

Abstract
Analysis of the intensely studied HIV-1 gp120 V3 protein region reveals that the among-population mean site-specific frequency of an amino acid is a measure of its relative marginal fitness. This surprising result may arise if populations are displaced from mutation–selection equilibrium by fluctuating selection and if the probability of fixation of a beneficial amino acid is proportional to its selection coefficient.
KNOWING the effect on fitness of every amino acid at every site of a protein would greatly facilitate the study of adaptive evolution at the molecular level. Such data would help determine the frequency distribution of fitness effects of new beneficial mutations, which is key to understanding the dynamics of adaptation (Orr 2002), allow protein variation to be more easily interpreted, and allow the realistic simulation of protein evolution. But, such data are difficult to obtain empirically and are therefore not available for any protein. However, for human immunodeficiency virus type 1 (HIV-1) proteins, amino acid site-specific frequencies calculated across virus populations infecting different patients appear to correlate with the marginal fitness effects of the amino acids (Hung et al. 1999; Reza et al. 2003), suggesting a simple method of estimating the site-specific marginal fitnesses of amino acids.
HIV-1 third variable region:
I investigated this correlation using sequence data from the third variable region (V3) of the HIV-1 exterior envelope glycoprotein (gp120), located on the surface of the virus particle (virion). V3 is the main determinant of which one of two cell-surface chemokine receptors (CCR5 or CXCR4) is used by a virion as a coreceptor to enter a cell, which determines the type of cell infected (Speck et al. 1997). V3 also modulates the use of each coreceptor (de Jong et al. 1992; Hung et al. 1999) and thereby affects the rate-limiting step in cell entry (Platt et al. 2005). Finally, V3 is also the main target of antibodies that interfere with cell entry (Zolla-Pazner 2004). Because of its critical function and its exposure to neutralizing antibodies, V3 has been the focus of intense study, resulting in the data necessary to test the correlation.
Site-specific marginal fitness:
Data on V3 site-specific fitness effects of amino acids are available from a study that employed site-directed mutagenesis to modify the amino acid at V3 site 25 (Hung et al. 1999). This study measured the effects of different amino acids at site 25 on the number of cells infected (infectivity) in a standardized assay. Note that infectivity is the appropriate measure of the effect of V3 on fitness under the assay conditions; the sole function of V3 appears to be in cell entry, and this component of fitness is unlikely to trade off against the remaining components of fitness, such as the rate of viral genome integration into the host cell genome, the rate of provirus (integrated viral genome) replication, and virion budding, which are controlled by other viral proteins (Coffin 1999). The site-directed mutagenesis study was conducted using CCR5-utilizing HIV-1 of subtype B, the phylogenetic clade most common in Europe and North America and the most sequenced and studied.
Site-specific amino acid frequency:
I calculated site-specific frequencies of subtype B V3 amino acids for the viral population infecting a patient and then averaged frequencies across patients. Among-population means of frequencies were used because the site-specific frequency of an amino acid in any one population is expected to be dynamic, being dependent on the population's level of adaptation, and thus a poor predictor of the amino acid's marginal fitness. This procedure was repeated for each of the three coreceptor usage viral phenotypes: exclusively CCR5 utilizing (R5) (269 sequences from 58 patients), exclusively CXCR4 utilizing (X4) (109 sequences from 11 patients), and dual coreceptor utilizing (R5X4) (60 sequences from 16 patients). The rank order of amino acid frequencies at most sites, including site 25, varies among coreceptor-usage phenotypes (Figure 1), as would be expected if the sites function in determining or modulating coreceptor usage.
Figure 1.—

V3 amino acids in order of decreasing (top to bottom) site-specific frequency for each coreceptor usage phenotype. Residues in boldface type are unique to a phenotype at that site. Site 25, which was the target of a site-directed mutagenesis experiment, is indicated. I downloaded V3 amino acid sequences from the HIV Sequence Database (http://hiv-web.lanl.gov) on May 2, 2005. Since V3 varies in sequence among subtypes (clades) of HIV-1 (Kuiken et al. 1999), I restricted the analysis to subtype B, which is the subtype used in the site-directed mutagenesis experiment. V3 also varies in sequence among coreceptor usage phenotypes. Therefore, I used only sequences from virus with known coreceptor usage, identified in the database as using CCR5 (R5), CXCR4 (X4), or both coreceptors (R5X4). Finally, since the objective was to calculate site-specific amino acid frequencies within patients (populations) and then average these values across patients, I considered only sequences associated with an identified patient. I omitted from the data set sequences identified as contaminants and sequences with unidentified residues or missing either conserved terminal cysteine residue (these residues are absolutely conserved and form a disulfide bond between them). Subtype B V3 is most commonly 35 amino acids long. To avoid uncertainties in sequence alignment (site homology), I used only sequences 35 amino acids long that aligned unambiguously (without gaps) with the vast majority of other sequences in the data set.
Site-specific fitness and frequency:
I regressed relative infectivity, scaled between 0 and 1, on the among-population mean relative frequency of the amino acid at site 25, also scaled between 0 and 1. This was done only for R5 virus because infectivity was measured for R5 virus. The regression shows a highly significant relationship (slope = 0.84; d.f. = 1, 5; P = 6.64 × 10−7), with frequency explaining 99% of the variance in infectivity (r2 = 0.99) (Figure 2). This surprisingly strong and linear relationship demonstrates that the among-population mean site-specific frequency of an amino acid is indeed a very good predictor of the amino acid's marginal fitness.
Figure 2.—

Simple least-squares linear regression of scaled virion relative infectivity on the scaled among-population mean relative frequency of the amino acid at site 25 of subtype B V3 from R5 virus. Data point labels are amino acid one-letter codes. The regression equation is y = 0.15 + 0.84x.
The intercept of the regression line (0.15) is also significantly different from zero (P = 1.18 × 10−4). Assuming that amino acids are not naturally observed at V3 site 25 if they render V3 nonfunctional, the 15% infectivity observed for amino acids with zero frequency suggests that some other factor accounts for this level of infectivity and that V3 accounts for only 85% of the infectivity component of fitness. Since we are interested only in the portion of fitness attributable to V3, infectivity can be scaled from the intercept to 1, giving a one-to-one relationship between marginal relative fitness (w) and relative frequency (f), both scaled between 0 and 1: w = f. The selection coefficient of amino acid j relative to any reference amino acid i at the same site can then be calculated by rescaling fitness to wi = 1 and calculating the absolute difference between fitnesses: sj = |1 − wj|. If the reference amino acid is the most common at a site, the selection coefficient is simply sj = 1 − fj.
Selection coefficients:
V3 site-specific amino acid selection coefficients were calculated relative to the most common residue at each respective site for the R5 phenotype. These calculations assume no linkage among sites, which is reasonable given the high rate of effective recombination (between viral variants) (1.38 × 10−4 recombination events/adjacent nucleotide site/generation) (Shriner et al. 2004a) relative to the mutation rate (2.4 × 10−5 point mutations/nucleotide site/generation) (Mansky and Temin 1995). Site-specific selection coefficients range from 0.180 to 0.999, with a mean of 0.923 (median = 0.970; Figure 3), suggesting strong selection. Even if a small intrapatient effective population size of Ne = 103 infected cells (Achaz et al. 2004; Shriner et al. 2004b) is assumed and the minimum selection coefficient is used, then 2Nes = 360 ≫ 1, indicating that selection will prevail over genetic drift.
Figure 3.—

Histogram of subtype B R5 V3 site-specific amino acid selection coefficients relative to the most common residue at each respective site. N = 71.
Selection:
Strong selection on V3 by coreceptors is consistent with a lack of genetic drift in this region in comparison to other similar-sized regions under severe population bottlenecks in culture (Yuste et al. 2000). It is also consistent with the convergent evolution, among patients shortly after infection, of subtype B V3 sequences toward the R5 sequence with the most common residue at each site (Figure 1) (Zhang et al. 1993). Given such apparent strong selection by the coreceptor, the high variability of V3 within and among patients suggests that equally strong forces displace sequences away from the sequence with the highest fitness relative to the interaction with CCR5. These forces may be migration, in the sense of repeat infections, or selection by either the alternative coreceptor or the immune system. Migration on its own may be excluded, since in the absence of strong opposing selection it would have no effect (all populations would consist of the sequence with the highest fitness). This leaves opposing selection by the alternative coreceptor, CXCR4, or the immune system. Numerous studies, using a wide variety of methods of comparative sequence analysis, have implicated coreceptors or the immune system or both as sources of positive selection of V3 (e.g., Bonhoeffer et al. 1995; Yamaguchi and Gojobori 1997; Nielsen and Yang 1998; Gerrish 2001; Williamson 2003; Templeton et al. 2004).
Mutation–selection balance:
Mutation–selection balance cannot explain the high frequencies of less favored amino acids. Given a genetically homogenous virus population at the time of infection (Derdeyn et al. 2004) and the early evolution of V3 toward the R5 sequence with the most common residue at each site (Zhang et al. 1993), and assuming no migration between patients, amino acid polymorphism within a population infecting a patient is assumed to arise via mutation. At mutation–selection equilibrium, the most favored amino acid at each site is the most frequent, with each alternative (deleterious) amino acid present at a mean frequency of p = μ/s, where μ is the rate of mutation to the deleterious amino acid and s is its selection coefficient relative to the most preferred amino acid (Crow and Kimura 1970). For example, in the case of glutamic acid (E), the second most common residue at site 25 of R5 V3 sequences (Figure 1), the mutation rate from the most common residue, aspartic acid (D), ignoring any mutation bias, is predicted to be 1.6 × 10−5/codon/generation and s = 0.18. This gives p = 8.9 × 10−5, which is much lower than the observed mean frequency of 0.35.
Episodic adaptation:
The high frequencies of less favored residues may be explained if V3 is displaced from mutation–selection balance, either at the time of infection, after which V3 evolves rapidly toward the R5 sequence with the most common residue at each site, or during chronic infection when it is subject to strong opposing selection, as argued above. Although the alternative coreceptor, CXCR4, may impose strong selection during the late stages of disease progression (Templeton et al. 2004), the immune system is expected to periodically select escape mutants throughout the chronic stage of infection (da Silva 2003; Wei et al. 2003; Williamson 2003; Templeton et al. 2004; Frost et al. 2005). For example, antibodies may impose frequency-dependent selection by targeting epitopes consisting of the most common residues at sites, but such sites may eventually become shielded from antibody surveillance by changes in other protein regions (Wei et al. 2003; Frost et al. 2005). This scenario could result in the episodic adaptation of V3 to CCR5.
The mutational landscape model:
Under weak selection and mutation (see Orr 2002), each step in an episode of adaptation can be described by the mutational landscape model of Gillespie (1984, 1991). In this model, a beneficial amino acid of fitness rank j spreads to fixation as the next step in adaptation, when an amino acid of fitness rank i is the current wild type at the site, with probability
 |
(1) |
where Π is the probability of fixation and the subscripts denote the marginal fitness ranks of the beneficial amino acids within one mutational step of the wild type, with 1 indicating the highest fitness and j < i. This model assumes no recombination and considers mutations at all sites of a sequence. However, the model can be applied to a single amino-acid site if free recombination is assumed or if it is assumed that the most preferred amino acid is present at all other sites (i.e., that there is no potential for adaptation at other sites). Since Πj ≈ 2sj for a new mutation (Haldane 1927) and Πj ≈ 2Nsjpj for standing variation (Orr and Betancourt 2001), Pij is, in either case, proportional to sj. For example, in the case of new mutations,
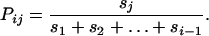 |
(2) |
Therefore, Pij is linearly related to sj and wj, and, after scaling both variables between 0 and 1, Pij ≈ wj regardless of which amino acid is the current wild type (Figure 4). This means that regardless of the level of adaptation of the population, a particular beneficial amino acid will be the next to spread to fixation with a probability that is proportional to its marginal fitness. If Pij is taken to be the proportion of populations fixed for an amino acid of fitness rank j at a particular site or the site-specific frequency of this amino acid averaged across many populations, then this relationship may explain why among-population mean site-specific frequencies of amino acids and their marginal relative fitnesses, both scaled between 0 and 1, are equal.
Figure 4.—

The probability that a beneficial amino acid of fitness rank j will be the next to spread to fixation, given that an amino acid of fitness rank i is the current wild type (Pij), plotted against the amino acid's marginal relative fitness (wj), both scaled between 0 and 1. Pij was calculated for V3 site 25 residues, using Equation 2 (see text) with selection coefficients calculated relative to residues K (circles), A (squares), and Q (triangles). One-letter amino acid codes are shown. The diagonal line is for y = x.
Assumptions of the model:
The mutational landscape model of adaptive evolution is based on two fundamental assumptions: that selection prevails over genetic drift (2Nes ≫ 1) and that mutation is rare (Neμ ≪ 1, where here μ is the number of mutations per nucleotide site per generation) (Orr 2002). Under these conditions, a single beneficial mutation is expected to spread to fixation before the next beneficial mutation begins spreading. Although the model was originally described as assuming strong selection (Gillespie 1984, 1991), selection in the model is strong only in relative terms, compared to 1/Ne; in absolute terms selection is assumed to be weak: 1/Ne ≪ s ≪ 1 (Orr 2002). HIV-1 clearly violates the assumptions of weak selection and mutation; V3 experiences strong selection, as shown above, and the point mutation rate for HIV-1 is high. Strong selection and mutation may mean that more than one beneficial mutation spread through the population simultaneously. Such clonal interference (Gerrish and Lenski 1998) among beneficial mutations at different amino acid sites on different genomes would tend to reduce probabilities of fixation. However, clonal interference is expected to be weak for HIV-1 because the effective recombination rate is more than five times the mutation rate and, therefore, amino acid sites can be considered unlinked.
The question then is, Is there clonal interference among beneficial mutations at a single amino-acid site? The answer depends on the probability that more than one beneficial mutation segregate at the same site. This, of course, depends on the beneficial mutation rate, which depends on the level of adaptation at a given site (the wild-type amino acid at the site). I calculated the proportion of nucleotide point mutations that produce a beneficial amino acid change using the rank order of fitnesses of amino acids at V3 site 25 (Figure 2). Excluding mutations at multiple codon positions, because the frequency of these is very low (μ2 and μ3) compared to the frequency of mutations at a single codon position (μ), and ignoring any mutation bias, the proportion of nucleotide point mutations that are beneficial, averaged among codons for each amino acid, ranges from 0.037 for arginine (R) to 0.389 for glycine (G) and is 0.182 averaged across all amino acids observed at site 25 [except aspartic acid (D), which has the highest fitness]. Then, the rate of beneficial mutations per codon is the product of the probability of a mutation in the codon (including synonymous mutations) and the proportion of these mutations that are beneficial. Allowing for the highest beneficial mutation rate at site 25, that is, with glycine as the wild-type amino acid, the rate of beneficial mutations per codon is 3 nucleotide sites/codon × 2.4 × 10−5 mutations/nucleotide site/generation × 0.389 beneficial mutations/mutation = 2.8 × 10−5 beneficial mutations/codon/generation. Therefore, with a census population size of ∼N = 107 infected cells (Chun et al. 1997) there will be a maximum total of 2.8 × 102 beneficial mutations at V3 site 25 each generation within a population. However, the probability that a new beneficial mutation becomes fixed is proportional to Ne/N (Kimura 1964), which for HIV-1 is 103/107 = 10−4. Therefore, at most 10−4 × 2.8 × 102 = 2.8 × 10−2 new beneficial mutations each generation ultimately become fixed. This is equivalent to one beneficial mutation that ultimately becomes fixed arising every 36 generations or more, on average, for the highest beneficial mutation rate at site 25. For the mean beneficial mutation rate at site 25 (1.3 × 10−5), one beneficial mutation that ultimately becomes fixed arises every 77 generations or more. To determine whether multiple beneficial mutations will segregate at the same site, we need to know how long it takes a single new beneficial mutation to spread to fixation. This can be calculated numerically from the frequency of a beneficial mutation after one generation of selection in a haploid organism (the HIV-1 provirus is haploid): p′ = p(1 + s)/(ps + 1), where p is the frequency in the current generation. With initial frequency p = 1/N = 10−7 for a new mutation, the number of generations taken to reach a frequency of 99% ranges from 6, for mutation G → D (s = 34.9), to 51, for mutation G → R (s = 0.5). Therefore, in the extreme case of the highest beneficial mutation rate and lowest selection coefficient, we can expect some clonal interference (on average, a new beneficial mutation fixes every 36 generations or more and it takes such a mutation 51 generations to spread to near fixation). However, with the average beneficial mutation rate, clonal interference is not expected regardless of the magnitude of the selection coefficient (on average, a new beneficial mutation fixes only every 77 generations or more).
A recent study with a DNA bacteriophage system under moderately strong selection (s = 0.11–0.39) and weak mutation is the only empirical examination of the mutational landscape model to date (Rokyta et al. 2005). The fitnesses of nine beneficial amino acid replacements that occurred as first steps in episodes of adaptation were measured, and the frequencies of the replacements across 20 replicate populations were compared to expectations under the model. Modifying the model to account for mutation bias and population bottlenecks resulting from the experimental protocol improved its fit to the observed distribution of amino acid replacement frequencies. In the case of HIV-1, population size is constant during chronic infection and mutation bias may be unimportant since selection is strong. The empirical support from the Rokyta et al. study suggests that the model is robust to violations of the assumption of weak selection. Indeed, the model requires only that fixation probabilities be proportional to s (Rokyta et al. 2005).
Most of the arguments above assume that the source of beneficial variants is new mutations within the population. However, variation may also be introduced by migration in the form of multiple infections of the same patient. Nevertheless, long-term studies of subtype B HIV-1-infected patients have not reported evidence of repeat infections (e.g., Shankarappa et al. 1999). Repeat infections may be rare because sexual transmission is the main route of infection in North America and western Europe (UNAIDS 2004), where subtype B predominates, and the probability of infection per coital act is <0.5% (Gray et al. 2001). And, perhaps more importantly, sexual transmission involves a severe bottleneck of the donor viral population, possibly due to strong selection by host target cells, resulting in typically only a single variant transmitting successfully (Derdeyn et al. 2004). Consequently, the impact of migration on genetic variation is expected to be minor compared to that of mutation and selection. For example, even with the successful transmission and integration of 102 distinct genomes, a migration event would contribute only a very small fraction of the 2.4 × 106 mutant genomes generated by mutation each generation (∼2 days) (Markowitz et al. 2003) in a population (for an HIV-1 genome size of ∼104 nucleotides). Therefore, mutation produces several orders of magnitude more variation in a population in a single generation than can be contributed by a rare migration event.
Another assumption made in applying the mutational landscape model to HIV-1 is that the HIV-1 populations studied here are true replicates. Strictly speaking, this assumption is violated because populations differ in a variety of ways, including differences in specific immunity (cytotoxic T-lymphocyte and antibody responses) among patients. However, the main source of selection of V3 of interest here is the chemokine receptor CCR5, which is expressed intact on cells targeted by HIV-1 in every infected individual. The intact, expressed protein appears to exhibit little adaptive variation within humans and among higher primates with respect to HIV-1 (Zhang et al. 2003). Therefore, CCR5 represents a homogenous source of selection among patients. Indeed, HIV-1 adapts to CCR5 shortly after infection, rapidly evolving the V3 sequence with the most common amino acid at each site for the CCR5-utilizing phenotype (Figure 1) (Zhang et al. 1993). Therefore, although the subtype B HIV-1 populations analyzed here are not true replicates, they are as similar with respect to the source of selection under study as one might expect to find in a nonexperimental setting.
Conclusion:
It will be interesting to see if the correlation between mean frequency and fitness observed here holds more generally, which should depend on whether fluctuating selection within populations combined with constant, strong selection across populations is a common condition. If so, the ability to estimate site-specific marginal fitnesses of amino acids from their among-population mean site-specific frequencies should open new avenues of research into the dynamics of adaptation at the molecular level.
Acknowledgments
I thank Alexei Drummond, for suggesting the interpretation of the intercept of the regression line in Figure 2, and two anonymous reviewers whose comments greatly improved the manuscript. I also acknowledge the support of the Discipline of Genetics and the School of Molecular and Biomedical Science of The University of Adelaide.
References
- Achaz, G., S. Palmer, M. Kearney, F. Maldarelli, J. W. Mellors et al., 2004. A robust measure of HIV-1 population turnover within chronically infected individuals. Mol. Biol. Evol. 21: 1902–1912. [DOI] [PubMed] [Google Scholar]
- Bonhoeffer, S., E. C. Holmes and M. A. Nowak, 1995. Causes of HIV diversity. Nature 376: 125. [DOI] [PubMed] [Google Scholar]
- Chun, T. W., L. Carruth, D. Finzi, X. Shen, J. A. DiGiuseppe et al., 1997. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 387: 183–188. [DOI] [PubMed] [Google Scholar]
- Coffin, J. M., 1999. Molecular biology of HIV, pp. 3–40 in The Evolution of HIV, edited by K. A. Crandall. Johns Hopkins University Press, Baltimore.
- Crow, J. F., and M. Kimura, 1970. An Introduction to Population Genetics Theory. Harper & Row, New York.
- da Silva, J., 2003. The evolutionary adaptation of HIV-1 to specific immunity. Curr. HIV Res. 1: 363–371. [DOI] [PubMed] [Google Scholar]
- de Jong, J. J., A. de Ronde, W. Keulen, M. Tersmette and J. Goudsmit, 1992. Minimal requirements for the human immunodeficiency virus type 1 V3 domain to support the syncytium-inducing phenotype: analysis by single amino acid substitution. J. Virol. 66: 6777–6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derdeyn, C. A., J. M. Decker, F. Bibollet-Ruche, J. L. Mokili, M. Muldoon et al., 2004. Envelope-constrained neutralization-sensitive HIV-1 after heterosexual transmission. Science 303: 2019–2022. [DOI] [PubMed] [Google Scholar]
- Frost, S. D. W., T. Wrin, D. M. Smith, S. L. K. Pond, Y. Liu et al., 2005. Neutralizing antibody responses drive the evolution of human immunodeficiency virus type 1 envelope during recent HIV infection. Proc. Natl Acad. Sci. USA 102: 18514–18519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrish, P., 2001. The rhythm of microbial adaptation. Nature 413: 299–302. [DOI] [PubMed] [Google Scholar]
- Gerrish, P. J., and R. E. Lenski, 1998. The fate of competing beneficial mutations in an asexual population. Genetica 102–103: 127–144. [PubMed] [Google Scholar]
- Gillespie, J. H., 1984. Molecular evolution over the mutational landscape. Evolution 38: 1116–1129. [DOI] [PubMed] [Google Scholar]
- Gillespie, J. H., 1991. The Causes of Molecular Evolution. Oxford University Press, New York.
- Gray, R. H., M. J. Wawer, R. Brookmeyer, N. K. Sewankambo, D. Serwadda et al., 2001. Probability of HIV-1 transmission per coital act in monogamous, heterosexual, HIV-1-discordant couples in Rakai, Uganda. Lancet 357: 1149–1153. [DOI] [PubMed] [Google Scholar]
- Haldane, J. B. S., 1927. A mathematical theory of natural and artificial selection. V. Selection and mutation. Proc. Camb. Philos. Soc. 28: 838–844. [Google Scholar]
- Hung, C. S., N. Vander Heyden and L. Ratner, 1999. Analysis of the critical domain in the V3 loop of human immunodeficiency virus type 1 gp120 involved in CCR5 utilization. J. Virol. 73: 8216–8226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, M., 1964. Diffusion models in population genetics. J. Appl. Probab. 1: 177–232. [Google Scholar]
- Kuiken, C. L., B. Foley, E. Guzman and B. T. M. Korber, 1999. Determinants of HIV-1 protein evolution, pp. 432–468 in The Evolution of HIV, edited by K. A. Crandall. Johns Hopkins University Press, Baltimore.
- Mansky, L. M., and H. M. Temin, 1995. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 69: 5087–5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz, M., M. Louie, A. Hurley, E. Sun, M. Di Mascio et al., 2003. A novel antiviral intervention results in more accurate assessment of human immunodeficiency virus type 1 replication dynamics and T-cell decay in vivo. J. Virol. 77: 5037–5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, R., and Z. Yang, 1998. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148: 929–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr, H. A., 2002. The population genetics of adaptation: the adaptation of DNA sequences. Evolution 56: 1317–1330. [DOI] [PubMed] [Google Scholar]
- Orr, H. A., and A. J. Betancourt, 2001. Haldane's sieve and adaptation from the standing genetic variation. Genetics 157: 875–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt, E. J., J. P. Durnin and D. Kabat, 2005. Kinetic factors control efficiencies of cell entry, efficacies of entry inhibitors, and mechanisms of adaptation of human immunodeficiency virus. J. Virol. 79: 4347–4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reza, S. M., L.-M. Shen, R. Mukhopadhyay, M. Rosetti, T. Pe'ery et al., 2003. A naturally occurring substitution in human immunodeficiency virus tat increases expression of the viral genome. J. Virol. 77: 8602–8606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokyta, D. R., P. Joyce, S. B. Caudle and H. A. Wichman, 2005. An empirical test of the mutational landscape model of adaptation using a single-stranded DNA virus. Nat. Genet. 37: 441–444. [DOI] [PubMed] [Google Scholar]
- Shankarappa, R., J. B. Margolick, S. J. Gange, A. G. Rodrigo, D. Upchurch et al., 1999. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J. Virol. 73: 10489–10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shriner, D., A. G. Rodrigo, D. C. Nickle and J. I. Mullins, 2004. a Pervasive genomic recombination of HIV-1 in vivo. Genetics 167: 1573–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shriner, D., R. Shankarappa, M. A. Jensen, D. C. Nickle, J. E. Mittler et al., 2004. b Influence of random genetic drift on human immunodeficiency virus type 1 env evolution during chronic infection. Genetics 166: 1155–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speck, R. F., K. Wehrly, E. J. Platt, R. E. Atchison, I. F. Charo et al., 1997. Selective employment of chemokine receptors as human immunodeficiency virus type 1 coreceptors determined by individual amino acids within the envelope V3 loop. J. Virol. 71: 7136–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Templeton, A. R., R. A. Reichert, A. E. Weisstein, X.-F. Yu and R. B. Markham, 2004. Selection in context: patterns of natural selection in the glycoprotein 120 region of human immunodeficiency virus 1 within infected individuals. Genetics 167: 1547–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UNAIDS, 2004. 2004 Report on the Global HIV/AIDS Epidemic: 4th Global Report. Joint United Nations Programme on HIV/AIDS (UNAIDS), Geneva.
- Wei, X., J. M. Decker, S. Wang, H. Hui, J. C. Kappes et al., 2003. Antibody neutralization and escape by HIV-1. Nature 422: 307–312. [DOI] [PubMed] [Google Scholar]
- Williamson, S., 2003. Adaptation in the env gene of HIV-1 and evolutionary theories of disease progression. Mol. Biol. Evol. 20: 1318–1325. [DOI] [PubMed] [Google Scholar]
- Yamaguchi, Y., and T. Gojobori, 1997. Evolutionary mechanisms and population dynamics of the third variable envelope region of HIV within single hosts. Proc. Natl. Acad. Sci. USA 94: 1264–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste, E., C. Lopez-Galindez and E. Domingo, 2000. Unusual distribution of mutations associated with serial bottleneck passages of human immunodeficiency virus type 1. J. Virol. 74: 9546–9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. Q., P. MacKenzie, A. Cleland, E. C. Holmes, A. J. Brown et al., 1993. Selection for specific sequences in the external envelope protein of human immunodeficiency virus type 1 upon primary infection. J. Virol. 67: 3345–3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y.-w., O. A. Ryder and Y.-p. Zhang, 2003. Intra- and interspecific variation of the CCR5 gene in higher primates. Mol. Biol. Evol. 20: 1722–1729. [DOI] [PubMed] [Google Scholar]
- Zolla-Pazner, S., 2004. Identifying epitopes of HIV-1 that induce protective antibodies. Nat. Rev. Immunol. 4: 199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]