

Abstract
We investigated DNA sequence diversity for loci on chromosomes 1 and 2 in six natural populations of Arabidopsis lyrata and tested for the role of natural selection in structuring genomewide patterns of variability, specifically examining the effects of recombination rate on levels of silent polymorphism. In contrast with theoretical predictions from models of genetic hitchhiking, maximum-likelihood-based analyses of diversity and divergence do not suggest reduction of diversity in the region of suppressed recombination near the centromere of chromosome 1, except in a single population from Russia, in which the pericentromeric region may have undergone a local selective sweep or demographic process that reduced variability. We discuss various possibilities that might explain why nucleotide diversity in most A. lyrata populations is not related to recombination rate, including genic recombination hotspots, and low gene density in the low recombination rate region.
A central issue in empirical population genetics concerns the genomic extent of positive and negative directional selection and the role of these processes in structuring patterns of genetic variation across the genome. Population genetic theory predicts that both the fixation of advantageous mutations (“selective sweeps”) (Maynard-Smith and Haigh 1974; Barton 2000) and the elimination of deleterious mutations (“background selection”) (Charlesworth et al. 1993) can lead to a reduction of linked neutral diversity. The degree of this reduction is expected to depend strongly on the rate of recombination, and only regions with very low recombination are expected to show detectably reduced diversity (Hudson and Kaplan 1995; Charlesworth 1996; Innan and Stephan 2003). A lower mutation rate in regions of low recombination could also reduce variability in these regions, so it is important to assess the effects of recombination rate on both patterns of diversity within species and levels of divergence between species.
There is consistent evidence for a strong correlation between recombination rate and DNA diversity in Drosophila melanogaster (Begun and Aquadro 1992; Andolfatto and Przeworski 2001), while no such effect is observed on divergence, suggesting the importance of natural selection in structuring genomewide patterns of polymorphism. However, in other organisms investigated, the evidence is less clear (Nachman et al. 1998; Nachman 2001; Lercher and Hurst 2002; Cutter and Payseur 2003; Hellmann et al. 2003, 2005). In particular, recombination appears to be correlated with divergence as well as diversity in a number of taxa, making it difficult to uncouple the effects of mutation and natural selection.
In plants, the influence of recombination rate on DNA sequence diversity has been investigated in detail only in maize (Tenaillon et al. 2002), tomato (Baudry et al. 2001; Roselius et al. 2005), and Arabidopsis thaliana (Nordborg et al. 2005). In maize, no correlation was found with recombination rates estimated from recombination nodule density—only with estimates based on linkage disequilibrium (LD)—most likely reflecting effects of demographic history causing both increased linkage disequilibrium and reduced neutral genetic diversity (Tenaillon et al. 2002). However, using an extensive polymorphism data set from maize and teosinte, Wright et al. (2005) showed that maize loci with a signature of positive selection tend to lie significantly closer to low-recombining centromeric regions than random loci do. The lack of any similar diversity reduction at these loci in the ancestral teosinte populations suggests that this pattern is a signature of selective sweeps during maize domestication, rather than differences in mutation rate.
In tomato, a consistent positive correlation was observed between recombination rate (estimated in Lycopersicon esculentum) and diversity in five species, but the relationship is weak and nonsignificant for several tests, suggesting that the action of linked selection may not have major effects on sequence diversity across the genome in these taxa (Baudry et al. 2001). Furthermore, no correlation with recombination was observed when silent polymorphism was scaled to silent divergence, suggesting that hitchhiking does not explain a significant amount of the variation in diversity in the tomato genome (Roselius et al. 2005).
In A. thaliana, polymorphism is not significantly correlated with recombination rate (Nordborg et al. 2005; Schmid et al. 2005), but a significant negative correlation was found between gene density and nucleotide polymorphism (Nordborg et al. 2005), consistent with stronger effects of selection in regions of high gene density. This is unlikely to be the result of mutagenic recombination; however, without divergence data from another species, neither heterogeneous mutation rates nor selective constraint on noncoding regions of the genome can be ruled out.
One possible explanation for the lack of a strong association between recombination and diversity is the effect of population subdivison and history on patterns of nucleotide variability. In structured populations, local adaptation may inflate between-population nucleotide variability (Charlesworth et al. 1997). Hitchhiking, particularly background selection, will predominantly affect within-population diversity, rather than species-wide diversity. It can thus leave a signature of elevated estimates of between-population differentiation in regions of reduced recombination, without reducing diversity in species-wide samples. Furthermore, population admixture and population size changes may increase the variance in diversity across loci and may complicate the expected effects of genetic hitchhiking. Tests of the role of recombination on patterns of diversity should thus include polymorphism data from both within- and between-population samples from across a species' range (Das et al. 2004; Takahashi et al. 2004). Such data are rarely available.
To investigate the relationship between sequence diversity and recombination, we studied nucleotide diversity in 26 loci on two chromosomes in samples from six natural populations of the outcrossing species Arabidopsis lyrata. The two linkage groups correspond to chromosome 1 of A. thaliana, originating from a fusion event of the two ancestral chromosomes (Hansson et al. 2006; Lysak et al. 2006). A. lyrata is excellent for investigating the role of recombination and selection because (1) the genomic resources from the closely related A. thaliana, combined with comparative mapping between the species, have provided information on rates of recombination in both species; (2) divergence estimates are readily obtained from loci across the genome; and (3) A. lyrata populations exhibit strong population subdivision, yet extensive polymorphism is found within populations, contrasting with the selfing congener A. thaliana (Savolainen et al. 2000; Wright et al. 2003b; Ramos-Onsins et al. 2004; Balana-Alcaide et al. 2006).
MATERIALS AND METHODS
Population samples:
A total of 71 diploid A. lyrata individuals from independent field-collected maternal families were used for this study. The samples originated from six natural populations, each collected at a single locality. The plants from which sequences were obtained include 15 individuals from Karhumaki, Russia (from O. Savolainen); 9 individuals from Stubbsand, Sweden (O. Savolainen); 12 individuals from Plech, Germany (T. Mitchell-Olds); 12 individuals from Esja Mountain, Iceland (E. Thorhallsdottir); 11 individuals from Indiana Dunes, Indiana (B. Mable), and 12 individuals from Rondeau Provincial Park, Ontario, Canada (B. Mable). DNA from a single plant from each maternal family was extracted using the QIAGEN (Chatsworth, CA) plant DNA extraction kit and used in PCR.
PCR and sequencing:
Table 1 shows the genomic location and size for the 26 loci surveyed in this study. With the exception of two of our five pericentromeric loci, we employed a direct sequencing strategy, using single large exons for PCR amplification and sequencing to minimize the chance of unreadable sequences caused by insertion/deletion (indel) variants. Exons from across chromosome 1 of A. thaliana were selected for direct sequencing in A. lyrata, using the annotation from the A. thaliana genome-sequencing project, and were chosen from genomic regions of contrasting recombination rates along chromosome I of A. thaliana (see Figure 1 and below). The exons selected for sequencing were all >800 bp, and, to ensure correct annotation, all encode genes with at least one full-length cDNA sequence. Each exon was submitted to a BLAST search (Altschul et al. 1997) of the genomic survey sequence database (http://www.ncbi.nlm.nih.gov/BLAST/) to check for the presence of orthologous regions in the shotgun genome sequence of Brassica oleracea. These orthologous regions were aligned to the A. thaliana genomic sequence to identify conserved regions for primer design. PCR primers were designed with the aid of PrimerQuest (Integrated DNA Technologies, http://biotools.idtdna.com/primerquest/) to amplify 650- to 750-bp fragments for sequencing using the same forward and reverse primers. Primers were also submitted to a BLAST search against the A. thaliana genome to ensure amplification of a single-copy region. To increase our sampling in regions expected to have reduced recombination, we designed primers for two additional pericentromeric loci, which included both coding and intron sites. The sequencing procedure for these two intron-containing loci was the same as for the other loci, except when two or more heterozygous indel variants were found. In such cases, the PCR products were cloned and sequences of at least two clones for both alleles were determined. A total of five to seven outbred individuals from the same populations were used for sequencing these latter two loci.
TABLE 1.
Loci surveyed in this study
| Locus name | Chromosome, A. lyrata | Physical position, A. thaliana chromosome 1a | Map position, A. lyratab | rthalc (cM/Mb) | rlyrc (cM/Mb) |
|---|---|---|---|---|---|
| AT1G01040 | 1 | 23,146 | 0 | 1.79 | 3.06 |
| AT1G03560 | 1 | 890,164 | 3.863 | 2.56 | 3.41 |
| AT1G04650 | 1 | 1,294,892 | 5.51 | 2.68 | 3.55 |
| AT1G06520 | 1 | 1,993,977 | 6.54 | 3.4 | 3.79 |
| AT1G10900 | 1 | 3,632,274 | 23.73 | 4.36 | 4.24 |
| AT1G10980 | 1 | 3,677,531 | 13.64 | 4.38 | 4.25 |
| AT1G11050 | 1 | 3,681,888 | 4.39 | 4.25 | |
| AT1G15240 | 1 | 5,243,445 | 17.8 | 4.97 | 4.55 |
| AT1G23200 | 1 | 8,227,167 | 5.23 | 4.77 | |
| AT1G31930 | 1 | 11,465,029 | 4.22 | 4.48 | |
| AT1G36310 | 1 | 13,670,700 | 65.111 | 0.05 | 0 |
| AT1G36370 | 1 | 13,697,254 | 65.111 | 0.05 | 0 |
| AT1G36730 | 1 | 13,899,422 | 65.111 | 0.05 | 0 |
| AT1G42470 | 1 | 16,138,292 | 65.074 | 0.5 | 0 |
| AT1G42990 | 1 | 16,688,272 | 66.25 | 0.5 | 0 |
| AT1G59720 | 2 | 21,943,533 | 13 | 4.53 | 4.5 |
| AT1G62310 | 2 | 23,039,704 | 7.225 | 4.77 | 4.5 |
| AT1G62390 | 2 | 23,087,975 | 4.78 | 4.5 | |
| AT1G62520 | 2 | 23,148,077 | 0 | 4.79 | 4.5 |
| AT1G64170 | 2 | 23,818,839 | 6.763 | 4.9 | 4.5 |
| AT1G65450 | 2 | 24,321,537 | 26.465 | 4.98 | |
| AT1G68520 | 2 | 25,712,777 | 5.08 | 8.82 | |
| AT1G68530 | 2 | 25,713,992 | 46.377 | 5.08 | 8.81 |
| AT1G72390 | 2 | 27,260,615 | 5.09 | 4.69 | |
| AT1G74600 | 2 | 27,996,107 | 59.555 | 5.05 | 5.29 |
| AT1G76550 | 2 | 29,741,292 | 73.742 | 4.84 |
Rows in italic are genes from the pericentromeric region.
Physical position on A. thaliana chromosome 1 from the The Arabidopsis Information Resource database (http://www.arabidopsis.org).
Genetic map position from A. lyrata genetic mapping of Hansson et al. (2006).
Recombination rate estimates in A. thaliana (thal) and A. lyrata (lyr).
Figure 1.—

Multi-locus-likelihood estimates of the population mutation parameter θ = 4Neu from six A. lyrata populations. The y-axis shows the difference in natural log likelihood from the maximum likelihood for the values of θ on the x-axis, shown on a log scale. Solid curves show the estimates from regions of normal recombination, lightly shaded curves are estimates from the pericentromeric regions of suppressed recombination, and darkly shaded curves show the pericentromeric regions, excluding the highly divergent locus AT1G36310. Dashed lines represent the 95% credibility interval using the chi-square approximation.
Sequencing reactions were conducted using the BigDye sequencing kit, and sequences were run on an ABI 3100 or an ABI 3700 capillary sequencer. Chromatograms were analyzed and managed using Sequencher 4.5, using the “call secondary peaks” option to aid in the identification of heterozygous sites. All chromatograms were carefully checked manually for heterozygous nucleotide positions, using the sequence from both strands to confirm putative heterozygous sites. Results from segregation analysis in the mapping population (Hansson et al. 2006) plus analysis of the segregation from population samples confirmed that we were amplifying single genomic regions, and not gene duplicates. All sequences were submitted to GenBank with accession nos. DQ885491–DQ885560 and BV683158–BV684553.
Recombination rate comparisons:
We tested the effect of recombination rate on nucleotide diversity using two approaches: (1) “regional comparisons” of diversity levels across large-scale chromosomal regions with contrasting recombination rates, comparing the surveyed pericentromeric region (suppressed recombination) with the chromosome arms (“normal” recombination), and (2) tests for correlations between diversity at each locus and local point estimates of recombination rates. Note that while the second test relies to some extent on the assumption of conserved physical distances between the two Arabidopsis species (see below), the first test assumes only suppressed recombination in the pericentromeric region in both species, which is supported by both linkage and cytological results. Since both background selection and selective sweeps are expected to strongly reduce diversity only when recombination rates are low (reviewed in Innan and Stephan 2003), our regional comparison (normal vs. suppressed recombination near the centromere) should be ideal for detecting any such effects in A. lyrata.
Given the low marker density around the centromere of chromosome I (CEN1), we used mapping data from the fine-scale centromere map of chromosome I of A. thaliana (Haupt et al. 2001) to select loci found in the region of suppressed recombination. On the basis of these results, our data set includes five loci from the region of suppressed recombination around CEN1; three loci (AT1G36310, AT1G36730, AT1G36730) are in a region with an estimated 50-fold reduction in recombination relative to the genome average, and two loci (AT1G42470, AT1G42990) are in a region with an estimated 10-fold reduction in recombination rates (Haupt et al. 2001). Importantly, the five loci from the region of suppressed recombination surrounding the centromere in A. thaliana were completely linked in the A. lyrata mapping family, strongly suggesting that they lie in a conserved region of suppressed recombination (Hansson et al. 2006). This is consistent with cytological observations showing the conserved position of the centromere between species (Lysak et al. 2006).
To obtain point estimates of recombination for each locus, genetic and physical distances of markers from chromosome 1 of A. thaliana were fit to a third-order polynomial (Wright et al. 2003a). In addition, we used the genetic map of A. lyrata (Kuittinen and Aguade 2000; Kuittinen et al. 2004; Hansson et al. 2006) to assess the similarity in recombination rates between species. Of our 26 genes, 20 have been genetically mapped in A. lyrata (Hansson et al. 2006) and the results confirm the general genome collinearity for the regions under study, with two exceptions discussed below (Hansson et al. 2006). Point estimates of recombination from A. lyrata mapping data were obtained in a similar way to A. thaliana, dividing the linkage groups into distinct regions, fitting third-order polynomials to the relationship between physical and genetic distance, and taking the first derivative of this polynomial at various physical positions. Note that our estimates assume that physical distances are similar between the species. Several genome size estimates have been made in A. lyrata and suggest a DNA content ∼1.5 times larger than that of A. thaliana (Johnston et al. 2005). If the difference in size is distributed uniformly across the genome, estimates of relative recombination rates in different genome regions will be unaffected by the overall difference in DNA content. This must be tested in the future, but there is some evidence suggesting that the difference in genome size may be due to a general difference in the relative rates of insertion and deletion. In particular, intron lengths in A. lyrata are consistently higher than in A. thaliana, on the basis of samples including 50 genes, mostly from A. thaliana chromosomes I and IV (Wright et al. 2002; A. Kawabe, unpublished results).
In the A. lyrata second linkage group, collinearity with chromosome 1 in A. thaliana is disrupted by an inversion in A. thaliana that encompasses four of our genes (Table 1). Furthermore, the location of the centromeric region for chromosome 2 is unknown, since this region was lost in the fusion that created the A. thaliana chromosome I (see Figure 4 of Hansson et al. 2006). Evidence from chromosome painting and genetic mapping suggests a location close to the inversion breakpoint (Lysak et al. 2006), bounded by the locus At1G59720 (Hansson et al. 2006), and we therefore estimated recombination rates separately for loci within the inversion and outside it. Several A. lyrata loci mapping to the ends of the inversion and the chromosome ends were excluded from the correlation analysis due to poorly estimated recombination rates. Although the departures from synteny on chromosome 2 make recombination rate estimates less certain, our results and interpretation remain unchanged when the analyses are restricted to the loci on chromosome 1 (data not shown), where there is no evidence for disruption of synteny, and recombination rate estimates and map distances are highly conserved. Additional information on recombination rate estimates across species is presented as supplemental material at http://www.genetics.org/supplemental/.
Data analysis:
Average numbers of silent pairwise differences within A. lyrata (π), levels of differentiation between populations (FST), and Jukes–Cantor estimates of the number of silent (Ks) and nonsynonymous (Ka) substitutions per site between A. lyrata and A. thaliana were calculated using DNASp. version 4.0. To compare diversity levels across regions with contrasting recombination rates, we used a maximum-likelihood estimate of θ = 4Neu, based on the numbers of segregating sites (see Wright et al. 2002) and using the ∼95% credibility intervals assuming a chi-square distribution of likelihood values. Note that this method assumes no recombination, and therefore our credibility intervals are conservative, particularly in regions of intermediate and high recombination rates. We tested for differences in diversity that might be caused by selection with a maximum-likelihood (ML) extension of the Hudson–Kreitman–Aguadé (HKA) test (Hudson et al. 1987). Using the MLHKA program (Wright and Charlesworth 2004), we tested for a diversity difference between the chromosome arm genes and the set of five genes in the regions near the centromere, using both polymorphism data and divergence from A. thaliana. Specifically, we tested a neutral null model in multi-locus tests, against an alternative allowing for the uncoupling of diversity and divergence (i.e., hitchhiking) on all centromeric loci. The alternative model estimates five independent parameters for the centromeric loci and thus has 5 d.f. This method assumes independent segregation of the loci, which is incorrect, because the centromere loci are partially linked. An alternative method, taking the centromeric region as a single locus in MLHKA with 1 d.f. for the selection model, yielded results and P-values similar to those presented.
Linkage disequilibrium among pairs of sites was calculated using Weir's (1996) EM algorithm for estimating the squared correlation coefficient, r2, from unphased diploid genotype data, as implemented by Macdonald et al. (2005). R code was kindly provided by S. Macdonald for r2 estimates.
RESULTS AND DISCUSSION
Levels of diversity across populations:
Supplemental Table S2 at http://www.genetics.org/supplemental/ summarizes estimates of nucleotide polymorphism for all loci in each of the six populations surveyed, and Figure 1 shows likelihood estimates of Watterson's (1975) estimator of the population mutation parameter θ = 4Neu at silent sites for each population. Levels of polymorphism are heterogeneous; the central European Plech, Germany, population has the highest diversity (maximum-likelihood estimate of 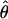 = 0.019 in regions of normal recombination), while the North American populations show an approximately fivefold reduction in diversity (Indiana
= 0.019 in regions of normal recombination), while the North American populations show an approximately fivefold reduction in diversity (Indiana 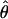 = 0.004 and Ontario
= 0.004 and Ontario 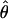 = 0.004), consistent with previous results suggesting a strong population bottleneck in North American populations (Wright et al. 2003b; Ramos-Onsins et al. 2004). In comparison with central Europe, diversity values are slightly reduced in Sweden (
= 0.004), consistent with previous results suggesting a strong population bottleneck in North American populations (Wright et al. 2003b; Ramos-Onsins et al. 2004). In comparison with central Europe, diversity values are slightly reduced in Sweden (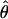 = 0.008), Russia (
= 0.008), Russia (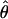 = 0.009), and Iceland (
= 0.009), and Iceland (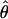 = 0.009), suggesting additional population bottlenecks associated with postglacial recolonization of northern Europe from an ancestral, central European source population (Clauss and Mitchell-Olds 2006; S. Wright, J. P. Foxe, A. Kawabe, J. Ross-Ibarra, L. DeRose-Wilson, G. Gos, D. Charlesworth and B. Gaut, unpublished results).
= 0.009), suggesting additional population bottlenecks associated with postglacial recolonization of northern Europe from an ancestral, central European source population (Clauss and Mitchell-Olds 2006; S. Wright, J. P. Foxe, A. Kawabe, J. Ross-Ibarra, L. DeRose-Wilson, G. Gos, D. Charlesworth and B. Gaut, unpublished results).
Levels of polymorphism and divergence in regions of normal vs. suppressed recombination
As explained above, our primary prediction is reduced nucleotide polymorphism in the region of clearly suppressed recombination, in comparison with regions of normal recombination (the regional test for hitchhiking). Figure 1 compares multi-locus likelihood estimates of θ for the five loci from the pericentromeric region of suppressed recombination (complete linkage in the A. lyrata mapping family and an estimated 10- to 50-fold reduction in recombination compared with average rates in A. thaliana) with those for the remaining 22 loci, from chromosome arm regions (normal recombination; the average recombination rate in A. lyrata, estimated from the chromosome 1 and chromosome 2 data, is 4.8 cM/Mb). In contrast with expectation, there is no general reduction in polymorphism in the pericentromere region. The single exception is in the Karhumaki population from Russia; in this population, diversity for the pericentromeric loci is significantly reduced, and we found no segregating synonymous sites in any of the five loci in the reduced recombination region. This finding supports a suppression of recombination in this region; if recombination is infrequent and selection has acted, the loci will have correlated evolutionary histories. For these regions, there is complete collinearity (no inversion or fusion events) between A. lyrata and A. thaliana and the recombination rate estimates are nearly identical in the two species. Note that the failure to find a general reduction in diversity in this pericentromere is unlikely to be due to lack of power in our study; in many populations, polymorphism in these genes is higher in these regions than in chromosome arm genes, although not significantly.
Similarly, except for the Karhumaki population, map-based estimates of individual locus recombination rates are uncorrelated with silent polymorphism within populations or with total diversity (Table 2), or with differentiation between populations (FST, data not shown). For the Karhumaki sample, the silent-site diversity values are positively correlated with the recombination rate estimates from either species (one-tailed P = 0.006 with A. lyrata recombination estimates, P = 0.006 for A. thaliana estimates; Table 2), and the one-tailed test remains significant after a Bonferroni correction for six tests (P < 0.05). Excluding the pericentromeric regions, no population shows a correlation between recombination and diversity. The primary effect in Karhumaki is thus a difference in polymorphism levels between pericentromeric regions of highly suppressed recombination and the rest of the genome. These patterns hold when only data from the 15 loci from chromosome 1 are considered; recombination is significantly positively correlated with synonymous diversity in Karhumaki samples (Spearman's r = 0.734 with A. lyrata recombination estimates, P < 0.01; r = 0.692 with A. thaliana recombination estimates, P < 0.01), but not in any other population. We checked the PCR failure rate for genes in the pericentromeric region in Russia, in case the low diversity was an artifact caused by failure to amplify a very divergent haplotype. As there is no difference in failure rates from that for other genes (supplemental Table S2 at http://www.genetics.org/supplemental/), this explanation is ruled out.
TABLE 2.
Spearman rank correlations between recombination rate estimates from A. thaliana and A. lyrata and synonymous nucleotide site diversity and diversity scaled to divergence
| rthalvs. πs | rlyrvs. πs | rthalvs. πs/Ks | rlyrvs. πs/Ks | |
|---|---|---|---|---|
| Plech | −0.381 | −0.282 | −0.282 | −0.172 |
| Stubbsand | −0.354 | −0.345 | −0.297 | −0.278 |
| Esja Mountain | −0.238 | −0.278 | −0.238 | −0.293 |
| Karhumaki | 0.481* | 0.506* | 0.456* | 0.475* |
| Indiana Dunes | −0.281 | −0.232 | −0.292 | −0.235 |
| Rondeau | −0.189 | −0.205 | −0.121 | −0.185 |
| Species wide | −0.251 | −0.230 | −0.188 | −0.173 |
thal, A. thaliana; lyr, A. lyrata; πs, synonymous nucleotide diversity; πs/Ks, diversity scaled to divergence. *P < 0.05.
Testing for mutation rate heterogeneity and balancing selection:
We next consider the possibility that mutation rates could differ between genomic regions. In our data set, species-wide silent-site diversity, πs, is significantly correlated with silent-site divergence, Ks (Spearman's r = 0.4, P < 0.05), consistent with an effect of mutation rate variation on diversity levels. However, estimated recombination rates and Ks are uncorrelated in our data set (Table 2; Spearman's r = 0.02, P ≫ 0.05), so recombination rate variation does not explain the mutation rate heterogeneity. To further test for effects of hitchhiking in the pericentromere, controlling for divergence, we performed maximum-likelihood-ratio HKA tests for diversity differences using multi-locus polymorphism and divergence values (Wright and Charlesworth 2004). The Karhumaki population yielded significant support for diversity-reducing selection (selective sweeps) at pericentromeric loci (χ2 = 40.0, d.f. = 5, P < 0.001, significant following a Bonferroni correction for multiple tests, P < 0.01). In contrast, all other populations showed no significant evidence for hitchhiking at pericentromeric loci (P ≫ 0.05).
Although there is no general correlation between recombination and divergence, one of the most polymorphic loci, AT1G36310, lies in the pericentromeric region, and synonymous diversity and divergence are both high (supplemental Table S2 at http://www.genetics.org/supplemental/), suggesting a real difference in mutation rate. This locus is also unusually divergent between A. thaliana and B. oleracea (Ks = 0.82 average Ks between A. thaliana and Brassica spp., 0.474; Tiffin and Hahn 2002). This isolated case of high divergence of a gene in a putatively low recombination region contrasts with results from other taxa, suggesting generally positive correlations between recombination and mutation rates (Cutter and Payseur 2003; Hellmann et al. 2003). Locus AT1G36310, a methyltransferase, was also previously hypothesized to be subject to balancing selection on the basis of a smaller number of loci and a population sample from Iceland (Wright and Charlesworth 2004). This raises the possibility that balancing selection could obscure the expected positive correlation between recombination and diversity. Our previous inference of balancing selection acting on or near the highly polymorphic AT1G36310 locus was based on fewer reference loci than analyzed here and on a sample from Iceland only (Wright and Charlesworth 2004). Our new data set with additional loci gives no evidence for the action of balancing selection in any population, including Iceland, when MLHKA is used to test for selection specifically on this locus (P > 0.05 for all tests). Thus, neither mutation rate heterogeneity nor balancing selection can explain the variability observed in the region of suppressed recombination.
Levels of linkage disequilibrium along chromosome arms vs. pericentromere:
It is important to test whether the pericentromeric regions undergo recombination. If most recombination occurs within genes, our coding sequences in the pericentromeric regions may be hotspots of recombination. Even if recombination is suppressed across the region, local recombination within coding regions could then occur at a significant rate. A related issue is whether our estimates of diversity in the pericentromeric regions are nonindependent, due to low recombination rates. With our present sample we can analyze LD, although a detailed understanding of LD in our populations requires fuller analysis of coding and noncoding regions of more loci, particularly at an intermediate physical distance, taking into account population structure and formally considering the possibility of gene conversion at high rates in regions of low crossing over (e.g., Langley et al. 2000). As expected, average LD (measured by r2) declines with physical distance in this region, and the decline is slower for the pericentromeric region genes than for those in the chromosome arms (Figure 2). This is consistent with a reduced rate of recombination in the former regions and at least partial independence of our observed levels of polymorphism for different loci in the pericentromeric region. In addition, the decline in LD is clearly considerably faster than that estimated in the highly selfing A. thaliana (Nordborg et al. 2005).
Figure 2.—

Levels of linkage disequilibrium, measured by r2, among pairs of sites found in the pericentromeric region (top) and the regions of normal recombination (bottom). The x-axis shows physical distances on the basis of the A. thaliana genome sequence. The fit line shows mean values of r2 and physical distance from across different physical distances.
Levels of hitchhiking in the region surrounding the centromere:
Background selection should generally reduce diversity in regions of low recombination. Our observation that diversity at pericentromeric loci is reduced in only one of six populations thus suggests that background selection is weak in A. lyrata pericentromeric regions. The explanation for our observed reduction in diversity in one population (Karhumaki) may be a recent selective sweep or possibly a local demographic event. Analysis of population subdivision suggests considerable differentiation between Karhumaki and the other Northern European populations (S. Wright, J. P. Foxe, A. Kawabe, J. Ross-Ibarra, L. DeRose-Wilson, G. Gos, D. Charlesworth and B. Gaut, unpublished results), suggesting some degree of independence in their recent evolutionary histories. However, subdivision is generally high between the populations studied, and it is unlikely that gene flow can account for the absence of reduced diversity at pericentromeric region loci in most populations. The Plech, Germany, population is most likely to represent an ancestral equilibrium population (Clauss and Mitchell-Olds 2003), and we detect no effect of recombination rate in this population. This implies that departures from demographic equilibrium are unlikely to explain the lack of diversity reduction in most populations.
However, it is possible that recent between-species introgression from related taxa could be causing a departure from the pattern expected under background selection. Preliminary results suggest some shared polymorphisms between A. lyrata and its close relative, Arabidopsis halleri (Ramos-Onsins et al. 2004). Thus, a low rate of introgression into A. lyrata following a period of isolation could elevate levels of diversity in regions of low recombination. While this possibility should be investigated in the future by surveying diversity at these loci in closely related taxa, it remains unclear whether the shared polymorphism is due to recent divergence or to ongoing migration, and we therefore also explore further the possibility that background selection is weak in A. lyrata.
The strength of background selection is a function of the deleterious mutation rate, the strength of purifying selection, and the rate of recombination. If deleterious mutation rates are unusually low in the pericentromeric regions, polymorphism might not be detectably reduced, despite suppression of recombination. One possibility is that the genes located in regions of low recombination are subject to little or no selective constraints. We should then see a high ratio of amino acid to synonymous divergence in regions of low recombination. However, the ratio of nonsynonymous to synonymous divergence correlates positively with recombination rates in our sample of genes (A. lyrata-based recombination rate estimates: Spearman's R = 0.553, P < 0.01; A. thaliana-based rates: R = 0.499, P < 0.05). Thus, either selective constraints are unusually high for our sample of pericentromeric genes or adaptive amino acid substitutions are more common in the genes from higher recombination regions.
These possibilities can be distinguished by comparing the ratio of amino acid replacements to synonymous variants among polymorphisms and fixed differences, using the McDonald-Kreitman (1991) test. Compared with the other loci, the pericentromeric genes show many fewer nonsynonymous relative to synonymous differences among both polymorphisms and divergence (for the chromosome arm genes, we find a ratio of nonsynonymous to synonymous polymorphisms equal to 0.86 vs. 0.67 among fixations and, for the pericentromeric genes, the respective values are 0.1 and 0.07). Thus this global analysis gives no evidence for adaptive protein evolution in our chromosome arm genes (McDonald–Kreitman test P > 0.05). Since both the pericentromeric and the chromosome arm genes have higher ratios of nonsynonymous to synonymous polymorphism than fixed differences, there is a trend suggesting weak purifying selection. Thus, while we cannot rule out positive selection contributing to amino acid substitution for some genes, the general picture suggests stronger selective constraints on our pericentromeric genes. This contrasts with results from Drosophila, where there is evidence for most adaptive protein evolution in regions of high recombination and accumulation of slightly deleterious amino acid polymorphisms in regions of reduced recombination (Presgraves 2005).
Low gene density is another possibility that may cause hitchhiking to be weak in the pericentromeric region. There may simply be too few targets for purifying or positive selection for diversity to be generally reduced. Gene density is low in the pericentromeric regions of A. thaliana (Haupt et al. 2001; Wright et al. 2003a) and rice (Nagaki et al. 2004), so the strength of background selection may be low in the highly outcrossing A. lyrata. Using gene expression data from full-length cDNAs (Yamada et al. 2003), ESTs, whole-genome tiling arrays (Yamada et al. 2003), and massively parallel signature sequencing (Meyers et al. 2004), we estimate that there are only ∼40 genes in the region of suppressed recombination surrounding the centromere of chromosome 1 in A. thaliana, excluding transposable elements. Using the genome annotation, this totals ∼78 kb of coding sequence. Using the estimated number of deleterious amino acid mutations accumulated per site since the divergence of A. lyrata and A. thaliana (0.077; Wright et al. 2002), this translates to a deleterious mutation rate of ∼0.0024 in this region per generation, assuming 5 million years since the common ancestor of A. thaliana and A. lyrata (Koch et al. 2000) and an average generation time of 2 years during this period. Under background selection, the relative amount of diversity in a nonrecombining region follows the equation (Charlesworth et al. 1993)
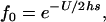 |
where U is the deleterious mutation rate, h is the dominance coefficient, and s is the selection coefficient against deleterious mutations. If the strength of selection against heterozygous mutations is similar to estimates from Drosophila (hs = 0.02; Charlesworth 1996), this predicts a slight reduction of diversity to 95% of that of regions experiencing no background selection; this would be difficult to detect, given the stochasticity of the coalescent process and the consequent variance in diversity estimates. Furthermore, this approximation assumes no recombination across the entire region, whereas the region encompasses almost a centimorgan of map distance. The approximation of Hudson and Kaplan (1995; Equation 8) predicts that a neutral site close to the center of the region would experience a diversity reduction of only 4%, with lesser effects closer to the edge of the region of suppressed recombination. Deleterious mutations other than substitutions affecting amino acids not considered in this approximation (e.g., mutations in noncoding sequences, insertions, deletions, transposable elements) might further reduce diversity and selection coefficients might be lower than those assumed here. However, overall, background selection alone clearly may not necessarily be expected to reduce silent nucleotide diversity in this pericentromeric region, largely due to low gene density.
To test for effects of hitchhiking acting in genes outside the pericentromeric regions, we investigated whether gene density per unit of genetic distance affects levels of genetic diversity, as was found in A. thaliana (Nordborg et al. 2005). In contrast with the A. thaliana results, we observe no correlation between gene density (measured as genes/centimorgan on the basis of the A. thaliana genome sequence) and levels of silent nucleotide polymorphism for the loci tested (P ≫ 0.05, all populations). If this result is confirmed with additional loci, it will suggest that regions of high gene density per centimorgan in A. lyrata may also experience little hitchhiking. This would be consistent with A. lyrata's higher effective rates of recombination and historical effective population sizes (given its outcrossing mating system), compared with A. thaliana. Hitchhiking causes only slight effects on diversity unless recombination is infrequent, even in high gene density regions. Thus, highly selfing species should experience such effects over a much larger portion of the genome. Recent theoretical work suggests that weak selection against deleterious mutations has a stronger diversity-reducing effect than previously thought, but very large data sets would be required to detect this (B. Charlesworth, personal communication).
Our results contrast with those in D. melanogaster, where reduced diversity has consistently been observed in all low crossing-over regions (reviewed in Charlesworth 1996). The D. melanogaster fourth chromosome has numbers of genes comparable to the pericentromeric region surveyed here and has reduced diversity (Jensen et al. 2002; Wang et al. 2004). It is possible that Drosophila experiences a higher deleterious mutation rate and/or a larger class of mutations subject to weak purifying selection in regions of low recombination, for example, through transposable element activity driving background selection (Charlesworth 1996). Our results thus suggest either that A. lyrata lacks a large class of slightly deleterious mutations that are segregating and being selectively removed from populations or that D. melanogaster experiences more frequent positive selection.
If background selection is generally weak in A. lyrata, the reduced polymorphism in the Karhumaki population may reflect a local selective sweep associated with recent directional selection. Alternatively, however, a population bottleneck or other demographic situation may have increased the variance in diversity across loci in the Russian population, by chance reducing polymorphism close to the centromere. If recombination is infrequent in this region, the loci will have correlated demographic history and should all have low polymorphism if a recent diversity-reducing event affected the region. Data from genes in the pericentromeric regions of other centromeres, combined with additional polymorphism data from the pericentromere surveyed here, should help distinguish between a population bottleneck and a selected substitution in a gene near the centromere of chromosome 1.
Acknowledgments
The authors thank S. Macdonald for providing R code and assistance in linkage disequilibrium analysis; O. Savolainen, T. Mitchell-Olds, E. Thorhallsdottir, and B. Mable for providing population samples; and P. Andolfatto and B. Charlesworth for helpful discussion. The research was supported by the National Science Foundation (B.G.), National Science and Engineering Research Council (Canada) (S.W.), and Natural Environment Research Council (UK) (D.C.).
References
- Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang et al., 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andolfatto, P., and M. Przeworski, 2001. Regions of lower crossing over harbor more rare variants in African populations of Drosophila melanogaster. Genetics 158: 657–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balana-Alcaide, D., S. E. Ramoa-Onsins, Q. Boone and M. Aguade, 2006. Highly structured nucleotide variation within and among Arabidopsis lyrata populations at the FAH1 and DFR gene regions. Mol. Ecol. 15: 2059–2068. [DOI] [PubMed] [Google Scholar]
- Barton, N. H., 2000. Genetic hitchhiking. Philos. Trans. R. Soc. Lond. B Biol. Sci. 355: 1553–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudry, E., C. Kerdelhue, H. Innan and W. Stephan, 2001. Species and recombination effects on DNA variability in the tomato genus. Genetics 158: 1725–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begun, D. J., and C. F. Aquadro, 1992. Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster. Nature 356: 519–520. [DOI] [PubMed] [Google Scholar]
- Charlesworth, B., 1996. Background selection and patterns of genetic diversity in Drosophila melanogaster. Genet. Res. 68: 131–149. [DOI] [PubMed] [Google Scholar]
- Charlesworth, B., M. T. Morgan and D. Charlesworth, 1993. The effects of deleterious mutations on neutral molecular variation. Genetics 134: 1289–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, B., M. Nordborg and D. Charlesworth, 1997. The effects of local selection, balanced polymorphism and background selection on equilibrium patterns of genetic diversity in subdivided populations. Genet. Res. 70: 155–174. [DOI] [PubMed] [Google Scholar]
- Clauss, M. J., and T. Mitchell-Olds, 2003. Population genetics of tandem trypsin inhibitor genes in Arabidopsis species with contrasting ecology and life history. Mol. Ecol. 12: 1287–1299. [DOI] [PubMed] [Google Scholar]
- Clauss, M. J., and T. Mitchell-Olds, 2006. Population genetic structure of Arabidopsis lyrata in Europe. Mol. Ecol. 15: 2753–2766. [DOI] [PubMed] [Google Scholar]
- Cutter, A. D., and B. A. Payseur, 2003. Selection at linked sites in the partial selfer Caenorhabditis elegans. Mol. Biol. Evol. 20: 665–673. [DOI] [PubMed] [Google Scholar]
- Das, A., S. Mohanty and W. Stephan, 2004. Inferring the population structure and demography of Drosophila ananassae from multilocus data. Genetics 168: 1975–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson, B., A. Kawabe, S. Preuss, H. Kuittinen and D. Charlesworth, 2006. Comparative gene mapping in Arabidopsis lyrata chromosomes 1 and 2 and the corresponding A. thaliana chromosome 1: recombination rates, rearrangements and centromere location. Genet. Res. 87: 75–85. [DOI] [PubMed] [Google Scholar]
- Haupt, W., T. C. Fischer, S. Winderl, P. Fransz and R. A. Torres-Ruiz, 2001. The centromere1 (CEN1) region of Arabidopsis thaliana: architecture and functional impact of chromatin. Plant J. 27: 285–296. [DOI] [PubMed] [Google Scholar]
- Hellmann, I., I. Ebersberger, S. E. Ptak, S. Paabo and M. Przeworski, 2003. A neutral explanation for the correlation of diversity with recombination rates in humans. Am. J. Hum. Genet. 72: 1527–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmann, I., K. Prufer, H. Ji, M. C. Zody, S. Paabo et al., 2005. Why do human diversity levels vary at a megabase scale? Genome Res. 15: 1222–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., and N. L. Kaplan, 1995. Deleterious background selection with recombination. Genetics 141: 1605–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., M. Kreitman and M. Aguade, 1987. A test of neutral molecular evolution based on nucleotide data. Genetics 116: 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innan, H., and W. Stephan, 2003. Distinguishing the hitchhiking and background selection models. Genetics 165: 2307–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen, M. A., B. Charlesworth and M. Kreitman, 2002. Patterns of genetic variation at a chromosome 4 locus of Drosophila melanogaster and D. simulans. Genetics 160: 493–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, J. S., A. E. Pepper, A. E. Hall, Z. J. Chen, G. Hodnett et al., 2005. Evolution of genome size in Brassicaceae. Ann. Bot. 95: 229–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch, M. A., B. Haubold and T. Mitchell-Olds, 2000. Comparative evolutionary analysis of the chalcone synthase and alcohol dehydrogenase loci among different lineages of Arabidopsis, Arabis and related genera (Brassicaceae). Mol. Biol. Evol. 17: 1483–1498. [DOI] [PubMed] [Google Scholar]
- Kuittinen, H., and M. Aguade, 2000. Nucleotide variation at the CHALCONE ISOMERASE locus in Arabidopsis thaliana. Genetics 155: 863–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuittinen, H., A. A. de Haan, C. Vogl, S. Oikarinen, J. Leppala et al., 2004. Comparing the linkage maps of the close relatives Arabidopsis lyrata and A. thaliana. Genetics 168: 1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley, C. H., B. P. Lazzaro, W. Phillips, E. Heikkinen and J. M. Braverman, 2000. Linkage disequilibria and the site frequency spectra in the su(s) and su(w(a)) regions of the Drosophila melanogaster X chromosome. Genetics 156: 1837–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lercher, M. J., and L. D. Hurst, 2002. Human SNP variability and mutation rate are higher in regions of high recombination. Trends Genet. 18: 337–340. [DOI] [PubMed] [Google Scholar]
- Lysak, M. A., A. Berr, A. Pecinka, R. Schmidt, K. McBreen et al., 2006. Mechanisms of chromosome number reduction in Arabidopsis thaliana and related Brassicaceae species. Proc. Natl. Acad. Sci. USA 103: 5224–5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald, S. J., T. Pastinen and A. D. Long, 2005. The effect of polymorphisms in the enhancer of split gene complex on bristle number variation in a large wild-caught cohort of Drosophila melanogaster. Genetics 171: 1741–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard-Smith, J., and J. Haigh, 1974. The hitch-hiking effect of a favorable gene. Genet. Res. 23: 23–35. [PubMed] [Google Scholar]
- McDonald, J. H., and M. Kreitman, 1991. Adaptive protein evolution at the Adh1 locus in Drosophila. Nature 351: 652–654. [DOI] [PubMed] [Google Scholar]
- Meyers, B. C., T. H. Vu, S. S. Tej, H. Ghazal, M. Matvienko et al., 2004. Analysis of the transcriptional complexity of Arabidopsis thaliana by massively parallel signature sequencing. Nat. Biotechnol. 22: 1006–1011. [DOI] [PubMed] [Google Scholar]
- Nachman, M. W., 2001. Single nucleotide polymorphisms and recombination rate in humans. Trends Genet. 17: 481–485. [DOI] [PubMed] [Google Scholar]
- Nachman, M. W., V. L. Bauer, S. L. Crowell and C. F. Aquadro, 1998. DNA variability and recombination rates at X-linked loci in humans. Genetics 150: 1133–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaki, K., Z. Cheng, S. Ouyang, P. B. Talbert, M. Kim et al., 2004. Sequencing of a rice centromere uncovers active genes. Nat. Genet. 36: 138–145. [DOI] [PubMed] [Google Scholar]
- Nordborg, M., T. T. Hu, Y. Ishino, J. Jhaveri, C. Toomajian et al., 2005. The pattern of polymorphism in Arabidopsis thaliana. PLoS Biol. 3: e196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presgraves, D. C., 2005. Recombination enhances protein adaptation in Drosophila melanogaster. Curr. Biol. 15: 1651–1656. [DOI] [PubMed] [Google Scholar]
- Ramos-Onsins, S. E., B. E. Stranger, T. Mitchell-Olds and M. Aguade, 2004. Multilocus analysis of variation and speciation in the closely related species Arabidopsis halleri and A. lyrata. Genetics 166: 373–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roselius, K., W. Stephan and T. Stadler, 2005. The relationship of nucleotide polymorphism, recombination rate and selection in wild tomato species. Genetics 171: 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savolainen, O., C. H. Langley, B. P. Lazzaro and H. Fr, 2000. Contrasting patterns of nucleotide polymorphism at the alcohol dehydrogenase locus in the outcrossing Arabidopsis lyrata and the selfing Arabidopsis thaliana. Mol. Biol. Evol. 17: 645–655. [DOI] [PubMed] [Google Scholar]
- Schmid, K. J., S. Ramos-Onsins, H. Ringys-Beckstein, B. Weisshaar and T. Mitchell-Olds, 2005. A multilocus sequence survey in Arabidopsis thaliana reveals a genome-wide departure from a neutral model of DNA sequence polymorphism. Genetics 169: 1601–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, A., Y. H. Liu and N. Saitou, 2004. Genetic variation versus recombination rate in a structured population of mice. Mol. Biol. Evol. 21: 404–409. [DOI] [PubMed] [Google Scholar]
- Tenaillon, M. I., M. C. Sawkins, L. K. Anderson, S. M. Stack, J. Doebley et al., 2002. Patterns of diversity and recombination along chromosome 1 of maize (Zea mays ssp. mays L.). Genetics 162: 1401–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiffin, P., and M. W. Hahn, 2002. Coding sequence divergence between two closely related plant species: Arabidopsis thaliana and Brassica rapa ssp. pekinensis. J. Mol. Evol. 54: 746–753. [DOI] [PubMed] [Google Scholar]
- Wang, W., K. Thornton, J. J. Emerson and M. Long, 2004. Nucleotide variation and recombination along the fourth chromosome in Drosophila simulans. Genetics 166: 1783–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson, G. A., 1975. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7: 256–276. [DOI] [PubMed] [Google Scholar]
- Weir, B. S., 1996. Genetic Data Analysis II. Sinauer Associates, Sunderland, MA.
- Wright, S. I., and B. Charlesworth, 2004. The HKA test revisited: a maximum-likelihood-ratio test of the standard neutral model. Genetics 168: 1071–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S. I., B. Lauga and D. Charlesworth, 2002. Rates and patterns of molecular evolution in inbred and outbred Arabidopsis. Mol. Biol. Evol. 19: 1407–1420. [DOI] [PubMed] [Google Scholar]
- Wright, S. I., N. Agrawal and T. E. Bureau, 2003. a Effects of recombination rate and gene density on transposable element distributions in Arabidopsis thaliana. Genome Res. 13: 1897–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S. I., B. Lauga and D. Charlesworth, 2003. b Subdivision and haplotype structure in natural populations of Arabidopsis lyrata. Mol. Ecol. 12: 1247–1263. [DOI] [PubMed] [Google Scholar]
- Wright, S. I., I. V. Bi, S. G. Schroeder, M. Yamasaki, J. F. Doebley et al., 2005. The effects of artificial selection on the maize genome. Science 308: 1310–1314. [DOI] [PubMed] [Google Scholar]
- Yamada, K., J. Lim, J. M. Dale, H. Chen, P. Shinn et al., 2003. Empirical analysis of transcriptional activity in the Arabidopsis genome. Science 302: 842–846. [DOI] [PubMed] [Google Scholar]