

Abstract
In the data collection of the QTL experiments using recombinant inbred (RI) populations, when individuals are genotyped for markers in a population, the trait values (phenotypes) can be obtained from the genotyped individuals (from the same population) or from some progeny of the genotyped individuals (from the different populations). Let Fu be the genotyped population and Fv (v ≥ u) be the phenotyped population. The experimental designs that both marker genotypes and phenotypes are recorded on the same populations can be denoted as (Fu/Fv, u = v) designs and that genotypes and phenotypes are obtained from the different populations can be denoted as (Fu/Fv, v > u) designs. Although most of the QTL mapping experiments have been conducted on the backcross and F2(F2/F2) designs, the other (Fu/Fv, v ≥ u) designs are also very popular. The great benefits of using the other (Fu/Fv, v ≥ u) designs in QTL mapping include reducing cost and environmental variance by phenotyping several progeny for the genotyped individuals and taking advantages of the changes in population structures of other RI populations. Current QTL mapping methods including those for the (Fu/Fv, u = v) designs, mostly for the backcross or F2/F2 design, and for the F2/F3 design based on a one-QTL model are inadequate for the investigation of the mapping properties in the (Fu/Fv, u ≤ v) designs, and they can be problematic due to ignoring their differences in population structures. In this article, a statistical method considering the differences in population structures between different RI populations is proposed on the basis of a multiple-QTL model to map for QTL in different (Fu/Fv, v ≥ u) designs. In addition, the QTL mapping properties of the proposed and approximate methods in different designs are discussed. Simulations were performed to evaluate the performance of the proposed and approximate methods. The proposed method is proven to be able to correct the problems of the approximate and current methods for improving the resolution of genetic architecture of quantitative traits and can serve as an effective tool to explore the QTL mapping study in the system of RI populations.
MOST biologically important traits show continuous variations and have poor heritability. Traditional study of quantitative genetics based on the phenotype evaluation to investigate quantitative trait loci (QTL) controlling these traits is difficult and limited. Recently, the advent of fine-scale molecular markers has provided researchers with an efficient tool for the detection of the underlying QTL. Most QTL detection experiments for producing marker genotypes and phenotypic traits in species have been conducted with populations derived from crosses between inbred lines, e.g., backcross, advanced backcross, F2, recombinant inbred (RI) populations, intermated recombinant inbred (IRI) populations, advanced intercross (AI) populations, advanced backcross populations, double haploid (DH) populations, and NC Design III, etc. (Comstock and Robinson 1952; Stuber et al. 1992; Beavis et al. 1994; Veldboom et al. 1994; Darvasi and Soller 1995; Austin and Lee 1996; Liu et al. 1996; Chapman et al. 2003; Winkler et al. 2003; Complex Trait Consortium 2004; Broman 2005). These different populations may show different properties in QTL mapping as they have different population structures, such as homozygosity, genotypic frequencies, and linkage disequilibrium (Weir 1996, Chap. 5). In principle, the use of the information about genotypes and phenotypes of individuals in these populations has become a key approach to detect the underlying QTL for the understanding of the genetic basis and the improvement of important traits in genetic study.
In the data collection of these QTL experiments, when individuals are genotyped for markers in a population, the trait values (phenotypes) can be recorded on the genotyped individuals (on the same population) or on some progeny of the genotyped individuals (on the progeny population). Fisch et al. (1996) illustrated the situations of data collection by Fu/Fv, where Fu is the genotyped population and Fv (v ≥ u) is the phenotyped population, in the system of RI populations (see recombinant inbred populations for the population structure of RI populations). For example, F2/F2 denotes the typical F2 design, where genotypes and phenotypes are obtained from the same individuals in the F2 population, and F2/F4 denotes the design to genotype F2 individuals and phenotype their progeny in the F4 population. Although the (Fu/Fv, u = v) designs are typical (Doerge et al. 1997; Lynch and Walsh 1998, Chap. 15), the (Fu/Fv, u < v) designs are also very popular and important for QTL detection in the genetic analysis of complex traits. For example, the F3/F3, F2/F3, F2/F4, F4/F4, F5/F5, and F6/F7 designs have been used to detect QTL in maize by Stuber et al. (1992), Beavis et al. (1994), Veldboom et al. (1994), Austin and Lee (1996), Mihaljevic et al. (2004, 2005), Zhang and Xu (2004), and Sala et al. (2006) and the F4/F6 design was used to study QTL in soybean by Chapman et al. (2003). There are some benefits of using the (Fu/Fv, u < v) with multiple phenotyping individuals and (Fu/Fu, u < 2) designs. For example, the cost can be economical for not genotyping the progeny for markers, the environmental variance can be reduced by phenotyping multiple progeny for trait measurement, and homozygotes can be accumulated so that QTL mapping may be improved (Cowen 1988; Lander and Botstein 1989; Knapp and Bridges 1990; Edwards et al. 1992; Austin and Lee 1996).
Traditional QTL mapping methods developed to date mostly assume that both marker genotypes and phenotypic traits are obtained from the same population [the (Fu/Fv, u = v) designs], and they especially focus on the F2/F2 and backcross designs (Lander and Botstein 1989; Jensen 1993; Zeng 1994; Satagopan et al. 1996; Kao et al. 1999; Nakamichi et al. 2001; Sen and Churchill 2001; Kao and Zeng 2002; Yi et al. 2003; Carlborg and Haley 2004; Zou et al. 2004). Some researchers have applied these traditional (approximate) methods to QTL mapping study by regarding the traits (trait means) of progeny as the traits of genotyped individuals, i.e., by treating (Fu/Fv, u < v) designs as (Fu/Fv, u = v) designs, in the analysis (Stuber et al. 1992; Beavis et al. 1994; Veldboom et al. 1994; Austin and Lee 1996; Chapman et al. 2003; Zhang and Xu 2004). Such application implicitly ignores the fact that the traits are controlled by the progeny (Fv) genomes, not by their ancestral (Fu) genomes, and that the segregation of heterozygotes will vary their population structures. Consequently, the power of QTL detection may be affected and the estimates of QTL effects can be biased by the approximate methods as shown in Zhang and Xu (2004) and in this article. Statistical methods are generally lacking or inadequate for the (Fu/Fv, u < v) designs. Fisch et al. (1996) suggested to propose an adequate model for the (Fu/Fv, u ≤ v) designs, and Zhang and Xu (2004) considered the nature of segregation to propose a one-QTL model for the F2/F3 design in QTL mapping. As shown in this article, the one-QTL model by Zhang and Xu (2004) and the approximate method may have confounding problems in the estimation of QTL parameters and lose power of QTL detection. Ideally, we would like to extend the one-QTL model to a multiple-QTL model and the F2/F3 design to the more general (Fu/Fv, u ≤ v) designs for more practical and broad use in a way that multiple QTL and their possible epistasis can be considered in the model to correct the problems and the benefit of other RI populations as mentioned can be utilized to further improve and study QTL mapping. In this article, a statistical method considering the differences in population structures between different RI populations is developed on the basis of a more complete multiple-QTL model for the more general (Fu/Fv, u ≤ v) designs. In addition, the QTL mapping properties of the proposed and approximate methods in the different (Fu/Fv, u ≤ v) designs are also derived and discussed. A simulation study was performed for evaluating the relative efficiencies of different (Fu/Fv, u ≤ v) designs and comparing the performance of the proposed and current methods in these designs. The proposed method is capable of improving the resolution of the genetic architecture of quantitative traits and can serve as a tool to study QTL mapping in the (Fu/Fv, u ≤ v) designs.
RECOMBINANT INBRED POPULATIONS
Assume that two parental inbred lines, P1 and P2, differ substantially in the quantitative trait of interest and are fixed for alternative alleles at QTL and markers. A cross between the parental lines produces an F1 population with all the same heterozygous individuals. If the F1 individuals are selfed or intermated, it produces an F2 population. In the F2 population, the genotypic frequencies of P1 homozygote, heterozygote, and P2 homozygote are 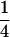 ,
, 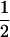 , and
, and 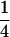 , respectively (the heterozygosity is 0.5), if one locus is considered. The frequency of recombinants (r) between any two loci in the F2 population is equivalent to the recombination fraction (c). If the F2 individuals are further selfed for t − 2 generations, it produces a so-called RI Ft population. For
, respectively (the heterozygosity is 0.5), if one locus is considered. The frequency of recombinants (r) between any two loci in the F2 population is equivalent to the recombination fraction (c). If the F2 individuals are further selfed for t − 2 generations, it produces a so-called RI Ft population. For 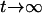 , the derived population is called recombinant inbred lines (RILs). In an Ft population, the frequencies of P1 homozygote, heterozygote, and P2 homozygote in a locus are expected to be
, the derived population is called recombinant inbred lines (RILs). In an Ft population, the frequencies of P1 homozygote, heterozygote, and P2 homozygote in a locus are expected to be 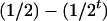 ,
, 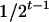 , and
, and 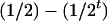 , respectively (the heterozygosity is
, respectively (the heterozygosity is 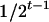 ), and the frequency of recombinants between two loci, denoted as rt, is increasing as t is increasing and can be obtained according to Haldane and Waddington (1931). Haldane and Waddington showed that
), and the frequency of recombinants between two loci, denoted as rt, is increasing as t is increasing and can be obtained according to Haldane and Waddington (1931). Haldane and Waddington showed that 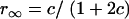 .
.
Genetic model:
In a RI population, any individual can have three possible QTL genotypes, QQ, Qq, and qq, if only one QTL, say Q, is considered. Let the genotypic value, Gi, of an individual i have the following relation with the genetic parameters as
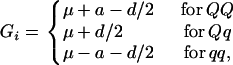 |
(1) |
where μ is the intercept, and a and d are the additive and dominance effects according to Cockerham's model (Kao and Zeng 2002). If multiple, say m, QTL are considered, the extension of the one-QTL genetic model in Equation 1 to a multiple-QTL model with epistasis is straightforward (Kao and Zeng 2002). If an individual i produces k progeny, the mean genotypic value of the k progeny, 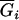 , is
, is
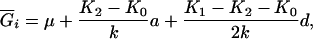 |
(2) |
where K2, K1, and K0 denote the numbers of progeny with QQ, Qq, and qq genotypes among the k progeny, respectively. If the genotype of the individual i is QQ (qq), all the k progeny have the same QQ (qq) genotype, i.e., K2 = k (K0 = k), and the mean genotypic value is μ + a − d/2 (μ − a − d/2). If the genotype of the individual i is Qq, the possible genotype of the progeny can be QQ, Qq, or qq, and the mean genotypic value depends on K2, K1, and K0. The possible allocations of (K2, K1, K0) have (k + 1)(k + 2)/2 combinations and follow a trinomial distribution with k trials and cell probabilities 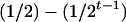 ,
, 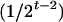 , and
, and 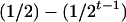 . The number of possible mean genotypic values corresponds to the number of possible allocations of (K2, K1, K0). Let
. The number of possible mean genotypic values corresponds to the number of possible allocations of (K2, K1, K0). Let 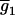 ,
, 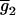 , … ,
, … , 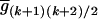 denote the (k + 1)(k + 2)/2 genotypic means. For simplicity, the mean genotypic value in Equation 2 is expressed as
denote the (k + 1)(k + 2)/2 genotypic means. For simplicity, the mean genotypic value in Equation 2 is expressed as
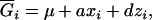 |
(3) |
where
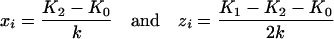 |
are to characterize the status of the additive and dominance effects in the genotypic means. Under the expression of Equation 3, the extension of the model for mean genotypic value from one QTL to multiple QTL is straightforward. For m QTL without epistasis, the genetic model can be written as
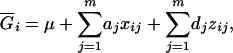 |
(4) |
where xij's and zij's are the coded variables for Qj's, j = 1, 2, … , m, and are defined similarly as xi and zj in Equation 3. The extension of this model to consider epistasis is straightforward by introducing the cross-product terms as the terms of epistasis.
Variance components:
When m QTL with complete marginal and epistatic effects are considered together, the genetic variances of a quantitative trait can be decomposed into 2m2 variances and 2m4 − m2 covariances in a RI population. Taking m = 2 as an example, there are 8 genetic variances and 28 genetic covariances. If the two QTL are unlinked, the genetic variance in an Ft population can be found as
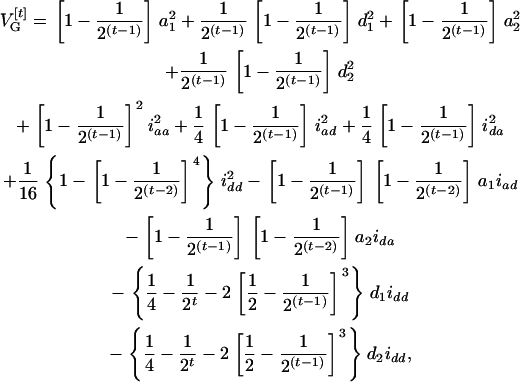 |
(5) |
where iaa, iad, ida, and idd are the additive-by-additive, additive-by-dominance, dominance-by-additive, and dominance-by-dominance epistatic effects, respectively, under the setting of the digenic model of Equation 1. Some of the covariances are zeros. For t = 2, there is no genetic covariance and the genetic variance reduces to eight independent components 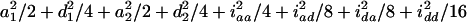 . As t is increasing, the additive variances are increasing due to the accumulation of homozygotes, and the dominance variances are decreasing for the loss of heterozygotes. For example, the additive and dominance variance components are
. As t is increasing, the additive variances are increasing due to the accumulation of homozygotes, and the dominance variances are decreasing for the loss of heterozygotes. For example, the additive and dominance variance components are 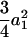 (
(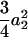 ) and
) and 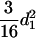 (
(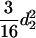 ) for the F3 population, and these components are
) for the F3 population, and these components are 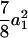 (
(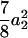 ) and
) and 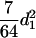 (
(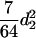 ) for the F4 population. These two components approach
) for the F4 population. These two components approach 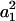 and zero for
and zero for 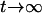 . This shows that the RI Ft, t > 2, populations can benefit the estimation of additive effects by cumulating the homozygotes, but may hurt the estimation of dominance effects due to the loss of heterozygotes. Also, the epistatic variances involving additive effects (iaa, iad, and ida) are increasing, and the dominance-by-dominance variance is decreasing. For example, the epistatic variances involving the additive effects are
. This shows that the RI Ft, t > 2, populations can benefit the estimation of additive effects by cumulating the homozygotes, but may hurt the estimation of dominance effects due to the loss of heterozygotes. Also, the epistatic variances involving additive effects (iaa, iad, and ida) are increasing, and the dominance-by-dominance variance is decreasing. For example, the epistatic variances involving the additive effects are 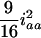 (
(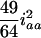 ),
), 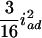 (
(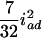 ), and
), and 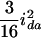 (
(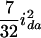 ), and the dominance-by-dominance variance is
), and the dominance-by-dominance variance is 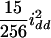 (
(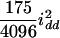 ) in the F3 (F4) population. The four variance components approach
) in the F3 (F4) population. The four variance components approach 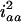 ,
, 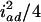 ,
, 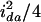 , and zero, respectively, as
, and zero, respectively, as 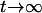 . Also, the covariances between genetic effects become present in the Ft, t > 2, populations, and they will cause confounding problems in estimation for the one-QTL approach or if epistasis is present and ignored in QTL mapping.
. Also, the covariances between genetic effects become present in the Ft, t > 2, populations, and they will cause confounding problems in estimation for the one-QTL approach or if epistasis is present and ignored in QTL mapping.
THE STATISTICAL METHODS
Data structure:
Consider a sample of size n from a (Fu/Fv, u < v) design or a (Fu/Fv, u = v) design. The n individuals from the Fu population are genotyped for markers (Xi, i = 1, 2, … , n). If the sample is from the (Fu/Fv, u = v) design, the n genotyped individuals are phenotyped to obtain the n trait values (yi's, i = 1, 2, … , n). If the sample is from the (Fu/Fv, u < v) design, each of the n genotyped individuals produces k progeny in the Fv generation for phenotyping, and their traits (yij's, j = 1, 2, … , k) or trait means (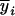 's) are recorded. For QTL mapping using the data from (Fu/Fv, u < v) designs, both the traditional (approximate) and the proposed QTL mapping methods have been used and are discussed here. When applying the traditional methods to (Fu/Fv, u < v) designs, one assumes that the mean trait is controlled by the QTL in the Fu individuals, referred to as Q[u]'s hereafter, rather than by the QTL in the Fv progeny, referred to as Q[v]'s hereafter (Q[u] and Q[v] have the same dimension). As a result, the problems, such as bias in estimation and loss in power of QTL detection, will occur in QTL mapping for the traditional methods. The proposed method intends to connect the trait of the Fv progeny with Q[v]'s using the marker information in the Fu individuals; hence it can correct the problems to improve QTL mapping in the (Fu/Fv, u < v) designs as shown below.
's) are recorded. For QTL mapping using the data from (Fu/Fv, u < v) designs, both the traditional (approximate) and the proposed QTL mapping methods have been used and are discussed here. When applying the traditional methods to (Fu/Fv, u < v) designs, one assumes that the mean trait is controlled by the QTL in the Fu individuals, referred to as Q[u]'s hereafter, rather than by the QTL in the Fv progeny, referred to as Q[v]'s hereafter (Q[u] and Q[v] have the same dimension). As a result, the problems, such as bias in estimation and loss in power of QTL detection, will occur in QTL mapping for the traditional methods. The proposed method intends to connect the trait of the Fv progeny with Q[v]'s using the marker information in the Fu individuals; hence it can correct the problems to improve QTL mapping in the (Fu/Fv, u < v) designs as shown below.
The proposed method:
Without loss of generality in inferring QTL mapping in the (Fu/Fv, u ≤ v) designs, consider that the sample is obtained from a (Fu/Fv, u < v) design and the trait means (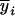 's) measured on the Fv progeny are used in the analysis. The proposed method attempts to relate the mean traits with the mean genotypic values at Q[v] using the marker information of the Fu individuals so that the genetic structure of the Fv population can be taken into account in modeling. If a quantitative trait is controlled by m nonepistatic QTL,
's) measured on the Fv progeny are used in the analysis. The proposed method attempts to relate the mean traits with the mean genotypic values at Q[v] using the marker information of the Fu individuals so that the genetic structure of the Fv population can be taken into account in modeling. If a quantitative trait is controlled by m nonepistatic QTL, 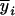 can be related to the m QTL by the model
can be related to the m QTL by the model
 |
(6) |
where 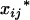 's and
's and 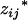 's, j = 1, 2, … , m, are the coded variables associated with the additive and dominance effects at
's, j = 1, 2, … , m, are the coded variables associated with the additive and dominance effects at 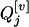 's, j = 1, 2, … , m, in the genotypic means, and they have the same definitions as
's, j = 1, 2, … , m, in the genotypic means, and they have the same definitions as 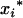 and
and 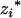 in Equation 4. The residual error εi is assumed to follow a normal distribution with mean zero and variance σ2. As multiple (m) intervals are used to infer the multiple QTL, this model is a multiple-interval mapping-based (MIM-based) method (Kao et al. 1999) for the (Fu/Fv, u < v) designs. A single-QTL model for the F2/F3 design was first proposed by Zhang and Xu (2004).
in Equation 4. The residual error εi is assumed to follow a normal distribution with mean zero and variance σ2. As multiple (m) intervals are used to infer the multiple QTL, this model is a multiple-interval mapping-based (MIM-based) method (Kao et al. 1999) for the (Fu/Fv, u < v) designs. A single-QTL model for the F2/F3 design was first proposed by Zhang and Xu (2004).
As QTL could be located in the marker intervals, the genotypic means (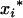 's and
's and 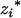 's) for the m QTL are unobservable and need to be inferred from the flanking marker genotype of the Fu individual. For k progeny, there are [(k + 1)(k + 2)/2]m genotypic means (possible values for
's) for the m QTL are unobservable and need to be inferred from the flanking marker genotype of the Fu individual. For k progeny, there are [(k + 1)(k + 2)/2]m genotypic means (possible values for 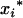 's and
's and 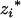 's) for m QTL. Given a sample with size n, the likelihood function of the model in Equation 6 for θ = (a1, d1, a2, d2, … , am, dm, σ2) is
's) for m QTL. Given a sample with size n, the likelihood function of the model in Equation 6 for θ = (a1, d1, a2, d2, … , am, dm, σ2) is
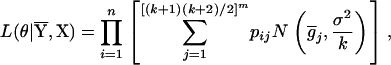 |
(7) |
where 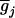 's are the [(k + 1)(k + 2)/2]m genotypic means, and the mixing proportions, pij's, are the conditional probabilities of the corresponding genotypic means given the marker genotype. The density of each individual is a mixture of [(k + 1)(k + 2)/2]m possible normals with different means,
's are the [(k + 1)(k + 2)/2]m genotypic means, and the mixing proportions, pij's, are the conditional probabilities of the corresponding genotypic means given the marker genotype. The density of each individual is a mixture of [(k + 1)(k + 2)/2]m possible normals with different means, 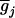 's, and mixing proportions, pij's. Note that the mixing proportions in the likelihoods can be obtained by Equation 9 and need not to be estimated at the tested positions. The EM algorithm (Dempster et al. 1977) is used for the estimation of the parameters in Equations 7 by treating the trait means and markers,
's, and mixing proportions, pij's. Note that the mixing proportions in the likelihoods can be obtained by Equation 9 and need not to be estimated at the tested positions. The EM algorithm (Dempster et al. 1977) is used for the estimation of the parameters in Equations 7 by treating the trait means and markers, 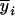 's and Xi's, as observed data and the coded variables of mean genotypic values,
's and Xi's, as observed data and the coded variables of mean genotypic values, 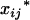 's and
's and 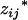 's, as missing data.
's, as missing data.
The EM algorithm and maximum-likelihood estimate:
The coded variables, 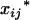 and
and 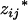 , associated with the additive and dominance effects in the mean genotypic values of
, associated with the additive and dominance effects in the mean genotypic values of 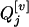 are determined by Kj2, Kj1, and Kj0. Therefore, inferring the distribution of
are determined by Kj2, Kj1, and Kj0. Therefore, inferring the distribution of 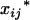 and
and 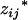 is equivalent to inferring the distribution of Kj2, Kj1, and Kj0. To infer the distribution of Kj2, Kj1, and Kj0 using the marker information from the Fu individuals, one may first infer the distribution of the
is equivalent to inferring the distribution of Kj2, Kj1, and Kj0. To infer the distribution of Kj2, Kj1, and Kj0 using the marker information from the Fu individuals, one may first infer the distribution of the 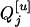 genotype given the marker information and then infer the distribution of Kj2, Kj1, and Kj0 given the QTL genotype of
genotype given the marker information and then infer the distribution of Kj2, Kj1, and Kj0 given the QTL genotype of 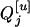 . That is,
. That is,
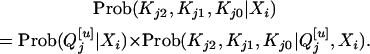 |
(8) |
For the flanking marker interval, Ij, in the RI populations, there are nine possible flanking marker genotypes. Given each of the nine marker genotypes, the conditional probabilities of the QTL genotypes QjQj, Qjqj, and qjqj for the within 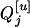 are different in different RI populations, and they depend on their population structure. If an F2 population (u = 2) is genotyped for markers, these conditional probabilities of
are different in different RI populations, and they depend on their population structure. If an F2 population (u = 2) is genotyped for markers, these conditional probabilities of 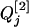 genotypes given the nine flanking marker genotypes have been provided by several researchers (see, e.g., Table 2 of Kao and Zeng 1997). If an Fu, u > 2, population is genotyped, the conditional probabilities are similar to those for the F2 population with rt substituted for r2 (see Haldane and Waddington 1931, for the derivation of rt). Due to segregation,
genotypes given the nine flanking marker genotypes have been provided by several researchers (see, e.g., Table 2 of Kao and Zeng 1997). If an Fu, u > 2, population is genotyped, the conditional probabilities are similar to those for the F2 population with rt substituted for r2 (see Haldane and Waddington 1931, for the derivation of rt). Due to segregation, 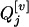 and
and 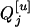 may have the same or different genotypes. If
may have the same or different genotypes. If 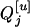 is QjQj (qjqj),
is QjQj (qjqj), 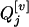 of each progeny is sure to be QjQj (qjqj). That is,
of each progeny is sure to be QjQj (qjqj). That is, 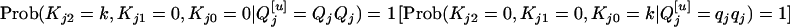 . If
. If 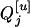 is Qjqj, Q[v] among the k progeny can be QjQj, Qjqj, or qjqj, and it will follow a multinomial distribution (see Genetic model); i.e.,
is Qjqj, Q[v] among the k progeny can be QjQj, Qjqj, or qjqj, and it will follow a multinomial distribution (see Genetic model); i.e.,
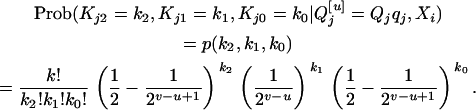 |
Taking all three genotypes of 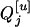 into consideration, it is straightforward to obtain
into consideration, it is straightforward to obtain
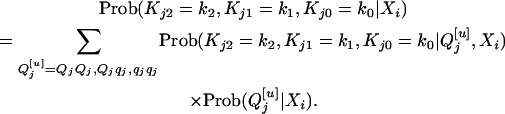 |
The possible number of allocations for each set of Kj2, Kj1, and Kj0 is (k + 1)(k + 2)/2. If all the m QTL are considered at a time, there are 9m possible flanking marker genotypes, and, for each marker genotype, there are totally [(k + 1)(k + 2)/2]m possible allocations for Kj2's, Kj1's, and Kj0's, j = 1, 2, … , m (genotypic means). The joint distribution of Kj2's, Kj1's, and Kj0's, j = 1, 2, … , m, is simply the product of the m individual multinomial distributions:
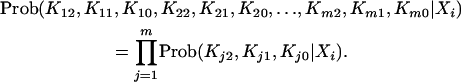 |
(9) |
Under such a setting, the proposed model can be statistically formulated as a two-stage hierarchical model for the use of the EM algorithm. First the random variables 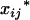 's and
's and 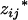 's, j = 1, 2, … , m, are sampled from a multinomial experiment
's, j = 1, 2, … , m, are sampled from a multinomial experiment
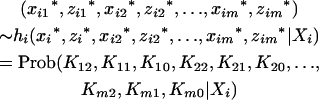 |
to determine the genotypic mean 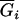 , and then a normal variable for that genotypic mean is generated from
, and then a normal variable for that genotypic mean is generated from
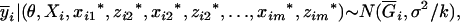 |
where 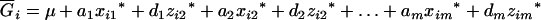 , belonging to one of
, belonging to one of 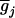 's, j = 1, 2, … , [(k + 1)(k + 2)/2]m, to produce the mean trait value,
's, j = 1, 2, … , [(k + 1)(k + 2)/2]m, to produce the mean trait value, 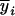 . Following the definition of the EM algorithm, the complete-data likelihood function can be written as
. Following the definition of the EM algorithm, the complete-data likelihood function can be written as
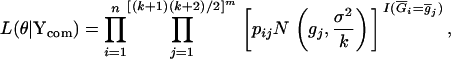 |
(10) |
where Ycom contains the missing and observed data to denote the complete data. Note that the mixing proportions, pij's, are not for estimation and can be determined by Equation 9. Following the definition of the EM algorithm. In the E-step, the conditional expected complete-data log-likelihood with respect to the conditional distribution of missing data given observed data and the current estimated parameter is computed. The M-step is to find θ to maximize the conditional expected complete-data log-likelihood. The maximization will become complicated as k or m increases. Although the derivations of the solutions in the M-step are complicated (not shown), these solutions can be regularized together in the form of the general formulas by Kao and Zeng (1997). The general formulas were originally devised to obtain the maximum-likelihood estimate (MLE) for the backcross and F2/F2 designs by constructing a genetic design matrix D to systematize the solutions into tidy formulations, and the elements in the D matrix are the coded variables associated with the genetic effects in all the possible genotypic values. For the (Fu/Fv, u < v) designs, the complicated solutions in the maximization step after regularization have the same forms of general formulas by assigning the coded variables associated with the genetic effects in the genotypic means to the elements of D. For example, when considering m = 1 and k = 3, there are two coded variables, one for the additive effect and another for the dominance effect, and 10 possible genotypic means. The solutions are equivalent to the general formulas by constructing D with dimension 10 × 2 as
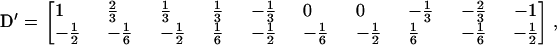 |
where the first column with elements 1 and 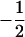 corresponds to the coded variables,
corresponds to the coded variables, 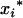 and
and 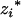 , in the first genotypic mean,
, in the first genotypic mean, 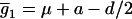 , for all the progeny with Q[v] = QQ (K2 = 3, K1 = 0, K0 = 0), and the second row with elements
, for all the progeny with Q[v] = QQ (K2 = 3, K1 = 0, K0 = 0), and the second row with elements 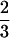 and
and 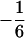 is for the coded variables in the second genotypic mean,
is for the coded variables in the second genotypic mean, 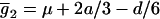 , for the progeny, two with QQ and one with Qq (K2 = 2, K1 = 1, K0 = 0). The remaining eight rows are for the other possible genotypic means,
, for the progeny, two with QQ and one with Qq (K2 = 2, K1 = 1, K0 = 0). The remaining eight rows are for the other possible genotypic means, 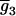 ,
, 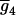 , … ,
, … , 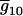 , corresponding with the allocations of different genotypes among progeny. For m QTL in the model, there are [(k + 1)(k + 2)/2]m possible genotypic means and 2m parameters (ignoring epistasis), and the genetic design matrix has a dimension of [(k + 1)(k + 2)/2]m × 2m. Each row of D is assigned to the values of the coded variables for the m QTL in each genotypic mean. The construction of the genetic design matrix for different m and k as well as for considering epistasis is straightforward, although the dimension expands dramatically as m or k becomes large. The E- and M-steps are iterated until convergence, and the converged values of the parameters are the MLE.
, corresponding with the allocations of different genotypes among progeny. For m QTL in the model, there are [(k + 1)(k + 2)/2]m possible genotypic means and 2m parameters (ignoring epistasis), and the genetic design matrix has a dimension of [(k + 1)(k + 2)/2]m × 2m. Each row of D is assigned to the values of the coded variables for the m QTL in each genotypic mean. The construction of the genetic design matrix for different m and k as well as for considering epistasis is straightforward, although the dimension expands dramatically as m or k becomes large. The E- and M-steps are iterated until convergence, and the converged values of the parameters are the MLE.
TABLE 2.
Simulation results of using different QTL mapping methods under the F2/F3 design with different numbers of phenotyping progeny
| QA
|
QB
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Design | Method | Posi = 25 | a1 = 2 | LRT | Power (%) | Posi = 25 | a2 = 2 | LRT | Power (%) | iad = 2 | σ2 = 85.5 | LRT | |
| F2/F3k = 1 | IM | e | 43.07 | 1.43 | 4.69 | 11 | 37.73 | 2.06 | 6.80 | 26 | |||
| (29.46) | (1.53) | (3.06) | (27.41) | (1.46) | (4.21) | ||||||||
| a | 43.55 | 1.45 | 4.67 | 10 | 36.78 | 2.05 | 6.71 | 24 | |||||
| (29.77) | (1.51) | (3.06) | (26.44) | (1.44) | (4.08) | ||||||||
| MIMa | e | 43.06 | 1.46 | 4.90 | 12 | 37.73 | 2.08 | 7.07 | 27 | 81.63 | 11.70 | ||
| (30.34) | (1.52) | (3018) | (27.41) | (1.46) | (4.48) | (9.99) | (5.69) | ||||||
| a | 43.00 | 1.48 | 4.84 | 11 | 36.78 | 2.06 | 6.94 | 25 | 84.42 | 11.58 | |||
| (29.91) | (1.50) | (3.26) | (26.44) | (1.45) | (4.36) | (9.90) | (5.52) | ||||||
| MIMb | e | 43.06 | 1.62 | 6.72 | 12 | 37.73 | 2.03 | 9.43 | 32 | 1.25 | 77.70 | 13.52 | |
| (29.23) | (2.01) | (3.39) | (27.41) | (1.44) | (5.04) | (5.48) | (10.22) | (5.93) | |||||
| a | 42.58 | 1.37 | 6.66 | 17 | 36.78 | 2.05 | 9.34 | 33 | 0.73 | 83.58 | 13.37 | ||
| (28.42) | (4.42) | (3.51) | (26.44) | (1.46) | (5.16) | (2.94) | (9.90) | (5.85) | |||||
| F2/F3k = 3 | IM | e | 30.12 | 1.56 | 8.31 | 37 | 28.16 | 2.04 | 13.89 | 79 | |||
| (19.09) | (0.25) | (4.29) | (14.47) | (0.49) | (5.90) | ||||||||
| a | 29.68 | 1.57 | 8.30 | 37 | 26.78 | 2.08 | 13.79 | 79 | |||||
| (18.71) | (0.58) | (4.30) | (13.52) | (0.19) | (5.89) | ||||||||
| MIMa | e | 32.22 | 1.51 | 8.77 | 40 | 28.16 | 2.09 | 14.44 | 80 | 83.95 | 22.66 | ||
| (22.61) | (0.68) | (4.66) | (14.47) | (0.49) | (6.15) | (10.62) | (4.21) | ||||||
| a | 31.16 | 1.51 | 8.75 | 39 | 26.78 | 2.08 | 14.34 | 79 | 85.90 | 22.54 | |||
| (20.97) | (0.68) | (4.65) | (13.52) | (0.48) | (6.15) | (10.59) | (7.20) | ||||||
| MIMb | e | 29.60 | 1.82 | 10.42 | 46 | 28.16 | 2.09 | 15.68 | 83 | 1.48 | 82.2 | 24.31 | |
| (22.43) | (1.04) | (4.44) | (14.47) | (0.49) | (6.81) | (2.96) | (10.59) | (4.13) | |||||
| a | 28.74 | 1.45 | 10.42 | 47 | 26.78 | 2.09 | 16.42 | 82 | 0.78 | 85.01 | 24.21 | ||
| (21.36) | (0.76) | (4.39) | (13.52) | (0.49) | (6.64) | (1.49) | (10.52) | (7.10) | |||||
| F2/F3k = 5 | IM | e | 28.64 | 1.56 | 12.33 | 67 | 25.22 | 2.02 | 20.22 | 91 | |||
| (16.09) | (0.49) | (5.89) | (8.25) | (0.47) | (8.90) | ||||||||
| a | 28.56 | 1.56 | 12.31 | 67 | 25.25 | 2.01 | 20.13 | 91 | |||||
| (16.07) | (0.48) | (5.87) | (8.23) | (0.16) | (8.86) | ||||||||
| MIMa | e | 27.12 | 1.51 | 12.92 | 70 | 25.22 | 2.00 | 21.06 | 92 | 85.99 | 33.14 | ||
| (13.04) | (0.49) | (6.06) | (8.25) | (0.45) | (9.09) | (10.53) | (9.67) | ||||||
| a | 27.04 | 1.52 | 12.97 | 70 | 25.25 | 2.00 | 20.98 | 92 | 87.68 | 33.14 | |||
| (12.90) | (0.49) | (6.08) | (8.23) | (0.45) | (9.01) | (10.52) | (9.66) | ||||||
| MIMb | e | 27.54 | 1.96 | 14.94 | 73 | 25.22 | 2.01 | 22.31 | 93 | 1.91 | 84.25 | 35.16 | |
| (15.82) | (0.68) | (6.40) | (8.25) | (0345) | (9.92) | (2.18) | (10.34) | (10.07) | |||||
| a | 27.10 | 1.48 | 15.00 | 72 | 25.25 | 2.01 | 23.54 | 93 | 0.98 | 84.25 | 35.16 | ||
| (16.03) | (0.49) | (6.46) | (8.23) | (0.45) | (9.79) | (1.10) | (10.36) | (10.12) | |||||
| F2/F3k = 10 | IM | e | 26.03 | 1.53 | 19.65 | 93 | 25.53 | 2.02 | 35.8 | 100 | |||
| (8.42) | (0.40) | (8.42) | (4.49) | (0.35) | (11.62) | ||||||||
| a | 26.21 | 1.54 | 19.64 | 93 | 25.52 | 2.01 | 35.65 | 100 | |||||
| (8.31) | (0.42) | (8.03) | (4.50) | (0.35) | (11.58) | ||||||||
| MIMa | e | 26.2 | 1.49 | 22.29 | 99 | 25.53 | 1.99 | 38.64 | 100 | 85.38 | 58.09 | ||
| (8.27) | (0.29) | (7.93) | (4.49) | (0.33) | (12.26) | (9.24) | (13.84) | ||||||
| a | 26.14 | 1.49 | 22.35 | 99 | 25.52 | 1.99 | 38.25 | 100 | 54.01 | 58.00 | |||
| (8.34) | (0.29) | (7.92) | (4.50) | (0.33) | (12.22) | (0.92) | (13.81) | ||||||
| MIMb | e | 27.04 | 1.89 | 24.67 | 99 | 25.53 | 2.00 | 40.15 | 100 | 1.62 | 83.74 | 60.47 | |
| (10.42) | (0.46) | (8.14) | (4.49) | (0.34) | (12.70) | (1.38) | (8.83) | (13.87) | |||||
| a | 27.30 | 1.49 | 24.73 | 99 | 25.52 | 2.00 | 41.25 | 100 | 0.81 | 85.37 | 60.38 | ||
| (10.39) | (0.31) | (8.13) | (4.50) | (0.34) | (12.49) | (0.69) | (0.89) | (13.83) | |||||
A total of 100 replicates, each with sample size 200, were analyzed with two unlinked epistatic QTL, QA and QB, controlling the trait variation. The heritability is 0.05 in the F2 population. The critical value for the methods of IM and MIM without epistasis is 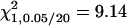 , and the value for MIM with epistasis is
, and the value for MIM with epistasis is 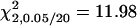 . Posi, position. k, the number of phenotyping progeny. e, proposed (exact) method. a, approximate method.
. Posi, position. k, the number of phenotyping progeny. e, proposed (exact) method. a, approximate method.
Without epistasis.
With epistasis.
The problems if epistasis is present and ignored:
Many current methods ignore epistasis in the analysis of QTL for simplicity. It is important to check the problems if epistasis is present and ignored and in addition to solve the problems in QTL mapping. Without loss of generality, consider that the quantitative trait is controlled by two unlinked epistatic QTL, QA and QB. If the trait value is regressed on QA (QB), the estimates of the additive and dominance effects can be found to be
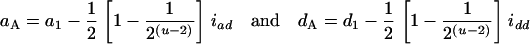 |
(11) |
in the Fu/Fu designs, where a1 (d1) is the additive (dominance) effect of QA, and iad and idd are their epistatic effects (Equations 11 can be obtained from Equations A2 and A3 by setting u = v in the appendix). It shows that the estimate of the aA can be confounded by a1 and iad, and dA is confounded by d1 and idd. Also, it is important to note that the epistatic effect iaa is not confounding in the estimation of the marginal effects. Therefore, in the F2/F2 design, aA = a1 and dA = d1, and there is no confounding problem as the model has the orthogonal property in this design. In the (Fu/Fu, u > 2) designs, the problem of confounding occurs. For example, 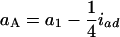 and
and 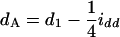 in the F3/F3 design,
in the F3/F3 design, 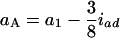 and
and 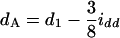 in the F4/F4 design, and
in the F4/F4 design, and 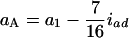 and
and 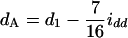 in the F5/F5 design. The fractions associated with the confounding epistatic effects are
in the F5/F5 design. The fractions associated with the confounding epistatic effects are 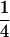 ,
, 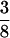 , and
, and 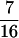 in the F3/F3, F4/F4, and F5/F5 designs, respectively, and the confounding problem is found to become more serious in the later RI populations. This fraction approaches
in the F3/F3, F4/F4, and F5/F5 designs, respectively, and the confounding problem is found to become more serious in the later RI populations. This fraction approaches 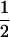 for the designs with large u. For large u, the dominance component may become diminished and hard to estimate, and the additive component plays the major role in estimation, due to the loss of heterozygotes and increase of homozygotes in the later Fu populations. But, for the early Fu populations, say the F2, F3, and F4 populations, the dominance components may not be negligible and should be considered in the model. In addition, ignoring epistasis can inflate the sampling variances of QTL effects and will reduce the power of QTL detection. Among the epistatic variance components, the component contributed by iaa is relatively larger than the components by other epistatic effects. According to Equation 5, the components contributed by iaa are
for the designs with large u. For large u, the dominance component may become diminished and hard to estimate, and the additive component plays the major role in estimation, due to the loss of heterozygotes and increase of homozygotes in the later Fu populations. But, for the early Fu populations, say the F2, F3, and F4 populations, the dominance components may not be negligible and should be considered in the model. In addition, ignoring epistasis can inflate the sampling variances of QTL effects and will reduce the power of QTL detection. Among the epistatic variance components, the component contributed by iaa is relatively larger than the components by other epistatic effects. According to Equation 5, the components contributed by iaa are 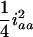 ,
, 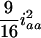 , and
, and 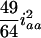 in the F2/F2, F3/F3, and F4/F4 populations, respectively. This component becomes greater for the later RI populations and approaches
in the F2/F2, F3/F3, and F4/F4 populations, respectively. This component becomes greater for the later RI populations and approaches 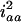 for
for 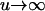 . The above implies that QTL mapping could be problematic, such as biasing the estimation of QTL parameters and reducing the power of QTL detection, if epistasis is ignored in QTL analysis. By taking epistasis into account, the variance components contributed by the epistatic effects can be controlled to enhance the power of QTL detection and the confounding problem can be avoided to improve QTL detection.
. The above implies that QTL mapping could be problematic, such as biasing the estimation of QTL parameters and reducing the power of QTL detection, if epistasis is ignored in QTL analysis. By taking epistasis into account, the variance components contributed by the epistatic effects can be controlled to enhance the power of QTL detection and the confounding problem can be avoided to improve QTL detection.
The traditional (approximate) method and its problems:
The approximate method is to model the relation between the mean trait of the Fv progeny and the QTL in their ancestral Fu individuals, Q[u]'s. It implicitly assumes that the traits measured on the Fv progeny are controlled by the genomes of the Fu individuals, rather than by those of the progeny. This assumption overlooks the differences between population structures, as the genotypic frequencies, heterozygosity, and linkage disequilibrium between the genotyped (ancestral) and phenotyped (progeny) populations are different. Consequently, some problems, such as less power and bias in estimation, will occur (see the appendix). By the approximate method, the estimate of the additive effect is confounded by the additive effect a1 and the epistatic effect iad, and the estimate of the dominance effect is confounded by d1 and idd. The confounding depends on u and v. For example, ba = a1 − iad/4 and bd = d1/2 − idd/8 in the F2/F3 design, 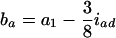 and
and 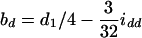 in the F2/F4 design, ba = a1 − 3iad/8 and bd = d1/2 − 3idd/16 in the F3/F4 design, and ba = a1 − 7iad/16 and bd = d1/4 − 7idd/64 in the F3/F5 design (see the appendix). The estimated additive effect is an unbiased estimate of a1, and the estimated dominance effect is only a fraction of d1. Using the approximate method, the confounding of iad and idd in the estimation of additive and dominance effects becomes more severe for the designs with a larger difference between u and v; moreover, the confounding problems remain unsolved if epistasis is taken into account (see the appendix). In addition to the confounding problem in estimation, the uncontrolled genetic variance will become a part of the genetic residual, causing loss of power in QTL detection. In general, the application of the approximate method to the QTL mapping in the (Fu/Fv, u < v) designs has the problems of confounding, estimating dominance effects, and controlling the genetic variances. To avoid the problems and to increase the power, it is desirable to consider the genome structure in the Fv population by using the proposed method for QTL mapping in the systems of the (Fu/Fv, u < v) designs.
in the F2/F4 design, ba = a1 − 3iad/8 and bd = d1/2 − 3idd/16 in the F3/F4 design, and ba = a1 − 7iad/16 and bd = d1/4 − 7idd/64 in the F3/F5 design (see the appendix). The estimated additive effect is an unbiased estimate of a1, and the estimated dominance effect is only a fraction of d1. Using the approximate method, the confounding of iad and idd in the estimation of additive and dominance effects becomes more severe for the designs with a larger difference between u and v; moreover, the confounding problems remain unsolved if epistasis is taken into account (see the appendix). In addition to the confounding problem in estimation, the uncontrolled genetic variance will become a part of the genetic residual, causing loss of power in QTL detection. In general, the application of the approximate method to the QTL mapping in the (Fu/Fv, u < v) designs has the problems of confounding, estimating dominance effects, and controlling the genetic variances. To avoid the problems and to increase the power, it is desirable to consider the genome structure in the Fv population by using the proposed method for QTL mapping in the systems of the (Fu/Fv, u < v) designs.
SIMULATION STUDIES
Simulations were performed to achieve three purposes: (1) to verify the derived mapping properties of the proposed and current methods, (2) to compare the performance of the proposed and current methods in different (Fu/Fv, v ≤ v) designs, and (3) to evaluate the relative mapping efficiency of different experimental designs. Two 100-cM chromosomes each with 11 equally spaced markers and one QTL were simulated. The two unlinked epistatic QTL, QA and QB, are assumed to be located at 25 cM on their chromosomes. The additive effects of QA and QB are assumed to be a1 = 2 and a2 = 2, respectively, and there is no dominance effect. Their additive-by-dominance effect is assumed to be iad = 2, and the other three epistatic effects are assumed to be zero. With these parameter settings, the marginal effects of the two QTL contribute 44.44% and 44.44% to the total genetic variance, respectively, and epistasis contributes 11.11% to the total genetic variance in the F2/F2 design. The environmental variance is assumed to be 85.5 (the heritability, h2, is 0.05 in the F2/F2 design). Also, according to Equation 11, when ignoring epistasis in the estimation of a1 and a2, the estimate of a1 will be confounded by iad, and the estimate of a2 will not be confounded. The QTL, QA, will be referred to as the confounded QTL, and QB will be referred to as the unconfounded QTL. The sample size is 200, and the number of replicates is 100. The simulations include three parts. The first part is for the (Fu/Fv, u = v) designs. Six such designs, F2/F2, F3/F3, F4/F4, F5/F5, F6/F6, and F10/F10, are simulated. The second part is for the F2/F3 designs, and four different numbers of phenotyping progeny, k = 1, k = 3, k = 5, and k = 10, are assumed. The third part considers designs with other genotyping and phenotyping populations, including the F2/F4, F3/F4, F3/F5, and F4/F5 designs. The number of progeny for phenotyping is assumed to be k = 5. In each part, a stepwise selection procedure (Kao et al. 1999; Zeng et al. 1999) is adopted to detect QTL. Both the proposed and approximate interval mapping (IM)-based (one-QTL) and MIM-based (multiple-QTL) methods are used in the analysis. The critical value for claiming significance was chosen as 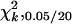 , where k is the number of parameters in testing (see discussion). The simulation results are shown in Tables 1–3.
, where k is the number of parameters in testing (see discussion). The simulation results are shown in Tables 1–3.
TABLE 1.
Simulation results of using different QTL mapping methods under different (Fu/Fv, u = v) designs
| QA
|
QB
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Method | Design | Posi = 25 | a1 = 2 | LRT | Power (%) | Posi = 25 | a2 = 2 | LRT | Power (%) | iad = 2 | σ2 = 85.5 | LRT |
| IM | F2 | 33.11 | 2.13 | 7.09 | 30 | 37.81 | 2.20 | 7.67 | 33 | |||
| (24.07) | (1.40) | (4.84) | (26.86) | (1.46) | (5.19) | |||||||
| F3 | 42.35 | 1.43 | 5.66 | 14 | 33.78 | 2.09 | 8.64 | 39 | ||||
| (31.10) | (1.25) | (4.11) | (21.60) | (1.06) | (4.74) | |||||||
| F4 | 38.73 | 1.40 | 5.74 | 17 | 30.34 | 2.06 | 9.27 | 44 | ||||
| (26.19) | (1.11) | (3.7) | (17.84) | (0.88) | (5.18) | |||||||
| F5 | 42.35 | 1.10 | 5.26 | 18 | 32.37 | 2.07 | 9.96 | 50 | ||||
| (30.26) | (1.24) | (3.84) | (22.16) | (0.97) | (5.39) | |||||||
| F6 | 43.04 | 1.08 | 4.95 | 11 | 30.63 | 2.23 | 10.25 | 53 | ||||
| (28.33) | (1.1) | (3.27) | (16.20) | (0.65) | (5.39) | |||||||
| F10 | 43.52 | 1.02 | 5.25 | 17 | 32.76 | 2.14 | 10.16 | 54 | ||||
| (27.0) | (1.25) | (3.52) | (19.43) | (0.75) | (5.41) | |||||||
| MIM (without epistasis) | F2 | 32.20 | 2.09 | 7.31 | 31 | 39.81 | 2.20 | 7.94 | 33 | 81.04 | 14.98 | |
| (24.05) | (1.45) | (5.08) | (26.86) | (1.43) | (5.52) | (10.39) | (7.44) | |||||
| F3 | 42.96 | 1.40 | 5.92 | 15 | 33.78 | 2.12 | 9.02 | 45 | 3.04 | 14.56 | ||
| (30.45) | (1.30) | (4.15) | (21.60) | (1.06) | (4.90) | (9.72) | (6.42) | |||||
| F4 | 37.40 | 1.47 | 5.98 | 20 | 30.34 | 2.06 | 9.57 | 46 | 82.90 | 15.25 | ||
| (26.71) | (1.00) | (3.69) | (17.84) | (0.89) | (5.49) | (4.86) | (6.17) | |||||
| F5 | 39.84 | 1.16 | 5.28 | 18 | 32.37 | 2.06 | 10.09 | 50 | 83.88 | 15.24 | ||
| (28.29) | (1.15) | (3.79) | (22.16) | (0.98) | (5.51) | (10.21) | (6.05) | |||||
| F6 | 41.60 | 1.09 | 5.08 | 14 | 30.63 | 2.23 | 10.47 | 56 | 84.88 | 15.33 | ||
| (28.02) | (1.14) | (3.48) | (16.20) | (0.65) | (5.57) | (8.67) | (6.60) | |||||
| F10 | 44.04 | 1.02 | 5.39 | 18 | 32.76 | 2.14 | 10.42 | 58 | 83.26 | 15.54 | ||
| (28.30) | (1.24) | (3.61) | (19.42) | (0.75) | (5.46) | (9.53) | (5.81) | |||||
| MIM (with epistasis) | F2 | 33.88 | 2.06 | 9.17 | 35 | 37.81 | 2.20 | 10.32 | 41 | 1.33 | 80.23 | 16.24 |
| (25.32) | (1.33) | (5.10) | (26.86) | (1.44) | (5.82) | (2.63) | (10.44) | (7.22) | ||||
| F3 | 39.62 | 1.74 | 8.30 | 20 | 33.78 | 2.10 | 11.90 | 52 | 1.50 | 81.75 | 16.94 | |
| (27.43) | (1.26) | (4.39) | (21.60) | (1.05) | (5.72) | (3.01) | (9.74) | (6.73) | ||||
| F4 | 39.06 | 1.63 | 7.87 | 20 | 30.34 | 2.08 | 11.89 | 56 | 0.78 | 81.83 | 17.14 | |
| (26.99) | (1.74) | (4.06) | (17.84) | (0.88) | (5.53) | (3.42) | (7.67) | (6.02) | ||||
| F5 | 42.58 | 1.51 | 7.61 | 18 | 32.37 | 2.04 | 12.39 | 52 | 1.15 | 82.77 | 16.98 | |
| (29.25) | (2.11) | (3.79) | (22.16) | (0.97) | (6.17) | (4.39) | (10.24) | (6.14) | ||||
| F6 | 38.61 | 1.52 | 6.61 | 15 | 30.63 | 2.23 | 12.61 | 60 | 0.97 | 83.76 | 16.87 | |
| (27.58) | (2.22) | (3.77) | (16.20) | (0.65) | (6.10) | (4.67) | (8.63) | (6.80) | ||||
| F10 | 44.24 | 1.04 | 6.57 | 18 | 32.76 | 2.13 | 11.87 | 59 | 0.10 | 82.25 | 16.73 | |
| (28.43) | (2.42) | (3.75) | (19.42) | (0.75) | (6.18) | (4.76) | (9.60) | (6.13) | ||||
A total of 100 replicates, each with sample size 200, were analyzed with two unlinked epistatic QTL, QA and QB, controlling the trait variation. The heritability is 0.05 in the F2 population. The critical value for the methods of IM and MIM without epistasis is 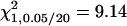 , and the value for MIM with epistasis is
, and the value for MIM with epistasis is 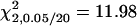 . Posi, position.
. Posi, position.
TABLE 3.
Simulation results of using different QTL mapping methods under different (Fu/Fv, u < v) designs with five phenotyping progeny
| QA
|
QB
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Design | Method | Posi = 25 | a1 = 2 | LRT | Power (%) | Posi = 25 | a2 = 2 | LRT | Power (%) | iad = 2 | σ2 = 85.5 | LRT | |
| F2/F4k = 5 | IM | e | 30.54 | 1.37 | 9.63 | 47 | 25.23 | 2.02 | 20.38 | 92 | |||
| (20.14) | (0.44) | (5.93) | (9.33) | (0.43) | (8.20) | ||||||||
| a | 30.37 | 1.37 | 9.62 | 47 | 25.58 | 2.00 | 20.24 | 92 | |||||
| (20.24) | (0.44) | (5.91) | (9.51) | (0.43) | (8.12) | ||||||||
| MIMa | e | 29.94 | 1.30 | 10.37 | 50 | 25.23 | 2.01 | 21.39 | 93 | 84.15 | 30.75 | ||
| (20.06) | (0.55) | (6.48) | (9.33) | (0.44) | (8.93) | (10.35) | (10.47) | ||||||
| a | 29.72 | 1.31 | 10.38 | 48 | 25.58 | 2.00 | 21.30 | 93 | 86.58 | 30.63 | |||
| (19.44) | (0.53) | (6.46) | (9.51) | (0.44) | (8.89) | (10.21) | (10.39) | ||||||
| MIMb | e | 29.02 | 1.80 | 11.77 | 52 | 25.23 | 2.01 | 22.12 | 94 | 1.36 | 82.25 | 32.15 | |
| (19.96) | (1.71) | (6.43) | (9.33) | (0.44) | (9.47) | (4.36) | (10.45) | (10.59) | |||||
| a | 29.42 | 1.30 | 11.73 | 50 | 25.58 | 2.00 | 22.82 | 93 | 0.34 | 85.87 | 31.98 | ||
| (19.84) | (0.54) | (6.42) | (9.51) | (0.44) | (9.12) | (1.11) | (10.16) | (10.47) | |||||
| F3/F4k = 5 | IM | e | 27.05 | 1.33 | 13.12 | 68 | 24.79 | 2.04 | 29.35 | 99 | |||
| (13.44) | (0.41) | (7.63) | (4.63) | (0.36) | (9.91) | ||||||||
| a | 27.02 | 1.33 | 13.12 | 68 | 24.75 | 2.03 | 29.25 | 99 | |||||
| (13.48) | (0.41) | (7.64) | (4.68) | (0.36) | (9.89) | ||||||||
| MIMa | e | 26.64 | 1.28 | 13.93 | 75 | 24.79 | 2.01 | 30.49 | 99 | 83.67 | 43.29 | ||
| (14.33) | (0.39) | (7.24) | (4.63) | (0.35) | (10.15) | (8.65) | (11.72) | ||||||
| a | 26.72 | 1.28 | 13.98 | 75 | 24.75 | 2.01 | 30.42 | 99 | 74.49 | 43.23 | |||
| (14.29) | (0.39) | (7.24) | (4.68) | (0.36) | (10.16) | (8.63) | (11.73) | ||||||
| MIMb | e | 25.60 | 1.92 | 16.53 | 84 | 24.79 | 2.01 | 32.44 | 99 | 1.81 | 81.78 | 45.89 | |
| (14.04) | (0.87) | (7.10) | (4.63) | (0.36) | (10.85) | (2.41) | (8.53) | (11.67) | |||||
| a | 25.78 | 1.48 | 16.63 | 84 | 24.75 | 2.01 | 33.40 | 99 | 0.94 | 82.98 | 45.88 | ||
| (14.04) | (0.43) | (7.14) | (4.68) | (0.36) | (10.69) | (1.22) | (8.46) | (11.72) | |||||
| F3/F5k = 5 | IM | e | 28.45 | 1.14 | 10.69 | 62 | 24.91 | 1.99 | 28.59 | 99 | |||
| (16.47) | (0.53) | (6.33) | (4.98) | (0.37) | (10.12) | ||||||||
| a | 28.47 | 1.14 | 10.69 | 61 | 24.95 | 1.99 | 28.53 | 99 | |||||
| (16.45) | (0.53) | (6.35) | (5.04) | (0.37) | (10.10) | ||||||||
| MIMa | e | 27.74 | 1.11 | 11.64 | 67 | 24.91 | 1.98 | 29.79 | 100 | 83.19 | 40.23 | ||
| (17.13) | (0.50) | (6.54) | (4.98) | (0.37) | (10.90) | (9.60) | (12.46) | ||||||
| a | 27.76 | 1.11 | 11.66 | 68 | 24.95 | 1.98 | 29.67 | 100 | 84.31 | 40.19 | |||
| (17.10) | (0.50) | (6.56) | (5.04) | (0.37) | (10.90) | (9.53) | (12.47) | ||||||
| MIMb | e | 28.88 | 1.49 | 13.17 | 71 | 24.99 | 1.98 | 30.72 | 100 | 0.97 | 81.75 | 41.76 | |
| (18.90) | (1.93) | (6.74) | (4.98) | (0.37) | (11.20) | (4.08) | (9.54) | (12.56) | |||||
| a | 28.38 | 1.17 | 13.36 | 71 | 24.95 | 1.98 | 31.84 | 100 | 0.34 | 83.35 | 41.89 | ||
| (18.38) | (0.63) | (6.67) | (5.04) | (0.37) | (10.98) | (1.23) | (9.42) | (12.44) | |||||
| F4/F5k = 5 | IM | e | 27.27 | 1.20 | 11.76 | 59 | 26.82 | 1.96 | 30.73 | 99 | |||
| (12.16) | (0.33) | (6.09) | (4.27) | (0.38) | (11.22) | ||||||||
| a | 27.28 | 1.20 | 11.76 | 59 | 26.83 | 1.96 | 30.67 | 99 | |||||
| (12.14) | (0.33) | (6.08) | (4.27) | (0.38) | (11.19) | ||||||||
| MIMa | e | 26.96 | 1.17 | 13.28 | 70 | 26.82 | 1.95 | 32.53 | 99 | 85.6 | 44.01 | ||
| (11.08) | (0.36) | (6.99) | (4.27) | (0.39) | (12.28) | (10.73) | (13.64) | ||||||
| a | 27.18 | 1.17 | 13.30 | 69 | 26.83 | 1.96 | 32.53 | 99 | 85.95 | 43.97 | |||
| (11.13) | (0.36) | (7.00) | (4.27) | (0.39) | (12.29) | (10.72) | (13.64) | ||||||
| MIMb | e | 26.86 | 1.86 | 15.18 | 75 | 26.82 | 1.95 | 33.37 | 99 | 1.65 | 84.07 | 45.90 | |
| (10.93) | (1.12) | (7.23) | (4.27) | (0.39) | (12.85) | (2.58) | (10.49) | (13.66) | |||||
| a | 26.94 | 1.47 | 15.28 | 75 | 26.83 | 1.95 | 34.59 | 99 | 0.87 | 84.68 | 45.95 | ||
| (10.92) | (0.56) | (7.30) | (4.27) | (0.39) | (12.41) | (1.35) | (10.51) | (13.68) | |||||
A total of 100 replicates, each with sample size 200, were analyzed with two unlinked epistatic QTL, QA and QB, controlling the trait variation. The heritability is 0.05 in the F2 population. The critical value for the methods of IM and MIM without epistasis is 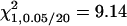 , and the value for MIM with epistasis is
, and the value for MIM with epistasis is 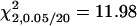 . Posi, position. k, the number of phenotyping progeny. e, proposed (exact) method. a, approximate method.
. Posi, position. k, the number of phenotyping progeny. e, proposed (exact) method. a, approximate method.
Without epistasis.
With epistasis.
Table 1 shows the results of the first part of the simulation. When the IM-based method is used to detect QTL, one can consider one (additive or dominance) effect or two effects in the search. The model considering dominance effect only did not detect any QTL, and the performance of the two-effect model is inferior as compared to that of the additive-effect model. Table 1 presents the QTL mapping result of the model considering the additive effect only. The powers for detecting the confounded QA are 30, 14, 17, 18, 11, and 17% in the F2/F2, F3/F3, F4/F4, F5/F5, F6/F6, and F10/F10 designs, respectively, and the powers for detecting the unconfounded QB are 33, 39, 44, 50, 53, and 54% in the six different designs, respectively (critical value 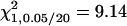 ). As compared to the QTL detection in the F2/F2 design, the confounded QA was detected with decreasing power, and the unconfounded QB was detected with increasing power by using the later RI populations. The reasons are that the estimation of the additive effect of QA is confounded by iad due to ignoring epistasis and such confounding becomes more severe in the F3, F4, F5, F6, and F10 populations and that the estimation of the additive effect of QB is not confounded so that the power can be increased due to the accumulation of homozygotes in the later RI populations (Equation 11). The estimated additive effects of the confounded QA by the IM method are 2.13 (SD 1.40), 1.43 (SD 01.25), 1.40 (SD 1.11), 1.10 (SD 1.24), 1.08 (SD 1.17), and 1.02 (SD 1.25), respectively, in the six designs (the predicted confounded estimates by the IM method are 2.0, 1.5, 1.25, 1.125, 1.0625, and 1.00394, respectively, according to Equation 11). Except for the F2/F2 design, the estimates of a1 by the IM method are poorly estimated and very far away from the true value a1 = 2 due to the confounding of iad = 2. The confounding problem is more severe for the confounded QA in the designs using the later RI populations if epistasis is ignored. The estimated effects of the unconfounded QB are 2.20 (SD 1.46), 2.09 (SD 1.06), 2.06 (SD 0.88), 2.07 (SD 0.97), 2.23 (SD 0.65), and 2.14 (SD 0.75), respectively. These estimates by the IM method are very close to the true value a2 = 2 as expected, because they are not confounded. The advantages of the MIM method include that the detected QTL can be fitted into the model for further QTL search and the epistasis between QTL can be considered. When the MIM method considers only one QTL in the model (m = 1), the mapping results are identical to those of the IM method. If the detected QA (QB) is fitted into the MIM model (m = 2 without epistasis) in the search for other QTL, both the powers of detecting QA and QB increase 1–6%. For example, the powers increase from 6% (1%) to 45% (15%) by using the MIM method without epistasis in the F3/F3 design. If epistasis is taken into account in the search, four different types of epistatic effects can be considered in the model. Among the four possible epistatic effects, only the model taking iad into account improves QTL detection, and the models fitting other epistasis become inferior as a higher critical value is used for claiming significance (critical value
). As compared to the QTL detection in the F2/F2 design, the confounded QA was detected with decreasing power, and the unconfounded QB was detected with increasing power by using the later RI populations. The reasons are that the estimation of the additive effect of QA is confounded by iad due to ignoring epistasis and such confounding becomes more severe in the F3, F4, F5, F6, and F10 populations and that the estimation of the additive effect of QB is not confounded so that the power can be increased due to the accumulation of homozygotes in the later RI populations (Equation 11). The estimated additive effects of the confounded QA by the IM method are 2.13 (SD 1.40), 1.43 (SD 01.25), 1.40 (SD 1.11), 1.10 (SD 1.24), 1.08 (SD 1.17), and 1.02 (SD 1.25), respectively, in the six designs (the predicted confounded estimates by the IM method are 2.0, 1.5, 1.25, 1.125, 1.0625, and 1.00394, respectively, according to Equation 11). Except for the F2/F2 design, the estimates of a1 by the IM method are poorly estimated and very far away from the true value a1 = 2 due to the confounding of iad = 2. The confounding problem is more severe for the confounded QA in the designs using the later RI populations if epistasis is ignored. The estimated effects of the unconfounded QB are 2.20 (SD 1.46), 2.09 (SD 1.06), 2.06 (SD 0.88), 2.07 (SD 0.97), 2.23 (SD 0.65), and 2.14 (SD 0.75), respectively. These estimates by the IM method are very close to the true value a2 = 2 as expected, because they are not confounded. The advantages of the MIM method include that the detected QTL can be fitted into the model for further QTL search and the epistasis between QTL can be considered. When the MIM method considers only one QTL in the model (m = 1), the mapping results are identical to those of the IM method. If the detected QA (QB) is fitted into the MIM model (m = 2 without epistasis) in the search for other QTL, both the powers of detecting QA and QB increase 1–6%. For example, the powers increase from 6% (1%) to 45% (15%) by using the MIM method without epistasis in the F3/F3 design. If epistasis is taken into account in the search, four different types of epistatic effects can be considered in the model. Among the four possible epistatic effects, only the model taking iad into account improves QTL detection, and the models fitting other epistasis become inferior as a higher critical value is used for claiming significance (critical value 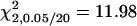 ). By considering epistasis, the values of the average partial LRT statistic increase by 1–2 and the powers of detecting QA and QB also increase as compared to the MIM method without epistasis. For example, the powers of detecting QA and QB increase 4% (5%) and 7% (7%) to 35% (20%) and 41% (52%) in the F2/F2 (F3/F3) design after taking epistasis into account. The increase in powers of detecting QA for the other designs is less notable. Also, by considering epistasis, the confounding problem in the estimation of a1 seems to be relieved by considering iad in the model. The means of the estimated a1 are 2.06 (SD 1.33), 1.74 (SD 1.26), 1.63 (SD 1.74), 1.51 (SD 2.11), 1.52 ( SD 2.22) and 1.04 (SD 2.42) in the designs, respectively, and the means of the estimated iad are 1.33 (SD 2.63), 1.50 (SD 3.01), 0.78 (SD 3.42), 1.15 (SD 4.39), 0.97 (SD 4.67), and 0.10 (SD 4.76), respectively. These estimates of a1 and iad in the later RI populations seem to be unsatisfactory, especially for the F10/F10 design, but they can be improved by increasing the sample size or as the heritability becomes higher (not shown) or by using the (Fu/Fv, u < v) designs with multiple phenotyping progeny (Table 3). The poor estimation of iad in the F10/F10 design may be attributed to the lack of heterozygotes. In the estimation of the QTL position, the means of the estimated positions of QA are 33.88 (SD 25.32), 39.62 (SD 27.43), 39.06 (SD 26.99), 42.58 (SD 29.25), 38.64 (SD 27.58), and 44.24 (SD 28.43), respectively, in the six different designs, and the means for QB are 37.81 (SD 26.86), 33.78 (SD 21.60), 30.34 (SD 17.84), 32.37 (SD 22.16), 30.63 (SD 16.20), and 32.76 (SD 19.42), respectively. The estimated QTL positions are found to be biased toward the center of chromosomes. The position of unconfounded QB (confounded QA) seems to be estimated with greater (reduced) accuracy as the later RIL populations are used in the design. The above results shows that the use of the RI population after F2 can improve the estimation of parameters and power of detection of the unconfounded QB, but it is difficult to improve the resolution of the confounded QA as compared to the use of the F2/F2 design.
). By considering epistasis, the values of the average partial LRT statistic increase by 1–2 and the powers of detecting QA and QB also increase as compared to the MIM method without epistasis. For example, the powers of detecting QA and QB increase 4% (5%) and 7% (7%) to 35% (20%) and 41% (52%) in the F2/F2 (F3/F3) design after taking epistasis into account. The increase in powers of detecting QA for the other designs is less notable. Also, by considering epistasis, the confounding problem in the estimation of a1 seems to be relieved by considering iad in the model. The means of the estimated a1 are 2.06 (SD 1.33), 1.74 (SD 1.26), 1.63 (SD 1.74), 1.51 (SD 2.11), 1.52 ( SD 2.22) and 1.04 (SD 2.42) in the designs, respectively, and the means of the estimated iad are 1.33 (SD 2.63), 1.50 (SD 3.01), 0.78 (SD 3.42), 1.15 (SD 4.39), 0.97 (SD 4.67), and 0.10 (SD 4.76), respectively. These estimates of a1 and iad in the later RI populations seem to be unsatisfactory, especially for the F10/F10 design, but they can be improved by increasing the sample size or as the heritability becomes higher (not shown) or by using the (Fu/Fv, u < v) designs with multiple phenotyping progeny (Table 3). The poor estimation of iad in the F10/F10 design may be attributed to the lack of heterozygotes. In the estimation of the QTL position, the means of the estimated positions of QA are 33.88 (SD 25.32), 39.62 (SD 27.43), 39.06 (SD 26.99), 42.58 (SD 29.25), 38.64 (SD 27.58), and 44.24 (SD 28.43), respectively, in the six different designs, and the means for QB are 37.81 (SD 26.86), 33.78 (SD 21.60), 30.34 (SD 17.84), 32.37 (SD 22.16), 30.63 (SD 16.20), and 32.76 (SD 19.42), respectively. The estimated QTL positions are found to be biased toward the center of chromosomes. The position of unconfounded QB (confounded QA) seems to be estimated with greater (reduced) accuracy as the later RIL populations are used in the design. The above results shows that the use of the RI population after F2 can improve the estimation of parameters and power of detection of the unconfounded QB, but it is difficult to improve the resolution of the confounded QA as compared to the use of the F2/F2 design.
Table 2 shows the QTL mapping results using the F2/F3 designs with different numbers of phenotyping progeny. If there is only 1 progeny (k = 1) for trait measurement, the IM and MIM (with or without epistasis) methods have less power to detect QA and QB as compared to the powers in the F2/F2 or F3/F3 designs. For example, the powers of detecting QB (QA) by the proposed MIM method are 32% (12%) in the F2/F3 design with k = 1, and they are 41% (35%) and 52% (20%) in the F2/F2 and F3/F3 designs. By increasing the number of phenotyping progeny, the powers of detecting QA and QB are enhanced. By using 3 (k = 3), 5 (k = 5), and 10 (k = 10) progeny for phenotyping, the powers of detecting the unconfounded QB increase to 83, 93, and 100%, respectively, and the powers of detecting the confounded QA become 46, 73, and 99%, respectively. As compared to the results of the IM method in the F2/F2 and F3/F3 designs, the use of three progeny for phenotyping can greatly enhanced the power of detecting the unconfounded QB from 33% (or 39%) to 79%, but it does not greatly increase the power of detecting the confounded QA [the powers of detecting QA in the F2/F2, F3/F3, and (F2/F3, k = 3) designs are 30, 14, and 37%, respectively]. The use of more progeny for phenotyping also improves the estimation of QTL effects and positions. For example, by using the proposed MIM method with epistasis, the means of the estimated QB (QA) positions are 37.73 cM with SD 27.41 cM (43.06 cM with SD 29.23 cM) and 25.22 cM with SD 8.25 cM (27.54 cM with SD 15.82 cM) for k = 1 and k = 5. The estimated a1, a2, and iad are 1.62 (SD 2.01), 2.03 (SD 1.44), and 1.25 (SD 5.48) for k = 1, and they are 1.96 (SD 0.68), 2.01 (SD 0.45), and 1.91 (SD 2.18) for k = 5. In addition, if epistasis is not taken into account or the approximate method is used, there will always be confounding problems in estimation as shown in Table 2. For example, the means of the estimated a1 and iad by the approximate MIM method are 1.48 (predicted value a1 − iad/4 = 1.5) with SD 0.49 and 0.98 (predicted value iad/2 = 1.0) with SD 1.10 for k = 5. By using 10 progeny for phenotyping, both QA and QB can almost be detected with power 1 and with good precision and accuracy. In general, the estimation becomes improved as more progeny are used for phenotyping. The performance of the MIM method is also better than that of the IM method as expected.
Table 3 shows the QTL mapping results of using the (Fu/Fv, v > u > 2) designs with k = 5. The means of the estimated additive effects by the proposed IM and MIM without epistasis are 1.33 (SD 0.41) and 1.28 (SD 0.39), respectively, in the F3/F4 design, and they are 1.20 (SD 0.33) and 1.17 (SD 0.36), respectively, in the F4/F5 design (the predicted estimated a1's by Equation 11 are 1.25 and 1.125 in the two designs). By taking epistasis into account, the confounding problem can be solved by the proposed method. The means of the estimated a1 by considering epistasis are 1.92 (SD 0.87) and 1.86 (SD 1.12) in the two designs, respectively. The means of the estimated iad's are 1.81 (SD 2.41) and 1.65 (SD 2.58), respectively. On the contrary, the approximate methods always have the confounding problem whether or not epistasis is taken into account. For example, the means of the estimated additive effects by the approximate MIM with epistasis are 1.48 (SD 0.43) and 1.47 (SD 0.56) for the two designs, respectively (the predicted a1 by the approximate method is 1.5). The powers of QTL detection are also increasing and the estimation of QTL positions is also improved by using the MIM approach and by taking epistasis into account. For example, the power increases from 68% (59%) by the IM method to 84% (75%) by the MIM method in the F3/F4 (F4/F5) design, and the mean of the estimated position of the confounded QA is improved from 27.05 with SD 13.44 (27.27 with SD 12.16) to 25.60 with SD 14.04 (26.86 with SD 10.93). In addition, the use of the F2/F4 and F3/F5 designs does not provide a better resolution of the confounded QA when compared to the use of F2/F3 and F3/F4 designs as expected. For example, the powers of detecting QA by the proposed MIM method are 52 and 71%, respectively, in the two designs, and the means of the estimated positions are 29.02 cM (SD 19.96 cM) and 28.88 cM (SD 18.90 cM), respectively. Across all different designs with k = 5, the unconfounded QB can be well detected with high power and great accuracy and precision as compared to the confounded QA.
DISCUSSION
The data required in QTL mapping analysis are usually composed of two parts, phenotypic trait values and marker genotypes. In data collection using the designs of RI populations, the trait values can be obtained from the same genotyped population by using the (Fu/Fv, u = v) designs or from the progeny of the genotyped population using the (Fu/Fv, u < v) designs. The great benefit of using the (Fu/Fv, u < v) designs in QTL mapping is not only through reducing the cost and environmental variance by phenotyping several progeny for each genotyped individual (Lander and Botstein 1989; Knapp and Bridges 1990), but also likely through taking advantage of the changes in population structures between different RI populations. Different RI populations have different homozygosities, genotypic frequencies, and proportions of recombinant genotypes. The increase of homozygosity may help the estimation of additive effects due to the accumulation of homozygotes, but it will hinder the estimation of dominance effects due to the loss of heterozygotes. Also, in modeling, the orthogonal property of the genetic model, which holds in the F2 population, will be lost in the other later RI populations as the genotypic frequencies have changed. Then, the confounding problem in the QTL estimation may occur if epistasis is present and ignored. Such a confounding problem cannot be relieved by enlarging the sample size or increasing heritability or using the approximate methods, and it becomes more severe for the later populations (see Equations 11 and Table 1). Therefore, the use of the later RI populations can greatly benefit the detection of unconfounded QTL with additive effects, but it may deter the detection of confounded QTL and the QTL with large dominance effects as compared to the QTL mapping using the F2 population. By taking epistasis into account, the confounding problem can be alleviated by the proposed method. The approximate method, however, always has the confounding problems. In addition, the (Fu/Fv, u < v) designs also allow for phenotyping more progeny for each genotyped individual to reduce environmental variance so that the resolution of QTL can be further enhanced. The resolution of the unconfounded QTL can be easily improved, but more progeny are needed to improve the resolution of the confounded QTL as compared to the Fu/Fu designs (comparing the results of Table 1 with those of Tables 2 and 3).
In statistical modeling, the relation between the phenotype and the underlying QTL genotype is relatively simple and can be modeled by a 3m normal mixture model for the (Fu/Fv, u = v) designs. However, for the (Fu/Fv, u < v) designs, when the phenotypic means of the k progeny from the genotyped individuals are used in QTL mapping, the relationship between the phenotypic means and the involved QTL genotypes becomes increasingly complicated and should be modeled by a [(k + 1)(k + 2)/2]m normal mixture model as discussed here. Such complication in statistical modeling arises mainly from the segregation of heterozygote into homozygotes and heterozygote and from the numerous possible combinations of different genotypes among the k progeny. Genetically, segregation will vary the homozygosity and linkage disequilibrium in different RI populations. It is possible to utilize different experimental designs of these RI populations to benefit QTL mapping by taking advantage of their specific population structures. To achieve this purpose, for QTL mapping in the (Fu/Fv, u < v) design, the proposed method is designed to take the population structures of phenotyping populations into account by modeling the relationship between the phenotypic means and the underlying QTL in the same populations. Then, the likelihood of the proposed method is a mixture of [(k + 1)(k + 2)/2]m normals with the number of mixture components and mixing proportions adjusted for the phenotyping population. The approximate method, however, ignores the fact of segregation and differences in population structure between different RI populations, and it relates the phenotypic traits of the progeny with the QTL in their ancestral populations. Therefore, the likelihood of the approximate method is always a mixture of 3m normals with constant mixing proportions derived from the genotyped population. Consequently, the approximate method may have the problems of confounding and estimating the dominance effect, and the proposed method can avoid the problems to improve the QTL mapping in (Fu/Fv, u < v) designs as shown in this article. In addition, it is straightforward to modify the proposed method for the (Fu/Fv, u < v) designs with each individual progeny trait (yij's, i = 1, 2, … , n, j = 1, 2, … , k) recorded. The mapping results by the approaches of using traits and trait means are similar, but the approach of using individual traits can be more computationally economical for its relatively simple likelihood (with a mixture of 3m normals).
The proposed method has a much more complicated mixture likelihood, and the mixture likelihood will have different numbers of components with different weights (mixing proportions) for different designs. Therefore, the determination of the critical value for the proposed method is challenging in the (Fu/Fv, u ≤ v) designs. It is well known that the critical value cannot be simply chosen from a χ2-distribution because of violation of the standard conditions of asymptotic theory for mixture models (Self and Liang 1987; Feng and McCulloch 1994) and that the determination of the critical values for claiming QTL detection may depend on the factors, such as heritability, marker density, size of the genomes, number of (linked or unlinked) QTL, and the direction of QTL effects (Jensen 1993; Zeng et al. 1999; Zou et al. 2004). Several methods, such as the method by Piepho (2001), the permutation tests (Churchill and Doerge 1994), residual bootstrapping (Zeng et al. 1999), and the resampling method by Zou et al. (2004), have been proposed to determine the values, but they generally require additional assumptions, such as a dense map with equally spaced markers, or are applicable only to some standard designs, such as a backcross or F2 design (the Fu/Fv, u = v design), or are restricted to the model with a 2- (3-) normal mixture (see Zou et al. 2004 for the discussion). In addition, the concept of the false discovery rate (Benjamini and Hochberg 1995) has been introduced to deal with the problem of statistical significance by the control of type II rather than type I errors in QTL mapping. As the proposed method considers a more complicated mixture of ([(k + 1)(k + 2)/2]m) normals with different numbers of components and mixing proportions varying with m, k, u, and v, and the different population structures may also affect the critical values, the issue of determining the critical values in the (Fu/Fv, u ≤ v) designs will become even more complicated and still needs to be unraveled. Here, Bonferroni argument based on χ2-distribution (Lander and Botstein 1989) is used to choose the critical values before the complicated issue is solved. Further research on the theoretical basis of determining the critical value is of great value to QTL mapping in the (Fu/Fv, u ≤ v) designs.
The RI populations have been very important and popular in the study of QTL for a long time (Haldane and Waddington 1931; Stuber et al. 1992; Beavis et al. 1994; Veldboom et al. 1994; Darvasi and Soller 1995; Austin and Lee 1996; Liu et al. 1996; Belknap 1998; Chapman et al. 2003; Complex Trait Consortium 2004; Broman 2005). As compared to the F2 population, the population structures in the later RI populations have some precious properties, such as larger additive genetic variance, higher homozygosity, and more recombinants. These properties may benefit the QTL resolution and should be well utilized in the study of QTL mapping. With the ability to consider the changes in population structures of different populations, the proposed method can serve as an effective tool to map for QTL in specific designs and evaluate the efficiency of QTL mapping among different experimental designs under the system of RI populations. Other important issues of QTL mapping by using the (Fu/Fv, u ≤ v) designs include the consideration of endosperm traits (Wu et al. 2002; Xu et al. 2003; Kao 2004) and the extension of the methods from the system of RI populations to the system of IRI populations. The IRI populations, which are derived by randomly mating for some generations after F2 and then followed by cycles of selfing, have the advantages of producing more recombinants as compared to the RI populations, and they can benefit the analysis of quantitative traits (Liu et al. 1996; Winkler et al. 2003). It is critical to provide adequate statistical methods for these designs by considering their specific population structures to explore their properties in the QTL mapping study.
Acknowledgments
The author thanks two anonymous reviewers for helpful comments. This study was supported by grants NSC94-2118-M-001-021 from the National Science Council, Taiwan, Republic of China.
APPENDIX
If m QTL without epistasis are considered, the model of the traditional method for the mean trait,  , of the k Fv progeny from each of the n genotyped Fu individuals and the QTL can be written as
, of the k Fv progeny from each of the n genotyped Fu individuals and the QTL can be written as
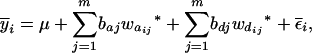 |
(A1) |
where 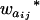 's and
's and 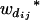 's, j = 1, 2, … , m, are the coded variables for the genotype of
's, j = 1, 2, … , m, are the coded variables for the genotype of 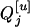 's, j = 1, 2, … , m, and they are coded as (1,
's, j = 1, 2, … , m, and they are coded as (1,  ), (0,
), (0, 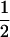 ), and (−1,
), and (−1, 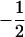 ) for QjQj, Qjqj, and qjqj, respectively. The mean residual error
) for QjQj, Qjqj, and qjqj, respectively. The mean residual error 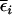 has a mean of zero and variance σ2/k, where σ2 is the residual variance of the trait on the basis of a single individual. As Q[u] may not be coincident with marker and could be QjQj, Qjqj, or qjqj, the likelihood of the model is a mixture of 3m normals. In parameter estimation, the general formulas by Kao and Zeng (1997) derived on the basis of the EM algorithm (Dempster et al. 1977) can be used for obtaining their MLE.
has a mean of zero and variance σ2/k, where σ2 is the residual variance of the trait on the basis of a single individual. As Q[u] may not be coincident with marker and could be QjQj, Qjqj, or qjqj, the likelihood of the model is a mixture of 3m normals. In parameter estimation, the general formulas by Kao and Zeng (1997) derived on the basis of the EM algorithm (Dempster et al. 1977) can be used for obtaining their MLE.
To show the problems of less power and bias in estimation for the approximate method, without loss of generality, again assume that the quantitative trait value yi is affected by two unlinked epistatic QTL, QA and QB. The variances of 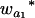 ,
, 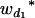 ,
, 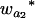 ,
, 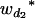 ,
, 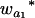 ×
× 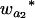 ,
, 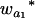 ×
× 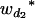 ,
, 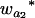 ×
× 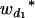 , and
, and 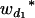 ×
× 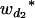 can be found to be 1 − (
can be found to be 1 − (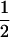 )u−1, (
)u−1, (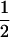 )u−1[1 − (
)u−1[1 − (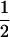 )u−1], 1 − (
)u−1], 1 − (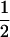 )u−1, (
)u−1, (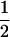 )u−1[1 − (
)u−1[1 − (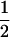 )u−1], [1 − (
)u−1], [1 − (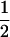 )u−1]2,
)u−1]2, 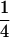 − (
− (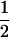 )(u+1),
)(u+1), 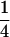 − (
− (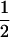 )(u+1), and
)(u+1), and 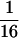 − [
− [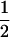 − (
− (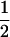 )(u−1)]4, respectively. The covariances between
)(u−1)]4, respectively. The covariances between 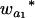 (
(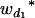 ) and the coded variables in Equation 6 are needed in the derivation. The covariances between
) and the coded variables in Equation 6 are needed in the derivation. The covariances between 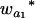 and x1 and between
and x1 and between 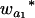 and wad are 1 − (
and wad are 1 − (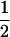 )u−1 and [
)u−1 and [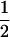 − (
− (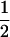 )u][(
)u][( )v−2 − 1], respectively. The covariances between
)v−2 − 1], respectively. The covariances between 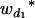 and z1 and between
and z1 and between 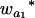 and wdd are (
and wdd are ( )v−1[1 − (
)v−1[1 − (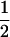 )u−1] and (
)u−1] and (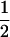 )v[1 − (
)v[1 − (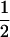 )v−2][(
)v−2][(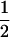 )(u−1) − 1], respectively. Therefore, when yi is regressed on QA with additive and dominance effects, the estimates of the additive and dominance effects by the approximate method can be found to be
)(u−1) − 1], respectively. Therefore, when yi is regressed on QA with additive and dominance effects, the estimates of the additive and dominance effects by the approximate method can be found to be
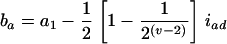 |
(A2) |
and
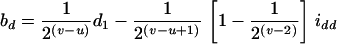 |
(A3) |
in the (Fu/Fv, u < v) design. It shows that the ba is confounded by the the additive effect a1 and the epistatic effect iad, and bd is confounded by d1 and idd. When multiple QTL and their epistasis are considered in the model, the estimates of their effects can be also derived. It can be found that the approximate methods also have the confounding problems.
References
- Austin, D. F., and M. Lee, 1996. Comparative mapping in F2:3 and F6:7 generations of quantitative trait loci for grain yield and yield components in maize. Theor. Appl. Genet. 92: 817–826. [DOI] [PubMed] [Google Scholar]
- Beavis, W. D., O. S. Smith, D. Grant and R. Fincher, 1994. Identification of quantitative trait loci using a small sample of topcrossed and F4 progeny from maize. Crop Sci. 34: 882–896. [Google Scholar]
- Belknap, J. K., 1998. Effect of within-strain sample size on QTL detection and mapping using recombinant inbred mouse strains. Behav. Genet. 28: 29–37. [DOI] [PubMed] [Google Scholar]
- Benjamini, Y., and Y. Hochberg, 1995. Controlling false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57: 289–300. [Google Scholar]
- Broman, K. W., 2005. The genomes of recombinant inbred lines. Genetics 169: 1133–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlborg, O., and C. S. Haley, 2004. Epistasis: too often neglected in complex trait studies??. Nat. Rev. Genet. 5: 618–625. [DOI] [PubMed] [Google Scholar]
- Chapman, A., V. R. Pantalone, A. Ustun, E. L. Allen, D. Landau-Ellis et al., 2003. Quantitative trait loci for agronomic and seed quality traits in an F2 and F4:6 soybean population. Euphytica 129: 387–393. [Google Scholar]
- Churchill, G. A., and R. W. Doerge, 1994. Empirical threshold values for quantitative trait mapping. Genetics 138: 967–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Complex Trait Consortium, 2004. The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat. Genet. 36: 1133–1137. [DOI] [PubMed] [Google Scholar]
- Comstock, R. E., and H. F. Robinson, 1952. Estimation of average dominance of genes, pp. 495–516 in Heterosis, edited by J. W. Gowen. Iowa State College Press, Ames, IA.
- Cowen, N. M., 1988. The use of replicated progenies in marker-based mapping of QTLs. Theor. Appl. Genet. 75: 857–862. [Google Scholar]
- Darvasi, A., and M. Soller, 1995. Advanced intercross lines, an experimental population for genetic fine mapping. Genetics 141: 1199–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster, A. P., N. M. Laird and D. B. Rubin, 1977. Maximum likelihood from incomplete data via the EM algorithm. J. R. Stat. Soc. 39: 1–38. [Google Scholar]
- Doerge, R. W., Z.-B. Zeng and B. S. Weir, 1997. Statistical issues in the search for genes affecting quantitative traits in experimental populations. Stat. Sci. 12: 195–219. [Google Scholar]
- Edwards, M. D., T. Helentjaris, S. Wright and C. W. Stuber, 1992. Molecular-marker-facilitated investigations of quantitative-trait loci in maize. IV. Analysis based on genome saturation with isozyme and restriction fragment length polymorphism markers. Theor. Appl. Genet. 83: 765–774. [DOI] [PubMed] [Google Scholar]
- Feng, Z. D., and C. E. McCulloch, 1994. On the likelihood ratio test statistic for the number of components in a normal mixture with unequal variances. Biometrics 50: 1158–1162. [PubMed] [Google Scholar]
- Fisch, R. D., M. Ragot and G. Gay, 1996. A generalization of the mixture model in the mapping of quantitative trait loci for progeny from a biparental cross of inbred lines. Genetics 143: 571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldane, J. B. S., and C. H. Waddington, 1931. Inbreeding and linkage. Genetics 16: 357–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen, R. C., 1993. Interval mapping of multiple quantitative trait loci. Genetics 135: 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao, C.-H., 2004. Multiple interval mapping for quantitative trait loci controlling endosperm traits. Genetics 167: 1987–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao, C.-H., and Z.-B. Zeng, 1997. General formulas for obtaining the MLEs and the asymptotic variance-covariance matrix in mapping quantitative trait loci when using the EM algorithm. Biometrics 53: 359–371. [PubMed] [Google Scholar]
- Kao, C.-H., and Z.-B. Zeng, 2002. Modeling epistasis of quantitative trait loci using Cockerham's model. Genetics 160: 1243–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao, C.-H., Z.-B. Zeng and R. D. Teasdale, 1999. Multiple interval mapping for quantitative trait loci. Genetics 152: 1203–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp, S. J., and W. C. Bridges, 1990. Using molecular markers to estimate quantitative trait locus parameters: power and genetic variances for unreplicated and replicated progeny. Genetics 126: 769–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander, E. S., and D. Botstein, 1989. Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121: 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, S. C., S. P. Kowalski, T. H. Lan, K. A. Feldmann and A. H. Paterson, 1996. Genome-wide high-resolution mapping by recurrent intermating using Arabidopsis thaliana as a model. Genetics 142: 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M., and B. Walsh, 1998. Genetics and Analysis of Quantitative Traits. Sinauer Associates, Sunderland, MA.
- Mihaljevic, R., H. F. Utz and A. E. Melchinger, 2004. Congruency of quantitative trait loci detected for agronomic traits in testcrosses of five populations of European maize. Crop Sci. 44: 114–124. [Google Scholar]
- Mihaljevic, R., C. C. Schon, H. F. Utz and A. E. Melchinger, 2005. Correlations and QTL correspondence between line per se and testcross performance for agronomic traits in four populations of European maize. Crop Sci. 45: 114–122. [Google Scholar]
- Nakamichi, R., Y. Ukai and H. Kishino, 2001. Detection of closely linked multiple quantitative trait loci using a genetic algorithm. Genetics 158: 463–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piepho, H. P., 2001. A quick method for computing approximate threshold for quantitative trait loci detection. Genetics 157: 425–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala, R. G., F. H. Andrade, E. L. Camadro and J. C. Cerono, 2006. Quantitative trait loci for grain moisture at harvest and field grain drying rate in maize (Zea mays, L.). Theor. Appl. Genet. 112: 462–471. [DOI] [PubMed] [Google Scholar]
- Satagopan, J. M., B. S. Yandell, M. A. Newton and T. C. Osborn, 1996. A Bayesian approach to detect quantitative trait loci using Markov chain Monte Carlo. Genetics 144: 805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Self, S. G., and K. Y. Liang, 1987. Asymptotic properties of maximum likelihood estimators and likelihood ratio tests under non-standard conditions. J. Am. Stat. Assoc. 82: 605–610. [Google Scholar]
- Sen, S., and G. A. Churchill, 2001. A statistical framework for quantitative trait mapping. Genetics 159: 371–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuber, C. W., S. E. Linncoln, D. W. Wolff, T. Helentjaris and E. S. Lander, 1992. Identification of genetic factors contributing to heterosis in a hybrid from two elite maize inbred lines using molecular marks. Genetics 132: 823–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldboom, L. R., M. Lee and W. L. Woodman, 1994. Molecular marker-facilitated studies in an elite maize population: I. Linkage analysis and determination of QTL for morphological traits. Theor. Appl. Genet. 88: 7–16. [DOI] [PubMed] [Google Scholar]
- Weir, B. S., 1996. Genetic Data Analysis II. Sinauer Associates, Sunderland, MA.
- Winkler, C. R., N. M. Jensen, M. Cooper, D. W. Podlich and O. S. Smith, 2003. On the determination of recombination rates in intermated recombinant inbred populations. Genetics 164: 741–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, R.-L., C.-X. Ma, M. Gallo-Meagher, R. C. Littell and G. Casella, 2002. Statistical methods for dissecting triploid endosperm traits using molecular markers: an autogamous model. Genetics 162: 875–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, C., X. He and S. Xu, 2003. Mapping quantitative trait loci underlying triploid endosperm traits. Heredity 90: 228–235. [DOI] [PubMed] [Google Scholar]
- Yi, N., S. Xu and D. B. Allison, 2003. Bayesian model choice and search strategies for mapping interacting quantitative trait loci. Genetics 165: 867–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Z.-B., 1994. Precision mapping of quantitative trait loci. Genetics 136: 1457–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Z.-B., C.-H. Kao and C. Basten, 1999. Estimating the genetic architecture of quantitative traits. Genet. Res. 74: 279–289. [DOI] [PubMed] [Google Scholar]
- Zhang, Y. M., and S. Xu, 2004. Mapping quantitative trait loci in F2 incorporating phenotypes of F3 progeny. Genetics 166: 1981–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou, F., J. P. Fine, J. Hu and D. Y. Lin, 2004. An efficient resampling method for assessing genome-wide statistical significance in mapping quantitative trait loci. Genetics 168: 2307–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]