. Insulin content was not determined.
Discussion
We have been able to expand human duct tissue and then to direct its differentiation to islet endocrine cells in vitro. The ability to cultivate human islets in vitro from digested pancreatic tissue that is usually discarded after islet isolation opens a new approach for β cell replacement therapy. Human islet isolations yield at best only 400,000–600,000 islets, which means that more than one donor may be required for a successful transplant (2). In the studies reported here, with modest expansion of tissue, insulin content was increased 10- to 15-fold and the endocrine tissue became organized into islet-like structures consisting of β and non-β endocrine cells. These experiments were designed to start with the nonislet ductal tissue with no effort made to salvage islet tissue from the nonislet layers; in fact, because islets rarely adhere to the nonsticky flasks, the conditions did not favor their inclusion. These data provide evidence of the potential to expand and differentiate human duct cells to islet cells, but further optimization of conditions are needed to generate yields of islet tissue that will make an impact on islet transplantation.
Optimization could include further expansion of the ductal tissue or higher efficiency in differentiating cells. Being able to use the pellet layer with higher efficiency would be particularly advantageous because this layer often contains 2–3 times more tissue than the middle layer. It is puzzling why the cultures of pellet layer had the same growth appearance (cobblestone plaques) and morphogenesis as the middle layer but had no increase in insulin content after application of the Matrigel. The adherent starting tissue was for the most part ductal epithelium, no matter which purification layer was being used. There were some fibroblasts, but the growth conditions favored the epithelial cells. The only noticeable difference in these two layers initially was that the middle had more sheets of duct epithelium from larger ducts whereas the pellet layer had mostly exocrine clumps consisting of small intercalated ducts continuing into acini. Although this difference may be key, the data from rodents suggest that culture of exocrine tissue (the ducts and acini) would result in ductal cultures. Mouse pancreatic exocrine (acinar and ductular) tissue gave rise to epithelial cultures that were indistinguishable from cultures of isolated duct, raising the possibility that acinar cells could dedifferentiate to form duct cells (23, 24). Other studies suggest that between 50% and 95% of the rodent exocrine cells die initially in culture with mainly the ductal cells left to replenish the cultures (21, 22). It is entirely possible that the cells from the smaller ducts/acini have little capacity to differentiate into endocrine cells.
In our study the adherent cells during the early culture period seem to be ductal cells. The large cytokeratin positive cells that form in cobblestone pattern are characteristic of pancreatic ductal epithelium. These large cells often had cytoplasmic and/or weak nuclear staining for the transcription factor IPF-1. In contrast, β cells were small in size, cytokeratin negative and insulin positive by immunostaining, and had strong nuclear staining for IPF-1. Although this transcription factor has been localized mainly to the embryonic duct cells and islet cells, particularly the β and some delta cells (25), we found in the adult rat that recently replicated duct cells also transiently express this protein in the nuclei (11). In these human cultures, the pattern of IPF-1 protein was variable but consistent with recent proliferation of the cells. In contrast, Beattie et al. (5) found IPF-1 stained cells only with expanded human islets and not with expanded human duct cells.
There are several lines of evidence supporting that islets are not a major component of the initial adherent cultures. First, there was little dithizone staining of the adherent cells, even from the tissue aliquots from the top layers in which the islets in suspension were strongly stained by dithizone. This is not unexpected because these nontreated flasks had been chosen originally to maintain isolated islets in suspension. In fact, the purer preparations had little adherent tissue; it was when the islet purity was lower (equal or less than 75%) that an appreciable amount of adherent tissue was found. Second, the initial insulin/DNA ratio was less than 2% that of our purest human islet preparations. Because the initial samples for insulin (at 2–4 days) were taken before there was much spreading or replication of the tissue and the suspended islets maintained their insulin content, it is unlikely that adherent islets had lost all of their insulin or became dedifferentiated. This is consistent with the low levels of insulin mRNA found in these early cultures. To obtain further insight into how many islets might have adhered in the early stages of culture, purified islets of pancreas H99–20 were extracted for insulin and DNA determination, with the finding of 5 ng insulin and 6.5 ng DNA per islet, indicating that each islet consisted of about 930 cells. The amount of insulin contained in the initial adherent tissue of a single flask from this pancreas was 174 ng, which is the equivalent of 35 islets. These 35 islets would contain 228 ng DNA which was 0.4% of the total from the adherent cells of the flask. After 8 days of Matrigel treatment, a flask that started with an identical aliquot of tissue contained 2,560 ng insulin or the equivalent of 512 islets or 7.1% of the final tissue, which was a 15-fold expansion.
Although theoretically it is possible to have increased insulin content and increased insulin-containing cells from replication of the few β cells that were in the initial adherent cell population, we think this is unlikely for several reasons. First, human β cells have been shown to have extremely low replication rate (less than 0.1% labeling for Ki67, a protein present in most cells that are in the cell cycle because it is expressed from mid-G1 through mitosis); this low level of replication was shown also in late fetal (26) and adult pancreas (27) as well as islet preparations that were cultured with hepatocyte growth factor and on extracellular matrix (8). Second, there is a parallel enrichment of glucagon during the culture as seen by the immunostaining and RT-PCR. Third, the pattern of budding of islet tissue is highly similar to that of in vivo neogenesis with the mix of β and non-β endocrine cells with immature endocrine cells as illustrated by both colocalization of islet hormones and ultrastructurally “undifferentiated” cells seen between the duct and endocrine cells. Additionally, the glucose-induced insulin response is immature as one would expect from newly formed islets. Thus, our data strongly favor neogenesis of islet tissue from ductal cells.
Our study differs from previous work in several ways. Most others have started with isolated adult human islets that were expanded on an extracellular matrix substrate (4–8). With expansion as monolayers, human islets were reported to lose their insulin expression but maintain IPF-1 expression (5). With culture in three-dimensional collagen gels human islets also lost insulin expression and endocrine phenotype, becoming duct-like (7). However, using very similar techniques of human islets embedded in three-dimensional collagen gels, Kerr-Conte et al. (6) reported proliferation of duct cells, formation of ductal cysts, and then appearance of single endocrine cells in the ductal walls. In the present study, the mainly ductal tissue remaining after islet isolation was expanded and then coated with extracellular matrix to induce differentiation of islet cells. The potential of extracellular matrix to induce differentiation in vitro has been shown for other epithelial cell types (12–14). Our technique, which uses Matrigel, a complex matrix with multiple components and growth factors (12), offers the opportunity to dissect the molecular mechanisms involved in the differentiation of the human islet.
The in vitro expansion of duct tissue is rapid and extensive, probably because the normal restraints found in vivo are removed (10). Because the default pathway of differentiation of embryonic pancreas is thought to be that of islet formation (28), such in vivo restraint might be protective and necessary to prevent excessive islet formation that could produce too much insulin and even hypoglycemia. In the present experiments we were able to generate new islet cells from duct cells in vitro but the quantities were limited. If the entire middle layer of pancreas H99–20 digest had been cultured, the equivalent of 32,000 new islets could have been generated in vitro. This amount would be expected to have little clinical impact although additional differentiation of islet cells from the duct cysts could occur in vivo. Despite the limitations at this early stage, these findings raise the tantalizing possibility that this in vitro approach, once optimized, might generate meaningful amounts of new human islet tissue from duct cells. This possibility has important implications for making β cell replacement therapy available to a larger number of people with type 1 and 2 diabetes mellitus.
Acknowledgments
We thank Michael Tenofsky, Jennifer Hollister Lock, and Maggie Merrill for expert technical help; Judy Lebet and Chris Cahill of the Juvenile Diabetes Foundation Center Islet Histology Core; and Richard Parent and Pete O'Neil of the Juvenile Diabetes Foundation Center Islet Isolation Core. The insulin RIAs were done by the Joslin Diabetes Endocrinology Research Center Core. This research was supported by the Juvenile Diabetes Foundation Center for Islet Transplantation at Harvard Medical School and the National Institute of Diabetes and Digestive and Kidney Diseases.
Abbreviations
- CHIBs
cultivated human islet buds
- RT-PCR
reverse transcription–PCR
- IPF-1
islet promoter factor-1
References
- 1.Weir G C, Bonner-Weir S. Diabetes. 1997;46:1247–1256. doi: 10.2337/diab.46.8.1247. [DOI] [PubMed] [Google Scholar]
- 2.Hering B J, Ricordi C. Graft. 1999;2:12–27. [Google Scholar]
- 3.Brelje T C, Scharp D W, Lacy P E, Ogren L, Talamantes F, Robertson M, Friesen H G, Sorenson R L. Endocrinology. 1993;132:879–887. doi: 10.1210/endo.132.2.8425500. [DOI] [PubMed] [Google Scholar]
- 4.Beattie G M, Cirulli V, Lopez A D, Hayek A. J Clin Endocrinol Metab. 1997;82:1852–1856. doi: 10.1210/jcem.82.6.4009. [DOI] [PubMed] [Google Scholar]
- 5.Beattie G M, Itkin-Ansari P, Cirulli V, Leibowitz G, Lopez A D, Bossie S, Mally M I, Levine F, Hayek A. Diabetes. 1999;48:1013–1019. doi: 10.2337/diabetes.48.5.1013. [DOI] [PubMed] [Google Scholar]
- 6.Kerr-Conte J, Pattou F, Lecomte-Houcke M, Xia Y J, Boilly B, Proye C, Lefebvre J. Diabetes. 1996;45:1108–1114. doi: 10.2337/diab.45.8.1108. [DOI] [PubMed] [Google Scholar]
- 7.Yuan S, Rosenberg L, Paraskevas S, Agapitos D, Duguid W P. Differentiation. 1996;61:67–75. doi: 10.1046/j.1432-0436.1996.6110067.x. [DOI] [PubMed] [Google Scholar]
- 8.Lefebvre V, Otonkoski T, Ustinov J, Huotari M, Pipeleers D, Bouwens L. Diabetes. 1998;47:134–137. doi: 10.2337/diab.47.1.134. [DOI] [PubMed] [Google Scholar]
- 9.Bonner-Weir S, Baxter L A, Schuppin G T, Smith F E. Diabetes. 1993;42:1715–1720. doi: 10.2337/diab.42.12.1715. [DOI] [PubMed] [Google Scholar]
- 10.Bonner-Weir S, Stubbs M, Reitz P, Taneja M, Smith F E. In: Pancreatic Growth and Regeneration. Sarvetnick N, editor. New York: Karger; 1997. pp. 138–153. [Google Scholar]
- 11.Cobb M H, Goldsmith E J. J Biol Chem. 1995;270:14843–14846. doi: 10.1074/jbc.270.25.14843. [DOI] [PubMed] [Google Scholar]
- 12.Streuli C H. Curr Opin Cell Biol. 1999;11:634–640. doi: 10.1016/s0955-0674(99)00026-5. [DOI] [PubMed] [Google Scholar]
- 13.Streuli C H, Bailey N, Bissell M J. J Cell Biol. 1991;115:1383–1395. doi: 10.1083/jcb.115.5.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Block G D, Locker J, Bowen W C, Petersen B E, Katyal S, Strom S C, Riley T, Howard T A, Michalopoulos G K. J Cell Biol. 1996;132:1133–1149. doi: 10.1083/jcb.132.6.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Linetsky E, Bottino R, Lehmann R, Alejandro R, Inveradi L, Ricordi C. Diabetes. 1997;46:1120–1123. doi: 10.2337/diab.46.7.1120. [DOI] [PubMed] [Google Scholar]
- 16.Yi E, Yin S, Harclerode D L, Bedoya A, Bikhazi N B, Housley R M, Aukerman S L, Morris C F, Pierce G F, Ulich T R. Am J Pathol. 1994;145:80–85. [PMC free article] [PubMed] [Google Scholar]
- 17.Montana E, Bonner-Weir S, Weir G C. J Clin Invest. 1993;91:780–787. doi: 10.1172/JCI116297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bouwens L, Braet F, Heimberg H. J Histochem Cytochem. 1995;43:245–253. doi: 10.1177/43.3.7532655. [DOI] [PubMed] [Google Scholar]
- 19.Sharma A, Zangen D H, Reitz P, Taneja M, Lissauer M E, Miller C P, Weir G C, Habener J F, Bonner-Weir S. Diabetes. 1999;48:507–513. doi: 10.2337/diabetes.48.3.507. [DOI] [PubMed] [Google Scholar]
- 20.Leong A S-Y, Milios J. Appl Immunohistochem. 1993;1:267–274. [Google Scholar]
- 21.Logsdon C D, Williams J A. Am J Physiol. 1986;250:G440–G447. doi: 10.1152/ajpgi.1986.250.4.G440. [DOI] [PubMed] [Google Scholar]
- 22.Elsasser H P, Lutcke H, Kern H F. The Exocrine Pancreas: Biology, Pathobiology, and Disease. New York: Raven; 1986. pp. 45–53. [Google Scholar]
- 23.Githens S, Schexnayder J A, Moses R L, Denning G M, Smith J J, Frazier M L. In Vitro Cell Dev Biol. 1994;30:622–635. doi: 10.1007/BF02631262. [DOI] [PubMed] [Google Scholar]
- 24.Githens S, Schexnayder J A, Desai K, Patke C L. In Vitro Cell Dev Biol. 1989;25:679–686. doi: 10.1007/BF02623720. [DOI] [PubMed] [Google Scholar]
- 25.Guz Y, Montminy M R, Stein R, Leonard J, Gamer L W, Wright C V E, Teitelman G. Development (Cambridge, UK) 1995;121:11–18. doi: 10.1242/dev.121.1.11. [DOI] [PubMed] [Google Scholar]
- 26.Bouwens L, Lu W G, Krijger R D. Diabetologia. 1997;40:398–404. doi: 10.1007/s001250050693. [DOI] [PubMed] [Google Scholar]
- 27.Bouwens L, Pipeleers D G. Diabetologia. 1998;41:629–633. doi: 10.1007/s001250050960. [DOI] [PubMed] [Google Scholar]
- 28.Gittes G K, Galante P E, Hanahan D, Rutter W J, Debase H T. Development (Cambridge, UK) 1996;122:439–447. doi: 10.1242/dev.122.2.439. [DOI] [PubMed] [Google Scholar]