

Abstract
Recombination decreases the association of linked nucleotide sites and can influence levels of polymorphism in natural populations. When coupled with selection, recombination may relax potential conflict among linked genes, a concept that has played a central role in research on the evolution of recombination. The sex determination locus (SDL) of the honeybee is an informative example for exploring the combined forces of recombination, selection, and linkage on sequence evolution. Balancing selection at SDL is very strong and homozygous individuals at SDL are eliminated by worker bees. The recombination rate is increased up to four times that of the genomewide average in the region surrounding SDL. Analysis of nucleotide diversity (π) reveals a sevenfold increase of polymorphism within the sex determination gene complementary sex determiner (csd) that rapidly declines within 45 kb to levels of genomewide estimates. Although no recombination was observed within SDL, which contains csd, analyses of heterogeneity, shared polymorphic sites, and linkage disequilibrium (LD) show that recombination has contributed to the evolution of the 5′ part of some csd sequences. Gene conversion, however, has not obviously contributed to the evolution of csd sequences. The local control of recombination appears to be related to SDL function and mode of selection. The homogenizing force of recombination is reduced within SDL, which preserves allelic differences and specificity, while the increase of recombination activity around SDL relaxes conflict between SDL and linked genes.
RECOMBINATION generates novel genomic variants through the rearrangement of existing ones and thus contributes to the evolution of new gene activity and function (Patthy 1996; Todd et al. 2001; Posada et al. 2002; Zhang et al. 2004). In addition, recombination decreases the association of linked nucleotide sites and thus reduces interference among selected sites (Felsenstein 1974). The latter concept has played a central role in research on the level of polymorphism in natural populations (Begun and Aquadro 1992; Charlesworth et al. 1993), as well as that on the origin and activity of recombination (Otto and Lenormand 2002; Rice 2002). When coupled with selection, recombination may influence the process of adaptation and have profound effects on the level and pattern of polymorphism at linked sites (Maynard Smith and Haigh 1974; Begun and Aquadro 1992; Andolfatto 2001; Meiklejohn et al. 2004). The latter may also prove a useful tool to locate genes that have experienced selection in the past (Kim and Stephan 2003; Nachman et al. 2003; Schlötterer 2003).
The sex determination locus (SDL) of the honeybee is an informative example for exploring the combined forces of recombination, linkage, and selection and their effects on the evolution of DNA sequences. Balancing selection operates strongly among sex-determining alleles. Homozygotes at SDL develop into diploid males that do not reproduce while heterozygotes at SDL differentiate into fertile females. As in all hymenopteran insects, males arise from hemizygous, unfertilized eggs. The great advantage of heterozygotes results in strong balancing selection at this locus. Rare alleles benefit from their increased representation in viable heterozygotes, while more common alleles have a selective disadvantage as they are present more often in nonviable homozygotes (Kimura and Crow 1964; Yokoyama and Nei 1979). The sex determination function within SDL is encoded by the complementary sex determiner (csd) gene (see Figure 1A for exon structure) (Beye et al. 2003; Hasselmann and Beye 2004).
Figure 1.—




Local recombination rates, nucleotide diversity (π) of the SDL, the flanking genomic regions, and csd. (A) Genomic organization of csd (redrawn after Beye et al. 2003). The start (exon 2) and the stop (in exon 9) of the ORF are shown. (B) Estimates of local recombination rate (in centimorgans per megabase) of genomic fragments as defined in C. The dashed line marks the average genomewide recombination rate of the honeybee. (C) Physical and genetic map of SDL and flanking regions. The number of recombination events between SDL and the genetic markers, which were estimated on the basis of 1000 meioses, are shown. Distances between markers were obtained from sequence and clone mapping data. Distance data on the right have been published previously (Beye et al. 2003). (D) Mean nucleotide diversity (π) and SD (shown as vertical bars) of up to eight chromosomes and of five loci (DF37; DF33; HF63; KF, and KF22) that have various distances (in kilobases) to the 3′-end of csd. Estimates of π within SDL (Ex2/3, Ex4/5, and Ex6/7) are based on silent sites within the ORF of csd (Hasselmann and Beye 2004), which excludes the type II sequence (see also Table 1). The clustering of exons reflects their genomic association as shown in A.
Previous genetic and physical mapping suggested a higher recombination rate in the genomic region surrounding SDL relative to genomewide average (Beye et al. 1999). This elevated rate may have adaptive significance (Beye et al. 1999); however, the low resolution of this study (a 360-kb fragment that composes SDL was analyzed) does not distinguish whether the recombination rate is elevated over the entire fragment, in close proximity to SDL, or whether it rises within SDL. An increase in recombination rate in the vicinity of SDL could reduce the association and potential selective conflicts between SDL and linked genes (Beye et al. 1999). Alternatively, recombination activity within the gene could mix existing variation in a combinatorial fashion to contribute to the rise of novel alleles. Such a mechanism has been suggested to explain the evolution of shared clusters among major histocompatibility complex (MHC) alleles (Hughes et al. 1993; Yuhki and O'Brien 1994; Zangenberg et al. 1995) and alleles of the gametophytic self-incompatibility (S) locus (Awadalla and Charlesworth 1999; Vieira et al. 2003). Recombination within the gene may also have an advantage if it increases the effectiveness with which deleterious mutations are eliminated (Kondrashov 1984; Rice 2002). Such an effect may be most relevant among interacting genes (Rice 1998) or among sex-determining alleles (Beye et al. 1999).
The complete isolation of SDL and its linked genomic sequences provides us with the power to determine the local recombination activity and the levels of polymorphism within SDL and its linked genomic regions. Recombination activity that increases with decreasing distance from SDL may be taken as evidence that recombination reduces the potential conflict between SDL and linked sites. Levels of polymorphism that decline with increasing distances from SDL may be a result of the combined forces of balancing selection at SDL, linkage, and recombination. Polymorphism differences among csd alleles could provide evidence of whether recombination has influenced sequence evolution within SDL (Beye et al. 2003), which is under strong balancing selection (Charlesworth 2004; Hasselmann and Beye 2004). Heterogeneity of nucleotide differences across csd sequences or a decay of linkage disequilibrium (LD) with physical distance may be taken as evidence of recombination, while shared clusters of polymorphic sites may indicate the occurrence of gene conversion resulting from recombination.
MATERIALS AND METHODS
Mapping procedures:
Mapping was conducted on 1000 female progeny of an inbred cross (Beye et al. 2003). Marker information of Q, Z, MEFAp-25, GF3MHi, and MKFHi-52 has been previously described (Beye and Moritz 1994; Hunt and Page 1994; Hasselmann et al. 2001; Beye et al. 2003), while markers A, B, and R were newly designed on the basis of sequence information from the honeybee genome assembly (the oligonucleotides used for A were 5′-AAGTGTACTTCATTGCAAAACAGGAG-3′ and 5′-TGTACTGTAACTAATCGTAACGCATCG-3′; for B, 5′-ACATAACTTGCATAATTCTCA AGGACG-3′ and 5′-CACTTAGGCAACGATAAACAATATTCG-3′; for R, 5′-ACACGTCCATTCGGAATTCGA-3′ and 5′-ACTGCCAGGATCTTCTGATTTAGATG-3′) (http://www.ensembl.org/Apis_mellifera/) according to procedures described elsewhere (Hasselmann et al. 2001). Recombination frequencies were converted into mapping distances by the Kosambi mapping function (Kosambi 1944). The estimate of the genomewide recombination rate (19 cM/Mb) is based on the cumulative genetic distance data of two independent maps (5000 cM; (Solignac et al. 2004; G. Hunt, personal communication) and a genome size of 240 Mb (genome assembly version 2.0).
Sequence and statistical analysis:
Fragments of loci DF37, DF33, HF63, KF, and KF22 that show linkage to SDL and loci of high (loci RR 21, RR11, RR17, RR28, RR7, and RR14) and low (loci RR 22, RR25, RR8, RR9, RR23, and RR19) recombining regions that show no linkage to SDL were amplified using standard protocols (Hasselmann et al. 2001) from a population sample of 10 chromosomes (haploid drones) collected in Davis, California. Estimates of local recombination rates were derived from genetic mapping and sequence data (Beye et al. 2006). Mean nucleotide differences (π) (Nei 1987) and standard deviations were calculated using the DnaSP 3.5.3 program (Rozas and Rozas 1999). Sequences were initially aligned using the ClustalX (Thompson et al. 1997) program and then manually edited by eye using the BioEdit program (Hall 1999). Gaps were excluded from the analysis. Sequence information from a chromosomal walk (Beye et al. 2003) was used to design oligonucleotides that amplify fragments of linked loci: DF37, 5′-TACGATTGGCAGACACGAAGG-3′ and 5′-CTGTGAAAGCAACACAAGTCC-3′; DF33, 5′-AGAATTGAATCTGTCACATCTGG-3′ and 5′-TTACCAATCGCCATTG AAATTC-3; HF63, 5′-CGTGTTCGTCAAATAATCGC-3′ and 5′-GCATACATGTTGAG AATCGAT-3; KF, 5′-TATTCGAAATAGAGAATCTTCACCG-3 and 5′-GATCGAATGTC CAAACTCGACG-3′; and KF22, 5′-TTGACACGTGCCTGAAGAAACAT-3′ and 5′-CGATGGTTGCTTGTCGACTGACA-3′. Sequence information from the honeybee genome was used to design oligonucleotides that amplify loci of high and low recombining regions (available from the authors upon request).
Distances between loci 3′ of csd rely on complete sequence information (for KF and KF22) and the restriction map (Beye et al. 2003). Specific PCR reaction conditions are 94° for 160 sec, followed by 35 cycles of 94° for 30 sec, 52° for 30 sec for primer combination DF33, or 55° for primers HF63 or 60° for primers DF37, KF, and KF22, followed by a final step of 72° for 30 sec. Amplified fragments were resolved on 1.3% agarose gels (Hasselmann et al. 2001), eluted, and cloned into the pGEM-T vector (Promega, Madison, WI). Sequencing was performed by MWG Biotech AG (Ebersberg, Germany).
Sequence characteristics, such as simple repeats, low-complexity DNA, and guanine–cytosine (GC) content of high and low recombining regions, were analyzed using the program Repeatmasker version 3.0.8 (http://repeatmasker.genome.washington.edu). Sequences were derived from contigs NW 629957.1; NW 627483.1, and NW 623616.1 of the honeybee genome.
Analyses of allelic csd sequences were performed only on the cDNA sequences of the type I class (Hasselmann and Beye 2004), as they show signatures of balancing selection. Heterogeneity across exon 2–3, exon 4–5, and exon 6–9 sequences (Beye et al. 2003; Hasselmann and Beye 2004) was calculated separately for the number of nonsynonymous (dn) and synonymous (ds) differences per site (Kimura 1980). Genetic distances were measured using Kimura's two-parameter model and used to construct a neighbor-joining tree implemented by the MEGA 2.1 program (Kumar et al. 2001). Bootstrap resampling of 1000 replications was applied. LD, R2, and D′ were estimated using the R2 program (http://www.daimi.au.dk/∼compbio/r2). To test whether R2 or D′ correlate with distance, levels of significance were determined from 5000 permutations that include either all polymorphic sites or only synonymous sites that have a frequency >10% (R2 program; http://www.daimi.au.dk/∼compbio/r2). The Stephens test (Stephens 1985), Sawyer's test (Sawyer 1989) (which is implemented in the GENECONV 1.81 program; http://www.math.wustl.edu/∼sawyer), and a maximum-composite-likelihood method (implemented in the MAXHAP program) were applied to identify the impact of short clusters of shared polymorphism among alleles on the overall variation. The Stephens test examines whether a collection of shared polymorphisms is consistent with a model of building two groups of sequences. Levels of significance were evaluated under the null hypothesis of a random distribution of sites. Sawyer's test identifies shared clusters of sites in pairwise alignments and evaluates their significance in the overall alignment by 10,000 random permutations of all polymorphic sites. Mismatches within the cluster of shared polymorphic sites were allowed and scaled by setting g at 2. The maximum-composite-likelihood method (Hudson 2001) estimates a gene conversion rate parameter (f), which is the ratio of gene conversion to crossing-over rate. The gene conversion model assumes different tract lengths (denoted as L in base pairs) while sites with more than two alleles are ignored. Analysis were performed by setting f = 0 (no gene conversion) and f > 0 (crossing over with gene conversion). Under the gene conversion model, values are obtained that maximize the composite likelihood over the set of f-values (fmin = 0.1 to fmax = 10).
RESULTS
Recombination rate is reduced within SDL but increased in the flanking fragments:
Local recombination rates were estimated (Figure 1B) from various fragments at distances ranging from 5 to 213 kb upstream and 1 to 53 kb downstream of SDL (Figure 1C). Homozygosity of genetic markers in 1000 females reflects recombination between markers and SDL [all females are heterozygous for SDL (Beye et al. 2003)]. These recombination fractions were used to calculate genetic distances (Kosambi 1944) (Figure 1C) and the local recombination rate of fragments. The recombination rate steadily increases approaching SDL from upstream flanking regions, from an average of 30 cM/Mb (213–15 kb from SDL) to 77 cM/Mb (fragment BR, 10–0 kb from SDL; Figure 1B). Similarly, the recombination rate steadily increases when approaching SDL from downstream flanking regions, from 10 cM/Mb (25–50 kb downstream of SDL) to 50 cM/Mb (fragment GK, 0–10 kb downstream of SDL). When the number of recombination events in SDL flanking regions (BR and GK) are combined, they deviate from the numbers expected under the genome average rate of 19 cM/Mb (χ2 = 4.03, P = 0.04). When a correction of small sample size of recombined genotypes is taken into account, the differences are marginally significant (Yates-corrected χ2 = 3.1, P = 0.08). In contrast to the finding of an elevated recombination activity in flanking regions, no recombination event was observed within a genomic region of ∼36 kb that encompasses SDL. This result is not expected under the genomewide recombination rate (χ2 = 6.02, P = 0.01; Yates-corrected χ2 = 4.18, P = 0.04) or under the recombination rate of the flanking regions (28.3 cM/Mb; χ2 = 8.03, P = 0.005; Yates-corrected χ2 = 6.2, P = 0.01), suggesting that recombination is reduced, or even suppressed, within SDL. An alternative explanation of this finding is that recombination within SDL results in nonfunctional sex-determining alleles that ensure male development (Beye et al. 2003). Eggs that develop into diploid males are not included in the analysis as they are eaten by worker bees shortly after they hatch (Mackensen 1955). This is an unlikely scenario, however, as the gene that initiates female development by heterozygous allelic composition, csd, is confined to only a part of the whole SDL region (9 of 36 kb), while recombination events are expected to occur, on average, every 3 kb, given the local recombination rate. We were not able to detect any sequence characteristics (simple repeats, low-complexity DNA, increased GC content) specifically associated with the high recombination rate in closest proximity to SDL (0–10 kb) when compared to regions of more distant sequences (50–150 kb) using sequence analysis software (data not shown). We detected two putative chi-like motifs (CGACCACC) (Cheng and Smith 1984), which are not present in the more distant sequences of 50–150 kb, as potential crossover instigators in the region of high recombination rate. This finding does not favor a simple, direct mechanistic interpretation that the local increase of recombination rate is solely based on distinct sequence differences. Even if differences in sequence composition or number of instigator motifs are mechanistically responsible for a higher recombination activity, the heterogeneity of potential facilitators across this sequence demands an explanation. Moreover, the finding that recombination rate steadily and independently increases on both sites of SDL in combination with recombination activity being reduced or even suppressed within SDL suggests a local control of recombination activity that is related to SDL function and to being a target of balancing selection.
Nucleotide diversity decreases with increasing distance from SDL:
To examine whether balancing selection and recombination leave their signatures at nucleotide sites linked to SDL, nucleotide diversity (π) was estimated at 5 linked and 12 unlinked loci that were compared to exon data of csd (Table 1). Linked loci are located at distances of 1.5, 2, 12, 28, and 45 kb downstream of csd, the gene under balancing selection. The unlinked loci were chosen to include regions of both high and low recombination. The loci themselves are assumed to not be direct targets of selection (i.e., neutral markers), as homology searches indicated no similarity to genes or regulatory elements (data not shown). Although several primer combinations were tested, not all chromosomes were amplified. The most plausible explanation is mismatches of primers to the designated binding sites in some haplotypes, but not in others, which is consistent with the finding that the number of nonisolated fragments increases with the number of nucleotide differences (see Table 1). When loci downstream of csd are compared, nucleotide diversity (π) sharply declines from 0.09 at exon 6/7 within the first 5 kb of distance (Figure 1D), but flattens within the subsequent 30 kb. KF22, a locus just 1.5 kb from the 3′-end of the csd gene, is 14 times higher when compared to DF37, a locus 43.5 kb downstream [π = 0.0233 vs. π = 0.00169 (Table 1); Z-test, Z = 2.9; two-tailed: P < 0.01)] that is 4 times higher than our genomewide average estimate [π = 0.0233 vs. π = 0.0055 (Table 1); Z-test, Z = 2.1; two-tailed: P < 0.05], suggesting that linkage to csd has a strong influence on sequence evolution. The severalfold increase of nucleotide diversity at linked downstream sites cannot be explained by the higher local recombination rate as diversity in regions of high recombination rates (51–72 cM/Mb) is still several magnitudes lower (Table 1). Although balancing selection substantially increases diversity at closely linked sites (<20 kb), this does not extend to larger distances. Within 45 kb, levels of polymorphism decline to the genomewide average estimate.
TABLE 1.
Nucleotide differences (π ± SD) of linked and unlinked loci to the SDL
| Recombination rate (cM/Mb) | Distance to 3′-end of csd (kb) | Fragments | Length (bp) | Polymorphic sites | π ± SD | |
|---|---|---|---|---|---|---|
| Loci linked to SDL | ||||||
| Ex2/3 | −7.3 | 14 | 456 | 27 | 0.0200 ± 0.0064 | |
| Ex4/5 | −4.7 | 14 | 201 | 10 | 0.0135 ± 0.0099 | |
| Ex6/7 | −0.5 | 14 | 288 | 51 | 0.0918 ± 0.021 | |
| KF22 | 50 | 1.5 | 3 | 372 | 13 | 0.0233 ± 0.0074 |
| KF2 | 50 | 2 | 4 | 294 | 7 | 0.0136 ± 0.0038 |
| HF63 | 25 | 12 | 8 | 298 | 8 | 0.0108 ± 0.0029 |
| DF33 | 10 | 28 | 5 | 268 | 4 | 0.0067 ± 0.0023 |
| DF37 | 10 | 45 | 7 | 338 | 2 | 0.0017 ± 0.0007 |
| Loci unlinked to SDL | Recombination rate (cM/Mb) | Fragments | Length (bp) | Polymorphic sites | π ± SD | |
| RR22 | 0 | 7 | 473 | 2 | 0.0012 ± 0.0008 | |
| RR25 | 7 | 7 | 503 | 4 | 0.0023 ± 0.0006 | |
| RR8 | 12 | 10 | 325 | 7 | 0.0092 ± 0.0024 | |
| RR9 | 19 | 9 | 699 | 9 | 0.0045 ± 0.0017 | |
| RR23 | 22 | 10 | 783 | 14 | 0.0062 ± 0.0012 | |
| RR19 | 25 | 10 | 532 | 2 | 0.0016 ± 0.0003 | |
| RR21 | 34 | 10 | 622 | 11 | 0.0045 ± 0.0012 | |
| RR17 | 51 | 9 | 482 | 4 | 0.0024 ± 0.0006 | |
| RR28 | 52 | 10 | 485 | 20 | 0.0173 ± 0.0021 | |
| RR7 | 56 | 10 | 416 | 14 | 0.0095 ± 0.0025 | |
| RR11 | 53 | 10 | 590 | 10 | 0.0051 ± 0.0007 | |
| RR14 | 72 | 10 | 884 | 7 | 0.0027 ± 0.0004 | |
| Average nucleotide diversity of unlinked loci | 0.0055 ± 0.0044 |
The number of polymorphic sites and π are obtained from the sequence alignment that exclude gaps and a chromosome sample of up to 10 sequences (see text). Synonymous sites of the csd coding sequence were used for calculations at Ex2/3, Ex4/5, and Ex6/7 and distances are calculated from the 3′-end of csd. Recombination rates for loci linked to SDL are based on mapping data (see Figure 1). For loci unlinked to SDL, recombination rates were estimated in 125-kb windows by a genomewide analysis of the honeybee sequence assembly 2.1. Putatively neutral loci were chosen on the basis of the region corresponding to those windows in the annotated genome.
The analysis of π at synonymous sites within csd (Figure 1A) shows a similar but weaker decline with distance (Figure 1D). The highest diversity is found in exon 6/7, which is 4 times higher than that of KF22 [π = 0.0918 vs. π = 0.0233 (Table 1); Z-test, Z = 3.1; two-tailed: P < 0.01)], 7 times higher than that of exon 4/5 [π = 0.0918 vs. π = 0.0135 (Table 1); Z-test, Z = 3.4; two tailed: P < 0.01], and 17 times higher than our estimate of the genomewide average [π = 0.0918 vs. π = 0.0055 (Table 1); Z-test, Z = 4.0; two tailed: P < 0.01]. The striking differences suggest that exon 6/7 is a strong target of balancing selection within the csd gene.
Signatures of intragenic recombination among csd sequences:
No recombination was observed within the region of SDL containing the sex determination gene csd (Figure 1B). To explore whether recombination has contributed to the evolution of sex-determining alleles in the past, we searched for signatures of recombination in the coding sequences of csd.
Signatures of recombination in long segments of the sequence:
Tests of heterogeneity were performed on the combined exons, exon 2+3, exon 4+5, and exons 6–9. These combined sequences represent the specific clustering of exons in the genome (Figure 1A). The rationale of combining these exon sequences is that they are separated by large introns, which enhances the potential to detect signatures of recombination. Heterogeneity of nucleotide differences across exons was explored using the neighbor-joining method (Figure 2). Differences in the lengths of terminal branches illustrate that some alleles are more similar when differences in exon 2+3 and 4+5 sequences are compared (Figure 2, A and B; indicated by a shaded bar), but are more diverged when exon 6–9 sequences are examined (Figure 2C).
Figure 2.—

Genealogies of exon 2+3 (A), exon 4+5 (B), and exon 6–9 (C) sequences of csd and type II (B2-17). The genealogy is based on a neighbor-joining analysis of differences calculated by the Kimura 2-parameter distance model. Bootstrap percentages >60 are indicated. Gaps in the overall sequence alignment were completely excluded in the analysis as they comprise ambiguous similarities. Sequences that are separated by short branch distances are apparently more related and marked with shaded bars (for further analysis see Table 2). The scale bar indicates nucleotide differences per site.
This preliminary analysis leads us to a closer examination of pairwise differences among silent, synonymous and nonsynonymous sites. When one region shows less divergence than expected based on other regions, this was taken as evidence of recombination. This analysis supports our preliminary findings. Pairwise differences of exon 2+3 sequences that are more similar in the above analysis (the so-called “similar” exon 2+3 group, which is marked by a shaded bar in Figure 2A: D1-22, D1-18, D2-38, S2-31, A5, A1-18, and S7-5) are significantly less diverged than sequences of exons 6–9, irrespective of whether synonymous or nonsynonymous sites are compared (see lines 1–8 in Table 2 for representative examples). Tests of heterogeneity that involve exon 2+3 sequences of the so-called “diverged” group (nonmarked alleles of Figure 2A: S2-33, A1-28, S7-16, B1-4, B2-25, D1-16, and A2-8) show an inconsistent pattern. Most sequences show no heterogeneity, irrespective of whether they are compared to sequences of the “similar” or the “diverged” group (representative examples lines 9–15 and lines 16–18, respectively; Table 2). Although a lack of heterogeneity is a trend for these groups of sequences, ∼30% of comparisons demonstrate heterogeneity, which again do not depend on whether synonymous or nonsynonymous sites are analyzed (e.g., B2-25 and D2-38; example 15 in Table 2), indicating a miscellaneous relation of these sequences. Heterogeneity is less frequent in exon 4+5 when compared to exon 2+3 comparisons (Table 2, lines 4 and 7). Other exon 4+5 sequences, however, are very similar to exon 2+3 sequences (see the shaded bars in Figure 2B and representative examples in Table 2, lines 1 and 6). Taken together, these analyses suggest that a considerable fraction of alleles share a more recent ancestral exon 2+3 sequence, an occurrence that is less frequently detected among exon 4+5 sequences. This finding is consistent with sporadic recombination, the likelihood of which increases with larger distances from exons 6–9, where balancing selection operates most strongly.
TABLE 2.
Examples of pairwise differences of exon 2+3, 4+5, and 6–9 sequences and tests of heterogeneity
| Exon 2+3
|
Exon 4+5
|
Exons 6–9
|
||||||
|---|---|---|---|---|---|---|---|---|
| Allele pair | ds | dn | ds | dn | ds | dn | ||
| 1 | D1-22 | D2-38 | 0.0 ± 0.0** | 0.0056 ± 0.004** | 0.0 ± 0.0** | 0.0 ± 0.0** | 0.0698 ± 0.0267 | 0.0711 ± 0.015 |
| 2 | A-5 | S7-5 | 0.0102 ± 0.0102* | 0.0028 ± 0.0028** | 0.0 ± 0.0** | 0.0197 ± 0.0114** | 0.0656 ± 0.026 | 0.0564 ± 0.0132 |
| 3 | S2-31 | S7-5 | 0.0102 ± 0.0102** | 0.0028 ± 0.0028** | 0.0231 ± 0.023* | 0.0197 ± 0.0114* | 0.0967 ± 0.0319 | 0.0534 ± 0.0132 |
| 4 | D1-22 | A1-18 | 0.0 ± 0.0** | 0.0028 ± 0.0028** | 0.0233 ± 0.023** | 0.0331 ± 0.0149 | 0.0869 ± 0.0303 | 0.0564 ± 0.0132 |
| 5 | A-5 | S2-31 | 0.0 ± 0.0* | 0.0 ± 0.0** | 0.0229 ± 0.023 | 0.0 ± 0.0** | 0.0505 ± 0.0261 | 0.0421 ± 0.0113 |
| 6 | D1-18 | S2-31 | 0.0102 ± 0.0102** | 0.0 ± 0.0** | 0.0 ± 0.0** | 0.0 ± 0.0** | 0.0706 ± 0.027 | 0.033 ± 0.01 |
| 7 | D2-38 | A1-18 | 0.0 ± 0.0* | 0.0028 ± 0.0028** | 0.0233 ± 0.023 | 0.0331 ± 0.0149 | 0.0546 ± 0.0235 | 0.0502 ± 0.0125 |
| 8 | D2-38 | S2-31 | 0.0 ± 0.0** | 0.0028 ± 0.0028** | 0.0115 ± 0.016 | 0.0098 ± 0 008 | 0.0096 ± 0.0096 | 0.0301 ± 0.0096 |
| 9 | S2-33 | D1-18 | 0.0312 ± 0.0181 | 0.017 ± 0.0069 | 0.0229 ± 0.023* | 0.0 ± 0.0** | 0.07 ± 0.027 | 0.0362 ± 0.0105 |
| 10 | B1-4 | D1-22 | 0.0205 ± 0.0146 | 0.0227 ± 0.008 | 0.0 ± 0.0** | 0.0263 ± 0.0132 | 0.0543 ± 0.0234 | 0.0164 ± 0.007 |
| 11 | D1-16 | D1-22 | 0.0415 ± 0.0209 | 0.0199 ± 0.0075 | 0.0115 ± 0.0162 | 0.0098 ± 0.008* | 0.0594 ± 0.0245 | 0.0332 ± 0.01 |
| 12 | A2-8 | S7-16 | 0.042 ± 0.0212 | 0.0198 ± 0.0075 | 0.023 ± 0.023 | 0.0198 ± 0.0114* | 0.0642 ± 0.0255 | 0.041 ± 0.0112 |
| 13 | S7-16 | D1-16 | 0.0637 ± 0.0263 | 0.0227 ± 0.008 | 0.023 ± 0.023 | 0.0 ± 0.0** | 0.0492 ± 0.022 | 0.021 ± 0.008 |
| 14 | D1-16 | S2-31 | 0.0415 ± 0.0209 | 0.017 ± 0.007 | 0.0 ± 0.0** | 0.0 ± 0.0** | 0.07 ± 0.027 | 0.0301 ± 0.0096 |
| 15 | B2-25 | D2-38 | 0.0205 ± 0.0145* | 0.0227 ± 0.008** | 0.0 ± 0.0** | 0.0332 ± 0.0149 | 0.0952 ± 0.0314 | 0.0601 ± 0.0137 |
| 16 | D1-16 | B1-4 | 0.0416 ± 0.021 | 0.0141 ± 0.0063 | 0.0231 ± 0.023 | 0.0197 ± 0.0114 | 0.024 ± 0.015 | 0.023 ± 0.008 |
| 17 | B1-4 | A2-8 | 0.0206 ± 0.0146 | 0.017 ± 0.0069 | 0.0232 ± 0.023 | 0.0131 ± 0.0093 | 0.0688 ± 0.0263 | 0.021 ± 0.008 |
| 18 | S2-33 | D1-16 | 0.0205 ± 0.0146 | 0.0141 ± 0.0063 | 0.0229 ± 0.023 | 0.0 ± 0.0 | 0.0488 ± 0.022 | 0.021 ± 0.008 |
Mean pairwise differences were separately calculated for synonymous (ds) and nonsynonymous (dn) sites along with their standard errors. Heterogeneity of differences across these groups of exons was analyzed by the Z-test. Representative examples of the heterogeneity test are the following: lines 1–8 are examples of differences between sequences that have the most similar 5′-ends in the explorative data analysis (marked with a shaded bar in Figure 2A); lines 9–15 are examples of pairwise comparisons between the two groups that have more similar and more diverged exon 2+3 sequences in the genealogy analysis (shaded bar sequences vs. nonmarked sequences of Figure 2A). The final lines (16–18) are comparisons among the sequences that have diverged exon 2+3 sequences in the genealogy (nonmarked sequences of Figure 2A). Test of the hypothesis that ds or dn of exon 2+3 or exon 4+5 equals the corresponding value for exons 6–9; *P < 0.05 and **P < 0.01.
LD of polymorphic sites among csd alleles was evaluated using R2 and D′ estimates. The more distant two segregating sites are, the greater the probability that recombination has occurred. Under this model, we would expect to find a decay of LD with increasing distance between sites. The analysis (Table 3) was conducted on variants with a frequency of >10%, for all sites, and on synonymous and silent sites separately. When all sites are included, LD slightly decreases with physical distance for R2 (r = −0.13, P < 0.001), which is only marginally significant for D′ (r = −0.08, P < 0.08). If the analysis is restricted to only synonymous sites, a correlation is supported only for D′ (r = −0.4, P < 0.01). A previous study (Innan and Nordborg 2002) argued that heterogeneity across a sequence can mimic a decline of LD with distance. Thus, we analyzed exons 2–5 and exons 6–9 separately, as heterogeneity has been identified in these sequences (Hasselmann and Beye 2004). LD strongly decays with physical distance in exon 2–5 sequences when all differences are included (r = −0.5 for R2 and D′, P < 0.001). The four synonymous sites follow the strong negative trend, but are too few to support the finding. There is support for a weak decline of R2 with distance in exon 6–9 sequences when all sites are included (r = −0.18, P < 0.001), although this is not supported by D′ (r = −0.09, P < 0.1). LD decreases even more strongly in exons 6–9 when only synonymous sites are analyzed (r = −0.37, P < 0.05 for R2 and r = −0.56, P < 0.01 for D′); however, this estimate is based on only seven sites. Combined with the former results of the heterogeneity test, the LD analysis supports the operation of recombination among exon 2–5 sequences.
TABLE 3.
Pearson correlation coefficient (r) of LD, R2, and D′ and distances of sites
|
r (Pearson correlation coefficient)
|
||||
|---|---|---|---|---|
| Region | Sites | No. | R2 | D′ |
| Exons 2–9 | Polymorphic | 51 | −0.13*** | −0.08 |
| Synonymous | 13 | −0.06 | −0.4** | |
| Exons 2–5 | Polymorphic | 17 | −0.5*** | −0.5*** |
| Synonymous | 4 | −0.7* | −0.97 | |
| Exons 6–9 | Polymorphic | 34 | −0.18*** | −0.09 |
| Synonymous | 7 | −0.37* | −0.56** | |
The analysis was performed on polymorphic and synonymous sites and on different parts of the sequence (coding sequence of exons 2–9, exons 2–5, exons 6–9). Only polymorphic sites that have a frequency >10% were included in the analysis. The significance of r was determined on the basis of 5000 random permutations of polymorphic sites. The type II sequence was not included in the analysis. *P < 0.05, **P < 0.01, ***P < 0.001.
Signatures of recombination in short segments of the sequence:
We applied three tests: the Stephens test (Stephens 1985), Sawyer's test (Sawyer 1999), and a maximum-composite-likelihood method (Hudson 2001) to examine whether short clusters of polymorphisms are shared among alleles more often than expected. This would indicate potential gene conversion as a result of recombination. No clusters were detected in the Sawyer's test. The Stephens test, however, identified three clusters of shared polymorphic sites of exons 6–9 (clusters 1 and 2) and exon 2+3 (cluster 3). The specific sites are shown in italics within their inferred codons (Table 4). The rationale behind this test is to determine whether a collection of shared polymorphic sites is consistent with a model of building two groups of sequences, the majority and minority groups. Nine alleles share the majority motif of cluster 1, which consists of five nucleotides (position 815–819), while five alleles have the minority motif. Cluster 2 includes two polymorphic sites (908, 910) that are shared among eight and six alleles. Cluster 3 consists of three more distant sites (positions 20, 70, and 120) that are present in nine and five sequences. In total, 80% of shared polymorphic sites code for amino acid differences (noncoding sites are underlined in Table 4). The Stephens test indicates a minor role for gene conversion in sequence evolution as only 10% of all polymorphic were detected. Furthermore, we tested whether the three clusters are randomly associated across alleles. Table 5 shows the amino acids encoded by the clusters and their associations. Associations did not deviate from that expected at random (χ2 = 0.14, P = 0.7). Given that the clusters are not the result of recurrent mutations, but rather of a single event of gene conversion, random association of these clusters is a further sign that recombination has operated among alleles. The minor, if any, contribution of gene conversion is further supported by the maximum-composite-likelihood analysis (Hudson 2001). The maximum composite likelihood of exons 2–5 with the model of no gene conversion (f = 0) and the most likely gene conversion model (10% gene conversion: f = 0.1), were nearly identical (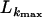 = −722; Table 6). The distribution of sites can be explained either by recombination alone or by recombination plus a small contribution of gene conversion, irrespective of the various trace lengths of the gene conversion model (L = 50, 100, and 500 bp). Similar results were obtained for exons 6–9, although the contribution of recombination was far less dominant (
= −722; Table 6). The distribution of sites can be explained either by recombination alone or by recombination plus a small contribution of gene conversion, irrespective of the various trace lengths of the gene conversion model (L = 50, 100, and 500 bp). Similar results were obtained for exons 6–9, although the contribution of recombination was far less dominant (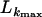 = −2955.35). Taken together, these analyses suggest that gene conversion has not played an important role in the sequence evolution of csd alleles.
= −2955.35). Taken together, these analyses suggest that gene conversion has not played an important role in the sequence evolution of csd alleles.
TABLE 4.
Clusters of shared polymorphic sites (Stephens test)
| Cluster | Nucleotide position | Deduced codons (majority/ minority) | Majority/ minority alleles | P |
|---|---|---|---|---|
| 1 | 815–819 | AGA GAA/AAT TCT | 9:5 | <0.0001 |
| 2 | 908, 910 | ATT AAT/AAT TAT | 8:6 | <0.005 |
| 3 | 20 | AG T/AA T | 9:5 | <0.05 |
| 70 | G AA/A AA | |||
| 120 | CGT/CGC |
Sites of the majority and minority motifs are shown in italics within their inferred codons. Synonymous sites within clusters are underlined. Positions of sites are assigned according to the overall sequence alignment.
TABLE 5.
Association of the Stephens clusters (the deduced amino acids are shown) across 14 csd haplotypes
| Stephens test cluster 3
|
Stephens test cluster 1
|
Stephens test cluster 2
|
||||||
|---|---|---|---|---|---|---|---|---|
| csd type I sequence | Amino acid position: | 7 | 24 | 40 | 272 | 273 | 303 | 304 |
| B2-25 | N | K | R | N | S | N | Y | |
| A2-8 | N | K | R | N | S | I | N | |
| B 1-4 | N | K | R | N | S | I | N | |
| S2-33 | N | K | R | R | E | I | N | |
| D1-16 | N | K | R | R | E | I | N | |
| S7-16 | S | E | R | R | E | N | Y | |
| A1-28 | S | E | R | R | E | N | Y | |
| A1-18 | S | E | R | R | E | N | Y | |
| D2-38 | S | E | R | R | E | N | Y | |
| A-5 | S | E | R | R | E | I | N | |
| S2-31 | S | E | R | R | E | I | N | |
| D1-18 | S | E | R | R | E | N | Y | |
| S7-5 | S | E | R | N | S | I | N | |
| D1-22 | S | E | R | N | S | I | N | |
Amino acids of the majority motif are in italics. Positions of the amino acids are assigned according to the protein alignment with gaps excluded.
TABLE 6.
Maximum-composite-likelihood analysis of exon 2–5 and exon 6–9 sequences without and with gene conversion models
| Region | L (bp) | f | 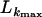 |
|---|---|---|---|
| Exons 2–5 | — | 0 | −722.33 |
| 50 | 0.1 | −722.74 | |
| 100 | 0.1 | −722.91 | |
| 500 | 0.1 | −722.67 | |
| Exons 6–9 | — | 0 | −2955.35 |
| 50 | 0.1 | −2955.46 | |
| 100 | 0.1 | −2955.41 | |
| 500 | 0.1 | −2955.38 |
Models of crossing over without (f = 0; no track length, L) and with gene conversion (f > 0; L = 50, 100, 500 bp) were applied. f-values that maximize the composite likelihood are shown. Polymorphic sites with more than two alleles were ignored in the analysis.
DISCUSSION
Recombination reduces association of linked sites:
The decline of nucleotide diversity with increasing distance from SDL (Figure 1D) suggests that linkage to SDL is the driving force maintaining higher levels of nucleotide polymorphism at closely linked sites. An alternative explanation that a higher recombination rate (Lercher and Hurst 2002) in proximity of SDL results in a higher mutation rate is an unlikely scenario. Regions unlinked to SDL that have the same high rate still show a fivefold lower nucleotide diversity than linked loci, indicating that recombination alone cannot explain this difference. In addition, nucleotide diversity is higher within SDL, although the recombination rate is substantially reduced at this locus. The most plausible explanation is that loci linked to csd have been maintained over extended periods of time as a consequence of balancing selection operating at this locus. As a result, linked loci have accumulated more differences over time in comparison to nonlinked, neutral variants (Hasselmann and Beye 2004). Consistent with this hypothesis, nucleotide differences at linked loci do not exceed the polymorphism levels found within csd. However, with increasing distance from SDL, recombination decouples and relaxes the impact of balancing selection, leading to a rapid decline of polymorphism within the first 45 kb downstream. Similar effects of a decline of nucleotide polymorphism at linked neutral sites have been documented at the HLA-B locus, a gene of the MHC complex that is under balancing selection (O'hUigin et al. 2000). This decay is consistent with the predictions of population genetics models that describe levels of polymorphism at linked sites of the MHC locus with respect to recombination rates (Takahata and Satta 1998b). Nucleotide polymorphism also declines within the csd gene with increasing distance to exon 6/7, but at a lower rate. More than fourfold more differences are found in exon 6/7 when compared to other parts of the csd. This suggests that this part has been maintained over an extended period of time compared to other exons. Balancing selection operates most strongly in this region, leading to the maintenance of polymorphism. Recombination relaxes linkage to exon 6/7, leading to a decline in silent differences when moving toward the 5′ part of the csd gene.
We have estimated local recombination activity within and around SDL on the basis of a large mapping population and defined physical fragments. The recombination rate increases from 2.5–5 times higher on both sides of SDL within a distance of 50–215 kb. This local increase is even more dramatic when the unobserved recombination within SDL and the high genomewide rates of recombination (Hunt and Page 1995; Solignac et al. 2004) are taken into account. These defined and pronounced differences suggest a position-specific control of recombination and demand an explanation related to SDL function and balancing mode of selection. Mechanistic explanations such as repair (Bernstein et al. 1981) or the stabilization of chromosome segregation (Baker et al. 1976; Hawley and Theurkauf 1993) cannot explain these distinct differences within several kilobases. No obvious sequence characteristics associated with the severalfold recombination differences have been detected, which would support a direct mechanistic interpretation of recombination activity. The strength of selection at SDL (Yokoyama and Nei 1979) combined with the finding that rates of recombination can change in response to selection (Michod and Levin 1988; Otto and Barton 2001) suggests instead an evolutionary explanation. Recombination could be selectively favored to reduce interference between SDL and linked sites or genes. On average, linked genes likely experience selective pressures other than balancing selection. Recombination reduces conflicts by relaxing linkage between SDL and flanking genes, which in turn may improve the response to selection.
The conflict of selection could be rather strong at csd as genetic drift, which generates more frequent and more rare sex-determining alleles, is a dominant evolutionary force in honeybees. Rates of genetic drift are dependent on the effective size of a population. Honeybees have small local breeding populations (the queen is the only reproductive female) when compared to nonsocial insects (Crozier 1979; Pamilo and Crozier 1997). Several identified genes in close linkage (data not shown), upstream and downstream of csd, could be a source of selective conflicts.
In agreement with the evolutionary interpretation of an increase of recombination activity to decrease the selective interference among loci that have different selection regimes, several hot spots of recombination have been identified in the human MHC class II gene cluster (Cullen et al. 2002). Recombination activity increases up to 100 times the genomewide average in several regions of this gene complex. In contrast to SDL, the increase of activity is found in different parts of the MHC gene cluster and is confined to small genomic regions of only several kilobases (Jeffreys et al. 2001). The evolutionary interpretation of the MHC study is obviously more complex, as several genes of the MHC are under balancing selection while others are not (the MHC class II complex comprises >15 genes) (Garrigan and Edwards 1999; Beck and Trowsdale 2000) and the strength of balancing selection differs among genes (Meyer and Thomson 2001).
No recombination activity was detected within SDL, suggesting that recombination is reduced or even suppressed. This finding contradicts a previous hypothesis (Beye et al. 1999) that an increase of recombination activity among SDL alleles could improve the effectiveness of removing deleterious mutations (Kondrashov 1984; Rice 2002). A reduction or even a suppression of recombination will preserve allelic differences, specificity, and function as recombination is a homogenizing force that will generate more similar sex-determining alleles over time. An unanswered question is, Why is the suppression of recombination not precisely restricted to the 9-kb genomic region of csd, the initial signal of complementary sex determination that is under balancing selection, but is also found in an additional 27 kb of genomic sequence?
Signatures of intragenic recombination in csd sequences:
Although we have no direct evidence of recombination activity within SDL, we have detected signatures of recombination among some 14 csd sequences. Tests of heterogeneity identified eight alleles most similar in exon 2+3 that extend in similarity to exon 4+5 for some allelic combinations. This is consistent with sporadic recombination that is more frequently detected with larger distances between exons. Concerted evolution and recurrent mutation could also lead to heterogeneity across haplotypes, although they are unlikely forces on the basis of the number of differences that were analyzed. In addition, weaker diversifying selection and stronger selective constraint on exon 2–5 sequences are unlikely alternative explanations as (i) silent sites show the same pattern; (ii) heterogeneity is absent in several alleles (e.g., Table 2, lines 16–18), arguing against a ubiquitous selective force but favoring a random process such as recombination; and (iii) the decline of LD with physical distance strengthens a model of recombination, but is not a plausible explanation under various models of selection. GC content or codon bias could homogenize sequences (Hughes et al. 1993), but should effect all alleles similarly. In contrast to exon 2+3 and exon 4+5 sequences, there is only weak support for recombination within exon 6–9 sequences; all exon 6–9 sequences are highly diverged. There is, however, support for a decay of LD with distance in some tests, which indicates a potential weak role of recombination within exons 6–9.
The Sawyer's test, the Stephens test, and the maximum-composite-likelihood analysis provide little evidence that gene conversion has substantially contributed to the evolution of csd alleles. The contribution is rather small, if any, comprising <10% of nucleotide differences as shown in the Stephens test analysis. In contrast to SDL, gene conversion has contributed substantially to the evolution of MHC (Hughes et al. 1993; Ohta 1997; Takahata and Satta 1998a; Hogstrand and Bohme 1999) and self-incompatibility alleles (S-alleles) (Kusaba et al. 1997; Wang et al. 2001). For example, almost all variation of HLA-DPB1 haplotypes in humans (Moonsamy et al. 1992; Zangenberg et al. 1995) is confined to short clusters that are possibly the result of gene conversion. Statistical analysis of mouse MHC class I genes using the Sawyer's and the Stephens test identified 25 clusters that could be the products of gene conversion (Kuhner et al. 1990). Even though signatures of gene conversion in these sequences are quite common, the question of whether gene conversion has adaptive significance in the rise of new, functional allelic MHC variants, or is instead the consequence of the basic genetic process of recombination, is not yet settled (Martinsohn et al. 1999).
In summary, the analyses of heterogeneity, LD, and shared sites suggest that recombination has contributed to the evolution of exon 2–5, but not exon 6–9 sequences. The finding that not all alleles are affected supports a suppression of recombination within SDL so that recombination operates only sporadically. Hence, recombination is not a dominant evolutionary force in the short term, but has contributed to the sequence evolution of some csd alleles in the long term. This interpretation is in agreement with functional considerations that higher levels of recombination are a strong homogenizing force that makes alleles more similar and thus less functional, or even nonfunctional. Exons 6–9 show no signatures of recombination that would favor the idea that this region encodes the allelic specificity, while the exon 2–5 sequences are less constrained. Although very few signatures of gene conversion have been detected, these clusters are randomly associated across alleles (Table 5 shows the associations and the different amino acids that the clusters encode) and could be a potential source of allelic differentiation. Further molecular studies could test whether these clusters contribute to the specificity and functioning of sex- determining alleles.
Acknowledgments
We thank John Baines, Morten Schioett, and two anonymous reviewers for critical comments on the manuscript and Irene Gattermeier and Stefan Sroka for cloning and sequencing help. This research was funded by grants from the Deutsche Forschungsgemeinschaft (2194/5, 2194/6) to M.B.
References
- Andolfatto, P., 2001. Adaptive hitchhiking effects on genome variability. Curr. Opin. Genet. Dev. 11: 635–641. [DOI] [PubMed] [Google Scholar]
- Awadalla, P., and D. Charlesworth, 1999. Recombination and selection at Brassica self-incompatibility loci. Genetics 152: 413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, B. S., A. T. C. Carpenter, M. S. Esposito, R. E. Esposito and L. Sandler, 1976. Genetic control of meiosis. Annu. Rev. Genet. 10: 53–134. [DOI] [PubMed] [Google Scholar]
- Beck, S., and J. Trowsdale, 2000. The human major histocompatability complex: lessons from the DNA sequence. Annu. Rev. Genomics Hum. Genet. 1: 117–137. [DOI] [PubMed] [Google Scholar]
- Begun, D. J., and C. F. Aquadro, 1992. Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster. Nature 356: 519–520. [DOI] [PubMed] [Google Scholar]
- Bernstein, H., G. S. Byers and R. E. Michod, 1981. Evolution of sexual reproduction: importance of DNA repair, complementation, and variation. Am. Nat. 117: 537–549. [Google Scholar]
- Beye, M., and R. F. A. Moritz, 1994. Sex linkage in the honeybee Apis mellifera detected by multilocus DNA fingerprinting. Naturwissenschaften 81: 460–462. [DOI] [PubMed] [Google Scholar]
- Beye, M., G. J. Hunt, R. E. Page, M. K. Fondrk, L. Grohmann et al., 1999. Unusually high recombination rate detected in the sex locus region of the honey bee (Apis mellifera). Genetics 153: 1701–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beye, M., M. Hasselmann, M. K. Fondrk, R. E. Page, Jr. and S. W. Omholt, 2003. The Gene csd is the primary signal for sexual development in the honey bee and encodes a SR-type protein. Cell 114: 419–429. [DOI] [PubMed] [Google Scholar]
- Beye, M., I. Gattermeier, M. Hasselmann, T. Gempe, M. Schioett et al., 2006. Exceptionally high levels of recombination across the honey bee genome. Genome Res. 16: 1339–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, B., M. T. Morgan and D. Charlesworth, 1993. The effect of deleterious mutations on neutral molecular variation. Genetics 134: 1289–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, D., 2004. Sex determination: balancing selection in the honey bee. Curr. Biol. 14: R568–R569. [DOI] [PubMed] [Google Scholar]
- Cheng, K. C., and G. R. Smith, 1984. Recombinational hotspot activity of chi-like sequences. J. Mol. Biol. 180: 371–377. [DOI] [PubMed] [Google Scholar]
- Crozier, R. H., 1979. Genetics of sociality, pp. 223–286 in Social Insects, edited by H. R. Hermann. Academic Press, New York.
- Cullen, M., S. P. Perfetto, W. Klitz, G. Nelson and M. Carrington, 2002. High-resolution patterns of meiotic recombination across the human major histocompatibility complex. Am. J. Hum. Genet. 71: 759–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein, J., 1974. The evolutionary advantage of recombination. Genetics 78: 737–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisse, L., R. R. Hudson, A. Bartoszewicz, J. D. Wall, J. Donfack et al., 2001. Gene conversion and different population histories may explain the contrast between polymorphism and linkage disequilibrium levels. Am. J. Hum. Genet. 69: 831–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrigan, D., and S. V. Edwards, 1999. Polymorphism across an exon-intron boundary in an avian Mhc class II B gene. Mol. Biol. Evol. 16: 1599–1606. [DOI] [PubMed] [Google Scholar]
- Hall, T. A., 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41: 95–98. [Google Scholar]
- Hasselmann, M., and M. Beye, 2004. Signatures of selection among sex-determining alleles of the honey bee. Proc. Natl. Acad. Sci. USA 101: 4888–4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselmann, M., M. K. Fondrk, R. E. Page, Jr. and M. Beye, 2001. Fine scale mapping in the sex locus region of the honey bee (Apis mellifera). Insect Mol. Biol. 10: 605–608. [DOI] [PubMed] [Google Scholar]
- Hawley, R. S., and W. E. Theurkauf, 1993. Requiem for distributive segregation: achiasmate segregation in Drosophila females. Trends Genet. 9: 310–317. [DOI] [PubMed] [Google Scholar]
- Hogstrand, K., and J. Bohme, 1999. Gene conversion can create new MHC alleles. Immunol. Rev. 167: 305–317. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., 2001. Two-locus sampling distributions and their application. Genetics 159: 1805–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, A. L., M. K. Hughes and D. I. Watkins, 1993. Contrasting roles of interallelic recombination at the HLA-A and HLA-B loci. Genetics 133: 669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt, G. J., and R. E. Page, 1994. Linkage analysis of sex determination in the honey bee (Apis mellifera). Mol. Gen. Genet. 244: 512–518. [DOI] [PubMed] [Google Scholar]
- Hunt, G. J., and R. E. Page, 1995. Linkage map of the honey bee, Apis mellifera, based on RAPD markers. Genetics 139: 1371–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innan, H., and M. Nordborg, 2002. Recombination or mutational hot spots in human mtDNA? Mol. Biol. Evol. 19: 1122–1127. [DOI] [PubMed] [Google Scholar]
- Jeffreys, A. J., L. Kauppi and R. Neumann, 2001. Intensely punctate meiotic recombination in the class II region of the major histocompatibility complex. Nat. Genet. 29: 217–222. [DOI] [PubMed] [Google Scholar]
- Kim, Y., and W. Stephan, 2003. Selective sweeps in the presence of interference among partially linked loci. Genetics 164: 389–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, M., 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16: 111–120. [DOI] [PubMed] [Google Scholar]
- Kimura, M., and J. F. Crow, 1964. The number of alleles that can be maintained in a finite population. Genetics 49: 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov, A. S., 1984. Deleterious mutations as an evolutionary factor. 1. The advantage of recombination. Genet. Res. 44: 199–217. [DOI] [PubMed] [Google Scholar]
- Kosambi, D. D., 1944. The estimation of map distances from recombination values. Ann. Eugen. 12: 172–175. [Google Scholar]
- Kuhner, M., S. Watts, W. Klitz, G. Thomson and R. S. Goodenow, 1990. Gene conversion in the evolution of both the H-2 and Qa class I genes of the murine major histocompatibility complex. Genetics 126: 1115–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S., K. Tamura, I. B. Jakobsen and M. Nei, 2001. MEGA2: molecular evolutionary genetics analysis software. Bioinformatics 17: 1244–1245. [DOI] [PubMed] [Google Scholar]
- Kusaba, M., T. Nishio and Y. Satta, 1997. Striking sequence similarity in inter- and intra-specific comparisons of class I SLG alleles from Brassica oleracea and Brassica campestris: implications for the evolution and recognition mechanism. Proc. Natl. Acad. Sci. USA 94: 7673–7678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lercher, M. J., and L. D. Hurst, 2002. Human SNP variability and mutation rate are higher in regions of high recombination. Trends Genet. 18: 337–340. [DOI] [PubMed] [Google Scholar]
- Mackensen, O., 1955. Further studies on a lethal series in the honeybee. J. Hered. 46: 72–74. [Google Scholar]
- Martinsohn, J. T., A. B. Sousa, L. A. Guethlein and J. C. Howard, 1999. The gene conversion hypothesis of MHC evolution: a review. Immunogenetics 50: 168–200. [DOI] [PubMed] [Google Scholar]
- Maynard Smith, J., and J. Haigh, 1974. The hitch-hiking effect of a favorable gene. Genet. Res. 23: 23–35. [PubMed] [Google Scholar]
- Meiklejohn, C. D., Y. Kim, D. L. Hartl and J. Parsch, 2004. Identification of a locus under complex positive selection in Drosophila simulans by haplotype mapping and composite-likelihood estimation. Genetics 168: 265–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, D., and G. Thomson, 2001. How selection shapes variation of the human major histocompatibility complex: a review. Ann. Hum. Genet. 65: 1–26. [DOI] [PubMed] [Google Scholar]
- Michod, R. E., and B. R. Levin, 1988. The Evolution of Sex. Sinauer Press, Sunderland, MA.
- Moonsamy, P. V., V. C. Suraj, T. L. Bugawan, R. K. Saiki, M. Stoneking et al., 1992. Genetic diversity within the HLA class II region: ten new DPB1 alleles and their population distribution. Tissue Antigens 40: 153–157. [DOI] [PubMed] [Google Scholar]
- Nachman, M. W., H. E. Hoekstra and S. L. D'Agostino, 2003. The genetic basis of adaptive melanism in pocket mice. Proc. Natl. Acad. Sci. USA 100: 5268–5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei, M., 1987. Molecular Evolutionary Genetics. Columbia University Press, New York.
- Ohta, T., 1997. Role of gene conversion in generating polymorphisms at major histocompatibility complex loci. Hereditas 127: 97–103. [DOI] [PubMed] [Google Scholar]
- O'hUigin, C., Y. Satta, A. Hausmann, R. L. Dawkins, and J. Klein, 2000. The implications of intergenic polymorphism for major histocompatibility complex evolution. Genetics 156: 867–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto, S. P., and N. H. Barton, 2001. Selection for recombination in small populations. Evolution 55: 1921–1931. [DOI] [PubMed] [Google Scholar]
- Otto, S. P., and T. Lenormand, 2002. Resolving the paradox of sex and recombination. Nat. Rev. Genet. 3: 252–261. [DOI] [PubMed] [Google Scholar]
- Pamilo, P., and R. H. Crozier, 1997. Population biology of social insect conservation. Mem. Mus. Victoria 56: 411–419. [Google Scholar]
- Patthy, L., 1996. Exon shuffling and other ways of module exchange. Matrix Biol. 15: 301–310. [DOI] [PubMed] [Google Scholar]
- Posada, D., K. A. Crandall and E. C. Holmes, 2002. Recombination in evolutionary genomics. Annu. Rev. Genet. 36: 75–97. [DOI] [PubMed] [Google Scholar]
- Rice, W. R., 1998. Requisite mutational load, pathway epistasis and deterministic mutation accumulation in sexual versus asexual populations. Genetica 102/103: 71–81. [PubMed] [Google Scholar]
- Rice, W. R., 2002. Experimental tests of the adaptive significance of sexual recombination. Nat. Rev. Genet. 3: 241–251. [DOI] [PubMed] [Google Scholar]
- Rozas, J., and R. Rozas, 1999. DnaSP vers. 3: an integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics 15: 174–175. [DOI] [PubMed] [Google Scholar]
- Sawyer, S., 1989. Statistical tests for detecting gene conversion. Mol. Biol. Evol. 6: 526–538. [DOI] [PubMed] [Google Scholar]
- Sawyer, S., 1999. GENECONV: A Computer Package for the Statistical Detection of Gene Conversion. http://www.math.wustl.edu/∼sawyer.
- Schlötterer, C., 2003. Hitchhiking mapping: functional genomics from the population genetics perspective. Trends Genet. 19: 32–38. [DOI] [PubMed] [Google Scholar]
- Solignac, M., D. Vautrin, E. Baudry, F. Mougel, A. Loiseau et al., 2004. A microsatellite-based linkage map of the honeybee, Apis mellifera L. Genetics 167: 253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens, J. C., 1985. Statistical methods of DNA sequence analysis: detection of intragenic recombination or gene conversion. Mol. Biol. Evol. 2: 539–556. [DOI] [PubMed] [Google Scholar]
- Takahata, N., and Y. Satta, 1998. a Footprints of intragenic recombination at HLA loci. Immunogenetics 47: 430–441. [DOI] [PubMed] [Google Scholar]
- Takahata, N., and Y. Satta, 1998. b Selection, convergence, and intragenic recombination in HLA diversity. Genetica 102/103: 157–169. [PubMed] [Google Scholar]
- Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin and D. G. Higgins, 1997. The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 24: 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd, A. E., C. A. Orengo and J. M. Thornton, 2001. Evolution of function in protein superfamilies, from a structural perspective. J. Mol. Biol. 307: 1113–1143. [DOI] [PubMed] [Google Scholar]
- Vieira, C. P., D. Charlesworth and J. Vieira, 2003. Evidence for rare recombination at the gametophytic self-incompatibility locus. Heredity 91: 262–267. [DOI] [PubMed] [Google Scholar]
- Wang, X., A. L. Hughes, T. Tsukamoto, T. Ando and T. H. Kao, 2001. Evidence that intragenic recombination contributes to allelic diversity of the S-RNase gene at the self-incompatibility (S) locus in Petunia inflata. Plant Physiol. 125: 1012–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama, S., and M. Nei, 1979. Population dynamics of sex determining alleles in honey bees and self-incompatibility in plants. Genetics 91: 609–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuhki, N., and S. J. O'Brien, 1994. Exchanges of short polymorphic DNA segments predating speciation in feline major histocompatibility complex class I genes. J. Mol. Evol. 39: 22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zangenberg, G., M. M. Huang, N. Arnheim and H. Erlich, 1995. New HLA-DPB1 alleles generated by interallelic gene conversion detected by analysis of sperm. Nat. Genet. 10: 407–414. [DOI] [PubMed] [Google Scholar]
- Zhang, J., A. M. Dean, F. Brunet and M. Long, 2004. Evolving protein functional diversity in new genes of Drosophila. Proc. Natl. Acad. Sci. USA 101: 16246–16250. [DOI] [PMC free article] [PubMed] [Google Scholar]