

Abstract
Cytomegalovirus (CMV) replicates rapidly in its human host with a doubling time of ≈ 1 day. Using simple mathematical models we show that the efficacy of the anti-CMV drug ganciclovir (GCV) against wild-type strains is 91.5% [95% confidence interval (CI) 89–94%] when given i.v. (5 mg/kg twice a day) but only 46.5% (95% CI 45–47.5%) when given orally (1 g three times a day) whereas the corresponding figures for a typical GCV-resistant virus are 62% (95% CI 57–66%) and 35% (95% CI 33–37%), respectively. During prolonged periods of GCV therapy we show that the apparent sudden appearance of GCV resistance is explained by the combination of two exponentially increasing populations (wild type and mutant) at doses of GCV that do not completely inhibit CMV replication. Cell culture methods to assess CMV drug resistance in vivo will underestimate its prevalence because of the fitness loss of resistant virus in the absence of therapy. The parameters determined from these models then were used to predict the likely viral load and resistance patterns in patients on prolonged therapy with GCV. The modeled and experimental data showed excellent agreement over extended time periods (up to 270 days of therapy) and provide a framework to predict the virologic course of patients at therapeutic initiation.
Keywords: PCR, mutation, fitness, mathematical model
Human cytomegalovirus (CMV) infection is widespread throughout the world. In the immunocompetent individual infection is usually asymptomatic but the virus can cause substantial pathology in the immunocompromised host (1, 2). Hence, CMV disease is a major problem after organ transplantation and in individuals infected with HIV. Consequently, antiviral agents able to inhibit CMV replication have been introduced into clinical practice with concomitant beneficial outcomes (3–5). The foremost agent used to control CMV replication in the immunocompromised host has been ganciclovir (GCV), although foscarnet and cidofovir also have been shown to be effective. In addition to the treatment of clinically overt disease, GCV has been used preemptively (at the initial signs of active infection), prophylactically (before evidence of active infection), and for long-term suppression of CMV replication subsequent to disease (6).
Long-term administration of antiviral agents may lead to the development of resistance. In the case of CMV, GCV resistance maps to two genetic loci, the UL97 protein kinase that phosphorylates GCV to its monophosphate form and the CMV DNA polymerase (7, 8). Temporally, mutations in UL97 usually occur first and give rise to low-level resistance (typically about 10-fold) followed by mutations in the DNA polymerase gene to yield high-level resistance (>30-fold). To date, patients who are treated with short-term i.v. GCV therapy [typically 14–21 days at a dose of 5 mg/kg twice a day (b.i.d.)] have not developed a high incidence of GCV-resistant strains. However, in patients requiring long-term treatment (>100 days) especially with lower doses of GCV, i.e., 1 g oral GCV, three times a day (t.i.d.), resistance has been observed with increasing frequency (7, 9, 10). Using conventional cell culture methods, resistance has been detected in approximately 8% of AIDS patients after extended GCV therapy (9), whereas with the advent of molecular approaches to detect resistant virus this frequency has increased to 20–25% (7, 10). We recently have shown that active CMV replication in the human host occurs dynamically with a doubling time of approximately 1 day and have defined relative fitness differences between wild-type and UL97 mutant strains of virus under different therapeutic conditions (11). In the present paper, we use an approach based on knowledge of CMV replication dynamics and viral fitness to provide: (i) a rationalization of the differential appearance of GCV-resistance strains of CMV in patients on long-term and short-term therapy, (ii) an explanation of the different detection rates of GCV resistance depending on the methodology used, (iii) an estimate of the efficacy of different doses of GCV against wild-type and UL97 mutant virus strains, and (iv) an accurate prediction of the CMV load profiles in patients on long-term GCV therapy.
Materials and Methods
Patients Investigated.
The data sets are derived from results of detailed investigations of AIDS patient cohorts whose clinical features have been described elsewhere (12–14) and from data relating to the replication dynamics of CMV in the human host (11). With the exception of data from a heart transplant recipient published in ref. 15 (kindly provided by the authors) all of the remaining patients included in the study were AIDS patients with CMV disease (predominately AIDS retinitis) who were treated with GCV.
Laboratory Measurements of CMV Load and Resistance.
With the exception of the patient described in ref. 15, DNA was extracted from whole blood samples (200 μl) by using ion-exchange chromatography with a Qiagen DNA extraction kit (Qiagen, Crawley, U.K.) according to the manufacturer's instructions. Primers directed to the glycoprotein B (UL55) gene of CMV were used for the qualitative and quantitative detection of CMV (16). The primers amplify a 149-bp fragment of gB and the conditions for PCR have been described by our group (16). The quantitative competitive-PCR method for assessing CMV load uses a control sequence consisting of a mutant version of the 149-bp authentic target sequence. The details of this method have been described (16–19). A point mutation assay was used to ascertain the distribution of wild-type and mutant alleles at codons of interest as described (14).
Fitness of Wild-Type and Mutant CMV UL97 Strains in the Presence or Absence of GCV.
Two equations have been used for the calculation of the fitness differences between wild-type and mutant strains of CMV in the presence or absence of GCV. The first approach (Eq. 1) provides the relative fitness, s, of the population (in this case, different UL97 variants) and assumes replication occurs in continuous time. This equation requires knowledge of the relative proportion of the most-fit variant (p) and the least-fit variant (q) at times 0 and t, respectively. These quantitative parameters were obtained from the distribution of nucleotides at the site of interest as determined by a point mutation assay described elsewhere (14). This equation previously has been used to calculate the fitness differences of CMV UL97 variants (11).
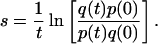 |
1 |
An alternative equation (Eq. 2) for calculating the fitness differences between two populations assumes evolution through discrete generations:
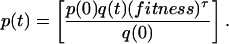 |
2 |
Similar to Eq. 1, this equation requires knowledge of the proportion of the least-fit and most-fit variants at times 0 and t but unlike Eq. 1 also requires knowledge of the number of generations that the population has traversed in the time period t. The doubling time of CMV in vivo was calculated in our previous work (11) and used here to determine the number of generations traversed in time t.
Simulated repopulation curves of mutant UL97 genotypes were generated by computing the proportion of p(t) at daily intervals with different starting populations of q in the presence or absence of GCV by using Eqs. 1 or 2 as appropriate (see Figs. 1 and 2).
Figure 1.

Proportion of UL97 single-site mutant within the population after the initiation of GCV therapy. Therapy lasts for 21 days and the fitness difference (s; Eq. 1) of mutant to wild type after cessation of therapy is the same as the fitness gain of mutant over wild type during therapy (5.6%). Three different starting concentrations of mutant within the population are modeled (0.015%, 0.15%, and 1.5%).
Figure 2.

Proportion of single-site (A) or double-site (B) mutant in the population after in vitro culture in the absence of GCV. The data were generated by using Eq. 2 (see Materials and Methods), and curves are shown for four different wild type/mutant ratios.
In the examples shown, a fitness difference of 5.6% between mutant and wild-type strains has been used and is typical of single-site UL97 mutants, e.g., L595F (11). To determine the effect of the fitness difference between wild-type and UL97 mutant viruses on the detection of resistant virus after in vitro passage in the absence of GCV, Eq. 2 was used. In these examples (see Fig. 2), two relative fitness values of mutant to wild type were used: 0.94 (a typical value for the L595F UL97 mutation) and 0.88 (the computed fitness difference for the double UL97 mutant M460V + L595F) and are derived from our previous data (11).
Estimating the Efficacy of Different Doses of GCV.
The basic models used to describe the viral dynamics of CMV are similar to those described for HIV and hepatitis C virus (HCV) (20–24) and have been described in detail (11). Although viral load decline after GCV approximates to an exponential decay process, the slope of decline is modulated by the efficacy of GCV. Similar to the data provided for HCV load decline after interferon therapy (22), the slope of decrease can be used to estimate drug efficacy. In 10 patients receiving i.v. GCV induction therapy, the log reduction in viral load from baseline to <200 genomes/ml blood was computed (ΔVL) and plotted against the time at which the viral load reached <200 genomes/ml blood. The resulting data set was subjected to linear regression, which then was used to estimate the likely efficacy of drug and superimposed on computed ΔVL time plots generated for different drug efficacy. These analyses were repeated for different doses of GCV and with different strains of virus (wild type and mutant). In some instances, e.g., during oral GCV therapy, the size of mutant and wild-type populations at the initiation of the drug regimen was computed by extrapolation of the linear regression line to the time point when therapy was commenced. All values quoted are the mean and 95% confidence intervals obtained from the linear regression model.
Predicting Viral Load Patterns of Resistant and Wild-Type Virus During GCV Therapy.
Data from AIDS patients and a heart transplant patient were used to assess the accuracy of the model to predict changes in viral load over time. Importantly these patients had not been used in the original calculations of the drug efficacy. Modeling was performed to generate a viral load time curve for wild-type and mutant virus and the experimental data points were superimposed on the resulting prediction.
Results
Appearance of GCV Resistance During Short- and Long-Term GCV Therapy.
Based on the fitness advantage of a single-site UL97 mutant in the presence of GCV and the doubling time of CMV in the host (approximately 1 day), an estimate of the proportional population size of mutant and wild-type strains of CMV can be obtained for different durations of GCV therapy. The results of these analyses are shown in Fig. 1. In patients exposed to 21 days of therapy, the mutant population increases approximately 3-fold; even if the UL97 mutant virus comprised 0.5% of the virus population at the initiation of therapy, it would still only represent ≈1.7% of the population at 21 days. Upon cessation of therapy, repopulation of wild-type virus occurs because it is now more fit than UL97 mutant in the absence of GCV. In the example shown where the fitness gain of wild type over mutant in the absence of GCV is equal to the fitness loss during GCV therapy, the mutant population has returned to baseline levels by day 42. Thus, on the basis of this model, short-term GCV therapy should not result in a substantial increase in the frequency of resistant virus during therapy nor will it persist within the infected individual after cessation of therapy. In contrast, on long-term GCV therapy, a UL97 mutant with the same fitness advantage (5.6%), continues to repopulate at the expense of wild-type virus such that by day 145, mutant virus accounts for 30% of the population and by day 150 accounts for >90% of the population when the initial mutant population size is 0.5% (data not shown). These analyses do not change dramatically whether they are computed by using Eqs. 1 or 2.
Detection of GCV-Resistant Virus in Vitro.
The predicted effects of in vitro culture in the absence of GCV on the strain composition of CMV are shown in Fig. 2. In the first case a single-site UL97 mutant is considered whereas in the second example a two-site mutant is modeled. In both cases, within 28 days of culture the composition of the viral population has changed in favor of wild type such that a 50:50 mixture of wild type/mutant at the initiation of in vitro culture would comprise 17% mutant at day 28 if the fitness difference was 0.94 and 2.7% if the fitness difference was 0.88. It is only when the mutant is present as a high proportion of the population at baseline, i.e., inoculation into culture, that the effects of the differential fitness of the two species are minimized. Again these profiles do not change substantially when computed with either Eq. 1 or 2.
Efficacy of Different Doses of GCV Against Wild-Type and UL97 Mutant Viruses in Vivo.
Using exponential decay models incorporating the efficacy of drug at inhibiting replication the second-phase decline of CMV load after GCV therapy can be used to provide an estimate of the efficacy of i.v. GCV against wild-type CMV. Using data derived from the CMV load measurement in relatively frequent blood samples of 10 AIDS patients during 21 days of i.v. GCV therapy and computing the log decline in CMV load from baseline (ΔVL; see Materials and Methods) produced a linear relationship (Fig. 3; r2 = 0.90; P < 0.0001). Superimposition of this line on the predicted viral load decay curves for CMV based on the previous replication dynamics data and different efficacy levels of i.v. GCV reveals that the best-line fit corresponds to an efficacy level of 91.5% [95% confidence interval (CI); 89–94%].
Figure 3.

Computation of the efficacy of i.v. GCV induction therapy (5 mg/kg b.i.d.). The linear regression line is shown (Lin Reg) superimposed on viral load decay curves generated for different efficacy of GCV together with the experimental data.
To ascertain the efficacy of lower doses of GCV, i.e., those used in oral GCV maintenance therapy, against wild-type and UL97 mutant strains of virus, viral loads for mutant and wild-type virus (see Materials and Methods) over time were plotted and linear regression analysis coupled with predictive modeling of the viral load patterns were computed for different efficacy levels. The results of a typical analysis are shown in Fig. 4. The data show that the efficacy of oral GCV against wild-type virus is 46.5% (95% CI 45–47.5%; P = 0.01; Fig. 4A) whereas for a UL97 single-site mutant (L595F;L595S; data not shown) the efficacy is approximately 35% (95% CI 33–37%; P = 0.01; Fig. 4B). In addition to providing an estimate of drug efficacy, extrapolation of the viral load line to the point of initiation of therapy allows an estimate of the likely viral load of mutant and wild type at this time point, i.e., after 21 days of i.v. GCV. In the example shown in Fig. 4B, the extrapolated viral load of wild type is 0.6 genomes/ml blood (95% CI, 0.01–33 genomes/ml). Because this patient possessed a CMV load of 4.75 log 10 genomes/ml blood at the initiation of i.v. GCV, the computed CMV load at the end of 21 days of therapy is consistent with the efficacy of i.v. GCV calculated from Fig. 3 (computed viral load at day 21 = 0.72 genomes/ml blood; 95% CI 0.3–1.71 genomes/ml). Similar data are obtained for the patient shown in Fig. 4A, where the extrapolated viral load of wild type and mutant are 9 genomes/ml blood (95% CI 0.14–363) and 0.45 genomes/ml blood (95% CI, 0.01–20), respectively.
Figure 4.

Computation of the efficacy of oral GCV (1 g t.i.d.) against wild-type (A) and single-site UL97 mutant viruses (B). The horizontal dotted line represents the level of sensitivity of viral load detection in the PCR assay.
The clinical management of recurrences in CMV retinitis involves reinduction courses with i.v. GCV, which has allowed the efficacy of i.v. GCV against single-site UL97 mutants to be determined in a similar fashion to that used in Fig. 3. Assimilation of data from four patients in whom UL97 mutant virus was present (>95% of the viral population) indicates that i.v. GCV has an efficacy of 62% (95% CI 57–66%; P = 0.02) against these single-site mutants.
Changes in Mutant and Wild-Type CMV Populations During Oral GCV Therapy.
The effect of i.v. GCV is to substantially inhibit both wild-type and mutant virus replication with an efficacy of 91.5% and 62%, respectively. Therefore at the end of 21 days of i.v. GCV therapy CMV load levels will be reduced substantially (see Fig. 3) although the relative proportion of mutant virus within the population will have increased (see above). The data shown in Fig. 4 illustrate that oral GCV (1 g t.i.d.) achieves levels of drug that are incapable of completely suppressing viral replication, and so both wild-type and mutant populations will increase during oral GCV therapy at the growth rates calculated from Fig. 4. The predicted viral load patterns of mutant and wild-type virus under oral GCV therapy is shown in Fig. 5. Because the wild-type virus is the dominant species at the initiation of oral GCV it continues to be the major contributor to the total viral load even when viral loads rise above the limit of detection in the currently available assays (ca. 200 genomes/ml). Thus, paradoxically, the kinetics of viral load after GCV therapy is determined not by mutant virus but by wild-type strains during the early phases of DNAemia. However, because of the differences in the relative efficacy of oral GCV against wild-type and mutant virus, this situation does not persist and it is the more rapidly growing mutant that eventually comprises the majority of the viral load. A feature of the data presented in Fig. 5 is that the simple exponential growth patterns of wild-type and mutant virus combine to give a more complex pattern to the overall viral load profile.
Figure 5.

Simulation of the viral load pattern of wild-type and single-site UL97 mutant CMV strains after initiation of oral GCV therapy. The total CMV load is shown and the proportion of mutant virus in the population at specific time points is superimposed as ⧫.
Predicting Viral Load Patterns in Patients on GCV Therapy.
Finally, we address the question of whether knowledge of the efficacy of different doses of GCV coupled with the exponential growth/decay models presented above can be used to predict the viral load patterns of patients on prolonged GCV therapy in whom resistance virus was detected. The first example of such modeling is shown in Fig. 5 and is an AIDS patient with CMV retinitis who was treated with i.v. GCV (5 mg/kg b.i.d.) for the first 21 days, oral GCV maintenance therapy (3 g/day) from days 22 to 126, i.v. GCV from days 127 to 134, i.v. GCV maintenance therapy (5 mg/kg per day) from days 135 to 179, oral GCV maintenance therapy from days 180 to 203, reinduction i.v. GCV from days 204 to 225, and finally oral maintenance from days 226 to 247. This patient developed an L595F mutation during therapy that was predominant in the population by day 111 (>95%). The model and superimposed experimental data points for this patient show good agreement over the entire period of therapy (linear regression analysis, r2 = 0.95; P < 0.0001).
Notwithstanding the close agreement between predicted and experimental viral loads shown for the patient in Fig. 6 it is important to determine whether this model is applicable to data sets obtained from other sources. Unfortunately, there is a paucity of published data where sequential, frequent viral load measurements together with mutational analyses have been affected. Nevertheless, the viral load and resistance profile of a heart transplant recipient prescribed extended periods of GCV is available (see Materials and Methods). This heart transplant recipient suffered a primary CMV infection posttransplant and was treated with multiple courses of i.v. GCV followed by a period of maintenance therapy with oral GCV (3 g/day). During maintenance therapy a UL97 GCV-resistant CMV strain with a mutation at codon 607 (C to Y) of the UL97 gene developed. On the basis of the data presented above we have modeled the viral load profile of this patient while on oral GCV by assuming that the efficacy of different doses of GCV against the C607Y mutant is comparable to that observed for other single-site UL97 mutants (L595F, L595S). The results shown in Fig. 7 model the response of wild-type and mutant viral loads to i.v. GCV therapy (initiated at day 89 posttransplant) and during the subsequent period of oral GCV maintenance therapy. There is a good agreement between the predicted and experimental derived data (linear regression analysis, r2 = 0.72; P = 0.0001), indicating that the parameter values used were appropriate. Indeed, using the viral loads of mutant and wild-type species predicted from the model, the composition of the viral population is 50% mutant at day 152, 88% mutant at day 173, and 94% mutant at day 180 compared with the experimental values of 35%, 85%, and 94%, respectively, at these time points determined by quantitative restriction digest analysis.
Figure 6.

Simulation of the CMV load profile of a patient who develops an L595F mutation after GCV therapy. The alterations in therapeutic dose are shown together with the experimental CMV loads from quantitative competitive-PCR analyses.
Figure 7.

Simulation of the CMV load profile of a heart transplant recipient during GCV induction therapy and oral GCV therapy. The experimentally determined CMV loads from ref. 15 are superimposed.
Discussion
Active CMV infection in the human host is a highly dynamic process (11). In the present paper we have used knowledge of the doubling time of CMV and the fitness differences between wild-type and mutant UL97 strains to investigate the quantitative appearance of GCV resistance in vivo and its detection in vitro. These data also have allowed an estimate of the efficacy of the different doses of GCV used in clinical practice. Based on population dynamics, the model predicts that short-term therapy (<3 weeks) should not result in a substantial increase in the proportion of GCV-resistant strains of virus within the population. This prediction is consistent with the clinical experience of short-term GCV therapy in which GCV resistance has not been reported. In contrast, extended continuous periods of GCV therapy results in a continued growth advantage for resistant strains such that these strains will predominate within the population at a finite time during therapy depending on their frequency at therapeutic initiation and the efficacy of GCV against these mutant strains (see below). In AIDS patients undergoing oral GCV maintenance after CMV retinitis, GCV-resistant UL97 strains of virus occur in approximately 22% of patients by 7 months (14). An important conclusion based on the fitness differences between wild-type and UL97 mutant viral strains relates to the kinetics of wild-type repopulation during in vitro culture in the absence of GCV. For example, even for relatively minor fitness differences, a clinical sample containing a 50:50 mixture of wild-type and single-site mutant viral strains would comprise 83% wild-type strains by 28 days of culture (the time period frequently used to culture CMV from clinical sources), assuming a doubling time in vitro of 1 day. Thus, it is likely that cell culture propagation of CMV ex vivo will lead to a selection bias against GCV-resistant viruses resulting in the under-recognition of resistance, especially when mutant virus is present as a minority species within the clinical sample. Such predictions are consistent with the discrepancies between data on the prevalence of GCV resistance using cell culture (9) and molecular-based approaches directly on clinical samples (7, 14). These observations parallel in vivo data showing that even when UL97 mutants comprise >99% of the population therapeutic alteration to a drug that does not require activation through UL97, e.g., cidofovir results in the rapid repopulation of the virus pool with wild-type UL97 strains (11, 25).
Previous studies have shown that high doses of GCV, e.g., 5 mg/kg b.i.d. i.v., act to rapidly inhibit CMV replication with concomitant declines in CMV load in blood and urine (11, 13). These data have been used to estimate the clearance rate and hence generation time of CMV in vivo (11). In the present study we have extended these models to assess the efficacy of different doses of GCV. Thus, i.v. GCV induction therapy has an efficacy of approximately 91.5% against wild-type virus but only 62% against single-site UL97 mutants and oral GCV (1 g t.i.d.) has an efficacy of 46.5% against wild-type virus. These differences in efficacy are consistent with pharmacokinetic studies showing that oral GCV (1 g t.i.d.) produces plasma GCV levels that are approximately 50% of those achieved with i.v. GCV (5 mg/kg b.i.d.). The efficacy of i.v. GCV against wild-type CMV is similar to the efficacy of IFN-α against hepatitis C virus (81–96% depending on dose; ref. 22) and lamivudine against hepatitis B virus (ca. 93%; range 83–97%; refs. 21, 26, and 27). In contrast, oral GCV is not sufficiently efficacious to cause elimination of either wild-type or mutant virus and hence the population of both species increases. The models provided here show that the early stage of DNAemia is largely determined by the growth pattern of wild-type virus because this species is present at a higher viral load compared with mutant at therapeutic initiation. Nevertheless, this effect is transient and within a short time period the viral load comprises predominately mutant GCV-resistant strains because of their growth advantage during GCV therapy. These predictions are consistent with the limited published data describing the quantitative alterations in the viral strain composition in patients on long-term GCV therapy (14, 15, 28). The early appearance of a predominately wild-type population and the relatively rapid transition to a mutant population parallels the predicted composition of the population based on the fitness differences between wild-type and mutant virus (11). The results of such models have implications for the frequency of monitoring of patients for CMV resistance and for the initiation of alternative therapeutic interventions and are reminiscent of the predicted HIV load and resistance patterns during AZT therapy (29).
The prevalence and persistence of mutant strains of virus within a population depends on many factors, including the error rate of the polymerase, the fitness loss of the mutant, and the number of generations traversed. Theoretically, a mutant will dominate a population on a time scale of −λ−1lnμ where λ is the growth rate of mutant and μ is the starting concentration of mutant. Thus, for a mutant frequency of 10−2-10−3 with a generation time of 0.25 per day, the time for a mutant to dominate would be 24–34.5 days, which is shorter than the observed time for mutant domination in the patients studied here (see Fig. 5 and ref. 14). This is probably accounted for by a combination of viral fitness and the competition between mutant and wild-type virus for infection of susceptible cells after i.v. GCV therapy. Estimates of the mutant pool in the AIDS patients included in the present analysis indicate a mutant frequency of approximately 0.5–1.5% and likely reflect the extensive periods of active CMV replication that precedes the development of CMV disease in this patient group. In contrast, in a transplant patient who suffers a primary CMV infection, the virus usually traverses far fewer generations before antiviral intervention. Hence, the frequency of mutant virus in this population is likely to be much lower (10−8-10−9), which correlates with a time to mutant dominance under GCV of 92–103 days. This time scale for mutant dominance is comparable to that observed for the transplant patient shown in Fig. 5. However, it should be noted that multiple doses of GCV interspersed with periods of active CMV replication are likely to increase the relative size of the mutant pool. Such an effect can be seen in Fig. 5 where the prevalence of the UL97 mutant is approximately 0.5% at the beginning of oral GCV therapy. This patient had been prescribed two previous courses of GCV interspersed between periods of active CMV replication (15).
The ability of a model based on GCV efficacy and CMV dynamics to predict viral load patterns over extended periods of time for an AIDS patient and a heart transplant recipient who developed GCV resistance was assessed. In both cases the computed viral load-time curve closely followed the viral load determined experimentally even during multiple alterations in GCV therapy (see Fig. 6). In addition, using data derived from a different group of investigators with an alternative quantitative PCR assay and a patient developing a single-site mutant that had not been previously modeled (C807Y) produced excellent agreement. Further examples are required to determine the influence of the immune system on the replication kinetics of wild-type and mutant strains of CMV and the effects of GCV and other anti-CMV agents against other single-site and double mutants at either the UL97 or UL97 + UL54 (DNA polymerase) loci. It is likely that such data will facilitate our ability to predict the viral load course of an individual prescribed an anti-CMV agent and to alter patient management as required and to evaluate the efficacy of newer antiviral agents as they become available.
Acknowledgments
We are grateful to our colleague Dr. Margaret Johnson (director HIV services, Royal Free Hospital National Health Service Trust) and to Professor G. Gerna and Dr. F. Baldanti (University of Pavia, Italy) for providing the data for the heart transplant recipient. This work was supported in part by the Wellcome Trust, the National Institutes of Health, and the United Kingdom Medical Research Council.
Abbreviations
- CMV
cytomegalovirus
- GCV
ganciclovir
- CI
confidence interval
- b.i.d.
twice a day
- t.i.d.
three times a day
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.140123497.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.140123497
References
- 1.Griffiths P D, Emery V C. In: Clinical Virology. Richman D D, Whitley R J, Hayden F G, editors. New York: Churchill Livingstone; 1997. pp. 445–470. [Google Scholar]
- 2.Fishman J A, Rubin R H. N Engl J Med. 1998;338:1741–1751. doi: 10.1056/NEJM199806113382407. [DOI] [PubMed] [Google Scholar]
- 3.Crumpacker C S. N Engl J Med. 1996;335:721–729. doi: 10.1056/NEJM199609053351007. [DOI] [PubMed] [Google Scholar]
- 4.Safrin S, Cherrington J, Jaffe H S. Rev Med Virol. 1997;7:145–156. doi: 10.1002/(sici)1099-1654(199709)7:3<145::aid-rmv196>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 5.Jacobsen M A. N Engl J Med. 1997;337:105–114. doi: 10.1056/NEJM199707103370207. [DOI] [PubMed] [Google Scholar]
- 6.Pillay D, Emery V C, Griffiths P D. In: Antiviral Chemotherapy. Jeffries D J, DeClerq E, editors. New York: Wiley; 1995. pp. 265–283. [Google Scholar]
- 7.Chou S, Guentzel S, Michels K R, Miner R C, Drew W L. J Infect Dis. 1995;172:239–242. doi: 10.1093/infdis/172.1.239. [DOI] [PubMed] [Google Scholar]
- 8.Chou S. Transplant Infect Dis. 1999;1:105–114. doi: 10.1034/j.1399-3062.1999.010204.x. [DOI] [PubMed] [Google Scholar]
- 9.Drew W L, Miner R C, Busch D F, Follansbee S E, Gullett J, Mehalko S G. J Infect Dis. 1991;163:716–719. doi: 10.1093/infdis/163.4.716. [DOI] [PubMed] [Google Scholar]
- 10.Bowen E F, Emery V C, Wilson P, Johnson M A, Davey C C, Sabin C A, Farmer D, Griffiths P D. AIDS. 1998;12:605–611. doi: 10.1097/00002030-199806000-00009. [DOI] [PubMed] [Google Scholar]
- 11.Emery V C, Cope A V, Bowen E F, Gor D, Griffiths P D. J Exp Med. 1999;190:177–182. doi: 10.1084/jem.190.2.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bowen E F, Sabin C A, Wilson P, Griffiths P D, Davey C C, Johnson M A, Emery V C. AIDS. 1997;11:889–893. doi: 10.1097/00002030-199707000-00008. [DOI] [PubMed] [Google Scholar]
- 13.Bowen E F, Wilson P, Cope A V, Sabin C A, Griffiths P D, Davey C C, Johnson M A, Emery V C. AIDS. 1996;10:1515–1520. doi: 10.1097/00002030-199611000-00009. [DOI] [PubMed] [Google Scholar]
- 14.Bowen E F, Johnson M A, Griffiths P D, Emery V C. J Virol Methods. 1997;68:225–234. doi: 10.1016/s0166-0934(97)00131-6. [DOI] [PubMed] [Google Scholar]
- 15.Baldanti F, Simoncini L, Sarasini A, Zavattoni M, Grussi P, Revello M G, Gerna G. Transplantation. 1998;66:324–329. doi: 10.1097/00007890-199808150-00008. [DOI] [PubMed] [Google Scholar]
- 16.Darlington J, Super M, Patel K, Grundy J E, Griffiths P D, Emery V C. J Gen Virol. 1991;72:1985–1989. doi: 10.1099/0022-1317-72-8-1985. [DOI] [PubMed] [Google Scholar]
- 17.Cope A V, Sweny P, Sabin C A, Rees L, Griffiths P D, Emery V C. J Med Virol. 1997;52:200–205. [PubMed] [Google Scholar]
- 18.Cope A V, Sabin C A, Burroughs A, Rolles K, Griffiths P D, Emery V C. J Infect Dis. 1997;176:1484–1490. doi: 10.1086/514145. [DOI] [PubMed] [Google Scholar]
- 19.Gor D, Vyas N, Prentice H G, Man S, Griffiths P D, Emery V C. Bone Marrow Transplant. 1997;21:597–605. doi: 10.1038/sj.bmt.1701139. [DOI] [PubMed] [Google Scholar]
- 20.Ho D D, Neumann A U, Perelson A S, Chen W, Leonard J M, Markowitz M. Nature (London) 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 21.Wei X, Ghosh S K, Taylor M E, Johnson V A, Emini E A, Deutach P, Lifson J D, Bonhoeffer S, Nowak M A, Hahn B H. Nature (London) 1995;373:117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- 22.Neumann A U, Lam N P, Dahari H, Gretch D R, Wiley T E, Layden T J, Perelson A S. Science. 1998;282:103–107. doi: 10.1126/science.282.5386.103. [DOI] [PubMed] [Google Scholar]
- 23.Bonhoeffer S, Coffin J M, Nowak M A. J Virol. 1997;71:3275–3278. doi: 10.1128/jvi.71.4.3275-3278.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonhoeffer S, May R M, Shaw G M, Nowak M A. Proc Natl Acad Sci USA. 1997;94:6971–6976. doi: 10.1073/pnas.94.13.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bowen E F, Cherrington J M, Lamy P D, Griffiths P D, Johnson M A, Emery V C. J Med Virol. 1999;58:402–407. [PubMed] [Google Scholar]
- 26.Burroughs N J, Rand D A, Pillay D, Elias E, Mutimer D. In: Therapies for Viral Hepatitis. Schinazi R F, Somadossi J-P, Thomas H C, editors. London: International Medical Press; 1998. pp. 335–343. [Google Scholar]
- 27.Nowak M A, Bonhoeffer S, Hill A M, Boehme R, Thomas H C, McDade H. Proc Natl Acad Sci USA. 1996;93:4398–4402. doi: 10.1073/pnas.93.9.4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jabs D A, Enger C, Dunn J P, Forman M. J Infect Dis. 1998;177:770–773. doi: 10.1086/514249. [DOI] [PubMed] [Google Scholar]
- 29.McLean A R, Frost S D W. Rev Med Virol. 1995;5:141–147. [Google Scholar]