

Abstract
Niddm1i, a 16-Mb locus within the major diabetes QTL in the diabetic GK rat, causes impaired glucose tolerance in the congenic NIDDM1I strain. Niddm1i is homologous to both human and mouse regions linked with type 2 diabetes susceptibility. We employed multiple QTL analyses of congenic F2 progeny selected for one recombination event within Niddm1i combined with characterization of subcongenic strains. Fine mapping located one hyperglycemia locus within 700 kb (Niddm1i4, P = 5 × 10−6). Two adjacent loci were also detected, and the GK allele at Niddm1i2 (500 kb) showed a glucose-raising effect, whereas it had a glucose-lowering effect at Niddm1i3 (400 kb). Most proximally, Niddm1i1 (800 kb) affecting body weight was identified. Experimental data from subcongenics supported the four loci. Sorcs1, one of the two known diabetes susceptibility genes in the region, resides within Niddm1i3, while Tcf7l2 maps outside all four loci. Multiple-marker QTL analysis incorporating the effect of cosegregating QTL as cofactors together with genetically selected progeny can remarkably enhance resolution of QTL. The data demonstrate that the species-conserved Niddm1i is a composite of at least four QTL affecting type 2 diabetes susceptibility and that two adjacent QTL (Niddm1i2GK and Niddm1i3GK) act in opposite directions.
TYPE 2 diabetes or noninsulin-dependent diabetes mellitus (NIDDM) is defined by chronic elevation of plasma glucose, but the underlying pathophysiology is complex and profoundly influenced by both polygenic background and environmental factors (such as dietary habits, smoking, and physical activity). Consequently, the phenotypes associated with type 2 diabetes susceptibility usually show a quantitative variation (Permutt et al. 2005).
The risk to type 2 diabetes in humans has a measurable genetic component as indicated by familial clustering and higher concordance rates in monozygotic twins compared with dizygotic twins (Medici et al. 1999; Poulsen et al. 1999) and by the high heritability of insulin secretion and insulin action (Iselius et al. 1985; Lehtovirta et al. 2000; Poulsen et al. 2005). Genetic studies of inbred animals raised in standardized environments facilitates the identification of disease mechanisms via identification of naturally occurring alleles capable of influencing the progression from health to diabetes (Aitman et al. 1999; Fakhrai-Rad et al. 2000). The GK rat was developed by selective breeding of the most hyperglycemic offspring of outbred Wistar rats during nine generations, followed by inbreeding to generate a strain with stably inherited and spontaneously developing diabetes without concurrent excessive obesity (Goto 1975). Progeny from F2 intercrosses arranged between GK and normoglycemic strains have been subjected to genomewide linkage analyses, and several significant quantitative trait loci (QTL) for diabetes-associated phenotypes have been identified (Galli et al. 1996; Gauguier et al. 1996). The Niddm1i locus on the telomeric end of rat chromosome 1q is a locus within the major glucose-controlling QTL (Niddm1) in F2 intercrosses between GK and the normoglycemic F344 rat. Studies of the congenic strain NIDDM1I demonstrated that Niddm1iGK encoded hyperglycemia and insulin secretion defects in pancreatic islets (Galli et al. 1999; Fakhrai-Rad et al. 2000; Lin et al. 2001).
Genomewide linkage analyses in humans (Duggirala et al. 1999; Reynisdottir et al. 2003), mice (Stoehr et al. 2000; Kim et al. 2001), and the OLETF rat model (Watanabe et al. 1999) have also located QTL for diabetes in chromosome regions homologous to Niddm1i, human chromosome 10q24.3–q26.11, and mouse chromosome 19. Recently, two genes residing within Niddm1i have been associated with diabetes in humans (Tcf7l2; Grant et al. 2006), and fasting insulin levels in an obesity-induced mouse model for diabetes (Sorcs1; Clee et al. 2006). The strong support for contributions to diabetes-associated phenotypes within the Niddm1i locus prompted us to undertake a high-resolution genetic study of glucose and body-weight regulation. We used a combination of two genotypically different sets of rats: (1) F2 progeny from normoglycemic F344 and congenic NIDDM1I selected for a single recombination event within Niddm1i and (2) subcongenic strains with homozygous GK genotype in different intervals of Niddm1i. QTL analyses of the F2 progeny were used to narrow down the confidence intervals for diabetes susceptibility genes, and five subcongenic strains substantiated the presence of four subloci within Niddm1i.
MATERIALS AND METHODS
Animals:
Inbred normoglycemic F344/DuCrl2Swe were originally purchased from Charles River Laboratories (Wilmington, MA). Diabetic GK/Swe was originally from Kyoto University. Generation of the congenic strain F344.GK-Niddm1i, or NIDDM1I in its shortened form, has been described (Galli et al. 1999). It is homozygous for GK from 252 Mb to the end of chromosome 1 at 268 Mb (16 Mb, with the last marker D1Rat90 at 257.1 Mb) on a homozygous F344 genetic background with mitochondrial DNA and chromosomes X plus Y from F344. The congenic F2 intercross between F344 and NIDDM1I was initiated from female F344 and male NIDDM1I. Markers D1Rat83 and D1Rat90 were genotyped in 1594 male progeny to select rats with a single recombination event within Niddm1i for further testing. This protocol was designed to generate a homogeneous set of animals with recombination events spread throughout the locus. In each marker locus, the expected Mendelian segregation ratio of 1:2:1 in our F2 population was not distorted by the genotypic selection procedure. At birth ∼10 males from different litters were pooled, and at 30 days of age they were weaned and five progeny from different litters were housed per cage. In the end, 210 male progeny had complete genotypic and phenotypic information. The subcongenic strains N1IREC6, N1I12, N1I3, N1IREC1, and N1IREC11 were generated from F2 progeny and carried homozygous GK genome in different segments of Niddm1i on a homozygous F344 background (Table 1). All strains were maintained by sister–brother breeding. Litter sizes and number of rats per cage were matched in all experiments with congenic strains. Rats were maintained at constant temperature and humidity in a 12-hr cycle of light and dark with free access to standard laboratory chow and water. The local ethics committees approved all experiments.
TABLE 1.
Microsatellite markers used for Niddm1i and genotypes of congenic NIDDM1I and subcongenic strains
| Marker | Chromosome 1 (Mb) | NIDDM1I | N1IREC6 | N1I12 | N1I3 | N1IREC1 | N1IREC11 |
|---|---|---|---|---|---|---|---|
| D1Rat83 | 252.1 | GK | F | F | F | GK | F |
| D1Arb46 | 252.3 | GK | F | F | F | GK | F |
| D1Rat452 | 252.6 | GK | F | F | F | GK | F |
| D1Rat175 | 252.9a | GK | GK | F | F | GK | F |
| D1Got249 | 253.7 | GK | GK | F | F | GK | F |
| D1Swe1 | 254.0 | GK | GK | F | F | GK | F |
| D1Got247 | 254.3 | GK | GK | F | F | GK | F |
| D1Swe2 | 254.4 | GK | GK | F | F | GK | F |
| D1Swe3 | 254.8 | GK | GK | F | F | GK | F |
| D1Swe4 | 255.4 | GK | GK | F | F | GK | GK |
| D1Got244 | 255.7 | GK | GK | F | F | GK | GK |
| D1Rat376 | 255.8a | GK | GK | F | F | GK | GK |
| D1Swe5 | 256.1 | GK | GK | F | F | F | GK |
| D1Swe6 | 256.2 | GK | GK | F | F | F | GK |
| D1Mit8 | 257.4 | GK | GK | F | F | F | GK |
| D1Rat477 | 257.6 | GK | GK | F | F | F | GK |
| D1Rat85 | 258.6 | GK | GK | F | F | F | GK |
| D1Got250 | 259.2 | GK | GK | GK | F | F | F |
| D1Swe7 | 259.6 | GK | GK | GK | F | F | F |
| D1Swe8 | 259.9 | GK | GK | GK | F | F | F |
| D1Smu2 | 260.2a | GK | GK | GK | F | F | F |
| D1Got251 | 260.6 | GK | GK | GK | F | F | F |
| D1Rat456 | 261.6 | GK | GK | GK | F | F | F |
| D1Mgh15 | 262.0 | GK | GK | GK | F | F | F |
| D1Smu1 | 263.0 | GK | GK | GK | F | F | F |
| D1Mgh14 | 263.7 | GK | GK | GK | GK | F | F |
| D1Got259 | 263.7a | GK | GK | GK | GK | F | F |
| D1Rat457 | 265.4 | GK | GK | GK | GK | F | F |
| D1Mit14 | 266.0 | GK | GK | GK | GK | F | F |
| D1Rat90 | 267.1 | GK | GK | GK | GK | F | F |
Genotypes of congenic strains are indicated as F344-derived genome (F) and GK-derived genome (GK).
Approximate position based of the number of F2 recombinants as the marker was not mapped in NCBI assembly 3.4 of the rat genome.
Phenotypic characterization:
To avoid effects of the estrus cycle and other minor gender-specific influences, only males were included in this study. No major sex-specific effect for Niddm1 was seen in the original genomewide scan between GK and F344 (Galli et al. 1996). Weight measurements and an intraperitoneal glucose tolerance test (IPGTT) with 2.0 g glucose/kg body weight were performed after 6 hr fasting without anesthesia at 95 days of age (Galli et al. 1999).
Genotype analysis:
Genomic DNA was extracted from both ear and tail biopsies. Biopsies were incubated at 55° overnight in 500 μl lysis buffer (100 mm Tris–HCl, pH 8.0, 5 mm EDTA, 0.2% SDS, 200 mm NaCl) and 0.1 mg/ml proteinase K. The supernatant was cleared by centrifugation at 12,000 × g for 10 min; an equal volume of isopropanol was added, and DNA was collected by centrifugation at 12,000 × g for 30 min, dried, and dissolved in 75 μl 10 mm Tris–HCl, pH 7.6, 0.1 mm EDTA. Genetic markers were selected from public databases and in-house information and were mapped within the Niddm1i region in 45 (GK × F344)F2 rats (Galli et al. 1996). Eight new microsatellite markers (D1Swe1–8, available at RatMap at http://ratmap.gen.gu.se/) were added in regions lacking informative markers. The PCR profile consisted of: 94° for 4 min, followed by 35 cycles of 94° for 40 sec, 55° for 40 sec, and 72° for 90 sec, with a final 7-min incubation at 72° (markers D1Swe7–8 annealed at 50°). PCR amplification was performed with one primer in each pair labeled with [γ-33P]ATP or fluorescence (hex or fam) (DNA technology A/S, Aarhus, Denmark). The locations of markers were taken from Ensembl Rattus Norvegicus version 40.34j based on RGSC 3.4 (http://www.ensembl.org) (see Table 1).
QTL analyses:
Single-marker QTL analysis was performed on the basis of linear regression using Minitab (Minitab, State College, PA). Conditional probabilities of the QTL genotypes, given the observed marker data, were estimated using the R/qtl package (Broman et al. 2003). These probabilities were used to calculate coefficients of additive and dominance components for putative QTL at each marker (Lynch and Walsh 1998). Phenotypic values were regressed onto the additive and dominance coefficients at each marker to compute likelihood ratios (LR) using the following equation:
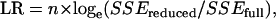 |
where SSEfull is the error sums of squares for the full regression model with a QTL at the marker locus, and SSEreduced is total sums of squares for the null model without a QTL. n denotes the number of observations. The genomic position with the highest LR was taken as the most likely position of a QTL. Multiple-marker QTL analysis was also performed to dissociate multiple linked gene effects during the identification of individual QTL. This was achieved by multiple-regression analyses testing the hypothesis of the existence of a QTL at a particular marker conditioned on selected markers as cofactors (Jansen 1993; Zeng 1993). The cofactors were chosen by a forward selection–backward elimination stepwise regression. The LR test statistic for the multiple-marker QTL analysis was calculated using the same equation as above, where SSEfull is the error sums of squares for the full model and SSEreduced is the error sums of squares for the reduced model incorporating all the cofactors except the QTL effect at the marker locus. Residuals of each trait were evaluated for normality by examining the normal probability plot of the residuals. Both the multiple regressions and the cofactor selection were conducted using the Minitab program. The numbers for the cofactors are 15-min glucose 3, 30-min glucose 3, and body weight 1.
Multiple testing issues in both the single- and the multiple-marker QTL analyses were addressed by calculation of experiment-wise empirical thresholds using a numerical method (Piepho 2001). Experiment-wise thresholds for significant linkage (α = 0.05), and highly significant linkage (α = 0.001) were employed. Experiment-wise 20% significance levels were used as the threshold for suggestive linkage. Thus, we applied a more conservative threshold for suggestive QTL compared with the suggestive threshold used in genomewide scans (Lander and Kruglyak 1995). Unless otherwise stated, the P-values are nominal. We applied the 1.5-LOD drop method to estimate support intervals for QTL (Sen and Churchill 2001).
Evidence for two-QTL interactions were investigated by two-dimensional scans for all marker pairs within Niddm1i using the scantwo function of the R/qtl package (Sen and Churchill 2001; Broman et al. 2003). Experiment-wise significance (α = 0.05) of a joint LOD was established by 1000 permutations of data (Churchill and Doerge 1994). The level of significance for an interaction LOD was set at P < 0.05.
RESULTS
The F2 progeny were established from a cross between normoglycemic F344 and congenic NIDDM1I with a 13.9-cM (16-Mb) genome segment on the telomeric part of chromosome 1 derived from the diabetic GK strain on a F344 genetic background. Only progeny with one recombination event within Niddm1i were included in the QTL analyses. Subsequently, subcongenic strains were established and genotyped (Table 1).
Single-marker QTL analysis:
Single-marker QTL analyses were performed to determine the presence and mode of inheritance of QTL by testing additive and dominant models against the null model (no QTL) at each marker position within Niddm1i. An experiment-wise highly significant QTL for postprandial glucose at 15 min (G15) during the intraperitoneal glucose tolerance test (IPGTT) was localized to the distal half of Niddm1i (Figure 1A; LR = 21.7, P = 3 × 10−6). This QTL colocalized with a highly significant QTL for postprandial glucose at 30 min (G30, Figure 1B; LR = 17.0, P = 4 × 10−5). Maximum LRs for both traits were obtained at marker D1Smu2. The two QTL were additive and explained 9.9 and 7.9% of the residual phenotypic variance. The GK allele was associated with higher postprandial glucose levels.
Figure 1.—

Single- and multiple-marker QTL analyses of diabetes-associated phenotypes within Niddm1i on chromosome 1. LR scores at each individual marker are indicated as thin lines for single-marker regression and as thick lines for multiple-marker regression. Experiment-wise significance thresholds (α = 0.05) are indicated as horizontal lines (thin line for single-marker and thick lines for multiple-marker regression). (A) Postprandial glucose at 15 min (G15) during IPGTT (B) Postprandial glucose at 30 min (G30) and (C) body weight.
Most proximal in Niddm1i, one significant QTL for body weight was mapped with a negative additive effect of the GK allele (Figure 1C; LR = 10.9, P = 1 × 10−3). The body weight of F2 progeny homozygous for the GK allele at this QTL was ∼5% lower than those homozygous for F344 at this locus. The residual variance was reduced by 5.1% by including the QTL in the model. Interestingly, all marker loci showed an additive inheritance since no dominant effect was observed; and all the identified QTL were best fitted by additive models (data not shown). Therefore we assumed strictly additive alleles in the further QTL analyses. Single-marker QTL analysis of postprandial insulin concentrations was unable to resolve conclusive evidence for distinct insulin loci within Niddm1i (data not shown).
Improved mapping resolution by multiple-marker QTL analysis:
Incorporation of markers as cofactors was used to enhance the ability to detect and locate closely linked QTL and to estimate their effects (Jansen 1993; Zeng 1993). The maximum LR test statistics and QTL map positions obtained with multiple-marker QTL analysis were very similar to those obtained with single-marker regression (Table 2). The explained residual variances were also of similar magnitude. However, the 1.5-LOD support intervals (SIs) for the QTL identified by multiple-marker regression were considerably narrower than those for the single-marker regression. Whereas SIs of the major loci at D1Smu2 for G15 and G30 based on single-marker QTL analysis were >6.0 Mb, they were reduced to 0.7 Mb by multiple-marker QTL analysis (Figure 1, A and B; Table 2). The location of the QTL for body weight was narrowed down to a 0.8-Mb interval between D1Rat83 and D1Rat175 (Table 1, Figure 1C). The QTL affecting body weight was designated Niddm1i1 and the major hyperglycemia QTL linked with D1Smu2 was denoted Niddm1i4.
TABLE 2.
Quantitative trait loci for diabetes-associated traits identified by multiple-marker QTL analysis
| QTL | Marker ± SI (Mb)a | Trait | LRb | Nominal P-value | Effectc ± SE | Vard % | No. of genese |
|---|---|---|---|---|---|---|---|
| Niddm1i1 | D1Rat83 + 0.8 | Bw | 11.0** | 1 × 10−3 | −6.8 ± 2.0 | 5.1 | 13 |
| Niddm1i2 | D1Swe2 ± 0.25 | G15 | 8.5* | 3 × 10−3 | +2.4 ± 0.8 | 4.0 | 2 |
| G30 | 4.1 | 0.04 | +1.4 ± 0.7 | 2.0 | |||
| Niddm1i3 | D1Got244 ± 0.2 | G15 | 8.3* | 4 × 10−3 | −2.6 ± 0.9 | 3.9 | 1 |
| G30 | 4.3 | 0.04 | −1.5 ± 0.7 | 2.1 | |||
| Niddm1i4 | D1Smu2 ± 0.35 | G15 | 20.9*** | 5 × 10−6 | +2.2 ± 0.5 | 9.6 | 7 |
| G30 | 15.6** | 8 × 10−5 | +1.5 ± 0.4 | 7.2 |
Bw, body weight (grams); G15, postprandial glucose at 15 min, and, G30 at 30 min in mmol/liter.
SI for QTL support intervals estimated by the 1.5-LOD drop method; Mb, megabase pair.
LR denotes the likelihood ratio test statistic for QTL and level of significance: *suggestive experiment-wise significance (α = 0.2); **significant experiment-wise significance (α = 0.05); ***highly significant experiment-wise significance (α = 0.001).
Additive effect defined as AA-BB/2, where AA is the genotypic value for GK homozygotes at the QTL, and BB is the genotypic value for F344 homozygotes; SE, standard error.
Var is percentage of phenotypic residual variance explained by the QTL.
The number of positional candidate genes within the QTL interval using NCBI assembly 3.4 of the rat genome.
Applying multiple-marker QTL analysis revealed two additional QTL for G15 at markers D1Swe2 and D1Got244, separate from the major hyperglycemia locus at D1Smu2 (Figure 1A). Surprisingly, while the GK allele at D1Swe2 was associated with higher glucose concentration, the opposite effect was seen for the GK allele at D1Got244 (Table 2, Figure 2). The effect sizes of the two opposing loci were of a similar magnitude (+2.4 and −2.6 mmol/liter) and comparable to that of the locus at D1Smu2 (Table 2, Figure 2). The hyperglycemia locus at D1Swe2 was denoted Niddm1i2, and the adjacent QTL with an opposite effect of the GK allele was denoted Niddm1i3. We found no evidence of interaction between any two loci after examining all pairwise combinations of marker loci for associations with the traits.
Figure 2.—

The effect of QTL on the 15-min postprandial glucose levels at Niddm1i2 (D1Swe2), Niddm1i3 (D1Got244), and Niddm1i4 (D1Smu2) jointly estimated by GLM procedure with MINITAB. Data are presented as least-squares mean ± SEM. Numbers of animals are given within parentheses. F/F denotes progeny homozygous for F344; GK/F, heterozygous progeny; and GK/GK, progeny homozygous for GK.
Analysis of subcongenic strains:
To substantiate the presence of multiple loci regulating diabetes-associated phenotypes within Niddm1i, a set of five subcongenic strains containing homozygous GK-derived segments covering parts of the locus were phenotypically investigated (Table 1). Glucose homeostasis during IPGTT in congenic NIDDM1I was, as previously described (Galli et al. 1999), impaired compared with F344 (Table 3). N1IREC6, with the entire Niddm1i locus except for 0.8 Mb in the most proximal end, was also hyperglycemic compared with F344. In addition, N1I12, harboring GK-derived genome in the 9-Mb distal end of Niddm1i, displayed a 13% increase in G15 and a 14% increase in G30. Three subcongenic strains did not show increased glucose levels compared with F344. These were N1IREC1, with a 4-Mb GK genotype most proximal in Niddm1i; N1IREC11, with a 3-Mb GK genotype within the proximal half of Niddm1i; and N1I3, with a 4-Mb GK genotype in the most distal end of Niddm1i (Table 1 and Table 3). This supported the presence of the major hyperglycemic locus Niddm1i4 (D1Smu2) in the genomic interval differing between N1I12 and N1I3. In fact, N1IREC11 with GK genotype in the proximal half of Niddm1i showed an improved glucose homeostasis compared with F344, which substantiated the location of Niddm1i3 (where the GK haplotype resulted in lower postprandial glucose concentration). The lower glucose levels of N1IREC11 were not simply an effect of the significantly lower body weight of N1IREC11, since the levels remained different after adjustment for the effect of body weight on glucose concentration in plasma (data nor shown). The N1IREC1 congenic strain covers both Niddm1i2 and Niddm1i3 (Tables 1 and 2); therefore the glucose-lowering effect of Niddm1i3 would be expected to counteract the glucose-elevating effect of Niddm1i2. This was also seen, since N1IREC1 did not display an improved glucose homeostasis compared with F344. There is no congenic strain with the GK genome solely in the Niddm1i2 locus; as a result we could obtain only indirect evidence for the presence of Niddm1i2 by comparing N1IREC1 with N1IREC11 (P = 0.02 for G15, P = 0.01 for G30).
TABLE 3.
Phenotypic characterization of F344, NIDDM1I, and subcongenic strains
| Trait | F344 | NIDDM1I | N1IREC6 | N1I12 | N1I3 | N1IREC1 | N1IREC11 |
|---|---|---|---|---|---|---|---|
| Bw | 289 ± 3 | 286 ± 3 | 307 ± 2 (P = 1 × 10−6) | 303 ± 3 (P = 1 × 10−3) | 286 ± 4 | 290 ± 5 | 279 ± 1 (P = 6 × 10−3) |
| I15 | 7.3 ± 0.4 | 5.5 ± 0.6 (P = 8 × 10−3) | 5.3 ± 0.5 (P = 4 × 10−3) | 7.5 ± 0.8 | 6.3 ± 0.9 | 7.2 ± 0.4 | 9.3 ± 1.0 (P = 0.04) |
| G15 | 18.1 ± 0.6 | 20.4 ± 0.5 (P = 4 × 10−3) | 22.0 ± 0.6 (P = 3 × 10−5) | 20.5 ± 0.5 (P = 3 × 10−3) | 18.5 ± 0.8 | 18.5 ± 0.4 | 17.0 ± 0.5 |
| G30 | 13.7 ± 0.5 | 16.5 ± 0.7 (P = 1 × 10−3) | 18.7 ± 0.6 (P = 1 × 10−7) | 15.5 ± 0.5 (P = 0.01) | 13.9 ± 0.6 | 13.5 ± 0.5 | 11.4 ± 0.6 (P = 5 × 10−3) |
Values are mean ± SEM at 95 days of age. Bw, body weight (grams) for 22–26 rats/strain; I15, postprandial insulin (μg/liter) at 15 min; G15, postprandial glucose at 15 min and 30 min (G30) in mmol/liter for 12–18 rats per strain during IPGTT. Phenotypes for the congenic strains were compared with those for F344 and evaluated by an unpaired Student t-test. Nominal P-values <0.05 are indicated.
There was no difference in body weight between NIDDM1I and F344 (Table 3). However, the presence of a body-weight-reducing QTL encoded by the GK haplotype was confirmed most convincingly by the difference in body weight between NIDDM1I and N1IREC6. NIDDM1I, differing from N1IREC6 only in the 0.8 Mb most proximal part of Niddm1i (Table 1), had 7% lower body weight compared with N1IREC6 (Table 3, P = 3 × 10−6). This confirmed the localization of Niddm1i1 to the small GK-derived segment most proximal in Niddm1i. In the congenics, the Niddm1i1GK effect is penetrant only in the two congenic strains (N1IREC6 and N1I12) that also harbor GK alleles in the region differing between N1I12 and N1I3 (D1Got250 to D1Smu1), since N1IREC1 with GK genotype at Niddm1i1 did not display lower body weight than F344. Hence, an explanation of this observation would be the presence of additional loci for body weight between D1Got250 and D1Smu1 (Niddm1i4). Indeed, in the F2 progeny there was an indication of three linked loci with opposite effects on body weight in this region. These three indicated body-weight QTL mapped within a genetic distance considerably shorter than for postprandial glucose at D1Swe2 and D1Got244 (Niddm1i2 and Niddm1i3). As a result, there were too few recombinants to significantly resolve and map these potential loci (data not shown). Furthermore, the N1IREC11 congenic strain was significantly lighter than F344, indicating the presence of an additional body-weight locus between D1Swe4 and D1Rat85 that was not observed in the F2 progeny.
NIDDM1I and N1IREC6 were the only strains that displayed significantly lower postprandial insulin levels compared with F344 (Table 3). N1IREC11, containing GK genome at the glucose-lowering locus Niddm1i3, displayed higher insulin levels compared with F344, which supported its improved glucose control. The phenotype displayed by N1IREC11 demonstrated that the GK haplotype between D1Swe4 and D1Rat85 encodes a complex pattern of diabetes-associated phenotypes (low glucose, high insulin, and low body weight).
Figure 3 portrays a summary of the location of the breakpoints for subcongenic strains and a summary of significant additive effects on diabetes-associated phenotypes within Niddm1i.
Figure 3.—

Congenic strains within Niddm1i and locations of mapped loci for diabetes-associated phenotypes. The solid bars designate known homozygous GK segments; the open ends of the bars designate intervals containing the recombinant endpoints. The locations and confidence intervals of loci influencing bodyweight (Niddm1i1, abbreviated N1i1) and postprandial glucose (Niddm1i2-4, abbreviated N1i2-4) are indicated as striped bars. The positions of the genes Sorcs1, Sorcs3, and Tcf7l2 are indicated. The locations of markers and genes were taken from Ensembl Rattus Norvegicus version 40.34j based on RGSC 3.4 (http://www.ensembl.org) and determined according to release 33.34c from ENSEMBL based on NCBI assembly 3.4 of the rat genome. The figure is drawn to scale.
DISCUSSION
Development of type 2 diabetes involves variations in a large number of genes, and although the identities of some have been elucidated, several more are likely to contribute to the loss of normal glucose control and type 2 diabetes. By studying animal models of type 2 diabetes, environmental factors affecting expression of disease-associated phenotypes can be better controlled. Construction of congenic strains and substrains has become a widely used method to isolate and narrow chromosomal regions containing susceptibility genes for genetically complex diseases. NIDDM1I harboring 16 Mb from the diabetic GK strain provides a unique possibility to combine mechanisms causing altered glucose homeostasis with detailed genetic information (Galli et al. 1999; Fakhrai-Rad et al. 2000; Lin et al. 2001). However, the map resolutions obtained from congenic strains are often not appropriate for identifying individual genes underlying a QTL. Theoretical approaches to improve the precision estimates of QTL position by inclusion of cosegregation at many genomic segments affecting the trait of interest have been reported (Jansen 1993; Zeng 1993). Also, improved map resolution can be achieved by selecting recombinants in the chromosome region (Darvasi 1998; Ronin et al. 2003; Jin et al. 2004; Xu et al. 2005). To refine genomic segments within Niddm1i coding for phenotypes relevant to type 2 diabetes, we performed multiple QTL analyses incorporating the effect of cosegregating QTL as cofactors using genetically selected progeny. In toto, one highly significant locus (Niddm1i4) and two suggestive loci (Niddm1i2 and Niddm1i3) were identified for postprandial glucose levels. The two suggestive QTL for glucose and one significant locus (Niddm1i1) for body weight were confirmed by independent characterization of the same phenotypes in subcongenics. The mapping data in this study confirm the theoretical predictions that data analyses based on more precise statistical genetic models for QTL analysis and a large number of recombinants are crucial factors for improving QTL mapping resolution for dissection of the genetic basis of quantitative traits such as type 2 diabetes susceptibility (Visscher et al. 2000). The mapping resolutions of Niddm1i1–Niddm1i4 were all <1 Mb (Figure1, Table 2).
The considerable genetic complexity displayed in Niddm1i is presumably a reflection of the selection protocol used to establish the diabetic GK strain (Goto 1975). Allelic fixation in genomic regions responding to strong selection is expected during establishment of GK. Closely linked QTL with opposite effects encoded by the same haplotype could occur, since selection operates on the net genotypic effect of several linked genes. Two inbred strains like GK and F344 represent only a limited fraction of the naturally occurring genetic variation (polymorphism) in the original population (Flint and Mott 2001). Therefore, it is tantalizing to note the species-conserved character of Niddm1i, emphasizing the relevance of genetic investigations of the region as a major type 2 diabetes locus. Despite the substantial complexity of the Niddm1i QTL, it is still readily amenable to achieving high-resolution mapping of QTL and to identifying genes that regulate mechanisms behind the phenotypic variation associated with common diseases such as type 2 diabetes.
A number of genes within Niddm1i are involved in pathways that may be important to energy metabolism, apoptosis, and insulin secretion, which are critical factors associated with the risk for diabetes. This high-resolution study has narrowed down the number of diabetes-associated candidate genes considerably. The 800-kb genome segment corresponding to Niddm1i1 is gene rich and harbors ∼13 annotated genes, with USMG5 (upregulated during skeletal muscle growth 5, or DAPIT/LZAP) as a possible candidate (Paivarinne and Kainulainen 2001). The two loci with opposing effect on glucose levels encoded by the GK alleles (Niddm1i2 and Niddm1i3) have exceptionally few identified protein-coding genes: Sorcs3 and a transposase from an L1 repeat in Niddm1i2, and Sorcs1 within Niddm1i3. Sorcs1 was recently identified as a type 2 diabetes susceptibility gene in the mouse (Clee et al. 2006). It is therefore conceivable that Sorcs3 also might be involved in the pathogenesis of diabetes. The Niddm1i4 hyperglycemia locus covers a 700-kb genome interval with 7 known genes, including the gene for programmed cell death 4, three genes without known functions, one microRNA gene, the leucine-rich repeat protein SHOC2 (Selfors et al. 1998), and the α-2-adrenergic receptor ADRA2A (Devedjian et al. 2000). The gene encoding TCF7L2 is located 1.4 Mb distally of Niddm1i4 and is not a probable candidate for the phenotype encoded by this QTL identified using regression models with cofactors selected by the stepwise regression procedure.
In conclusion, the combined analysis of genotypically selected F2 progeny and subcongenic rat strains has revealed an intricate pattern of genetic effects, which are amenable to experimental dissection and subsequent molecular identification of disease mechanisms. Four QTL for phenotypes highly relevant to type 2 diabetes were mapped to intervals <1 Mb and several positional candidate genes have been selected for studies of their relevance to this disease. The complex genetic interplay between several diabetes susceptibility loci under controlled environmental conditions, as reported here, emphasizes the need for caution when attempting to identify disease mechanisms and risk alleles in the genetically and environmentally heterogeneous human population.
Acknowledgments
We thank Hans-Peter Piepho for discussions regarding thresholds for QTL analysis; Annika Andersson, Cecilia Johansson, and Berit Rydlander for genotyping; Martha Nordberg for breeding and phenotyping; Johannes Luthman for contributions to the design and accomplishment of F2 progeny and congenic strains; and Agneta Petersson for phenotyping. Joakim Galli established the N1IREC series of congenics. This study was supported by grants from the Swedish Research Council, Swedish Diabetes Association, the Medical Faculty of Lund University, Malmö University Hospital, MAS Stiftelser och Gåvor, and Albert Påhlsson Research Foundation.
References
- Aitman, T. J., A. M. Glazier, C. A. Wallace, L. D. Cooper, P. J. Norsworthy et al., 1999. Identification of Cd36 (Fat) as an insulin-resistance gene causing defective fatty acid and glucose metabolism in hypertensive rats. Nat. Genet. 21: 76–83. [DOI] [PubMed] [Google Scholar]
- Broman, K. W., H. Wu, S. Sen and G. A. Churchill, 2003. R/qtl: QTL mapping in experimental crosses. Bioinformatics 19: 889–890. [DOI] [PubMed] [Google Scholar]
- Churchill, G. A., and R. W. Doerge, 1994. Empirical threshold values for quantitative trait mapping. Genetics 138: 963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clee, S. M., B. S. Yandell, K. M. Schueler, M. E. Rabaglia, O. C. Richards et al., 2006. Positional cloning of Sorcs1, a type 2 diabetes quantitative trait locus. Nat. Genet. 38: 688–693. [DOI] [PubMed] [Google Scholar]
- Darvasi, A., 1998. Experimental strategies for the genetic dissection of complex traits in animal models. Nat. Genet. 18: 19–24. [DOI] [PubMed] [Google Scholar]
- Devedjian, J. C., A. Pujol, C. Cayla, M. George, A. Casellas et al., 2000. Transgenic mice overexpressing alpha2A-adrenoceptors in pancreatic beta-cells show altered regulation of glucose homeostasis. Diabetologia 43: 899–906. [DOI] [PubMed] [Google Scholar]
- Duggirala, R., J. Blangero, L. Almasy, T. D. Dyer, K. L. Williams et al., 1999. Linkage of type 2 diabetes mellitus and of age at onset to a genetic location on chromosome 10q in Mexican Americans. Am. J. Hum. Genet. 64: 1127–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakhrai-Rad, H., A. Nikoshkov, A. Kamel, M. Fernström, J. R. Zierath et al., 2000. Insulin-degrading enzyme identified as a candidate diabetes susceptibility gene in GK rats. Hum. Mol. Genet. 14: 2149–2158. [DOI] [PubMed] [Google Scholar]
- Flint, J., and R. Mott, 2001. Finding the molecular basis of quantitative traits: successes and pitfalls. Nat. Rev. Genet. 2: 437–445. [DOI] [PubMed] [Google Scholar]
- Galli, J., L. S. Li, A. Glaser, C. G. Ostenson, H. Jiao et al., 1996. Genetic analysis of non-insulin dependent diabetes mellitus in the GK rat. Nat. Genet. 12: 31–37. [DOI] [PubMed] [Google Scholar]
- Galli, J., H. Fakhrai-Rad, A. Kamel, C. Marcus, S. Norgren et al., 1999. Pathophysiological and genetic characterization of the major diabetes locus in GK rats. Diabetes 48: 2463–2470. [DOI] [PubMed] [Google Scholar]
- Gauguier, D., P. Froguel, V. Parent, C. Bernard, M. T. Bihoreau et al., 1996. Chromosomal mapping of genetic loci associated with non-insulin dependent diabetes in the GK rat. Nat. Genet. 12: 38–43. [DOI] [PubMed] [Google Scholar]
- Goto, Y., M. Kakizaki and N. Masaki, 1975. Spontaneous diabetes produced by selective breeding of normal Wistar rats. Proc. Jpn. Acad. 51: 80–85. [Google Scholar]
- Grant, S. F., G. Thorleifsson, I. Reynisdottir, R. Benediktsson, A. Manolescu et al., 2006. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat. Genet. 38: 320–323. [DOI] [PubMed] [Google Scholar]
- Iselius, L., J. Lindsten, N. E. Morton, S. Efendic, E. Cerasi et al., 1985. Genetic regulation of the kinetics of glucose-induced insulin release in man. Studies in families with diabetic and non-diabetic probands. Clin. Genet. 28: 8–15. [DOI] [PubMed] [Google Scholar]
- Jansen, R. C., 1993. Interval mapping of multiple quantitative trait loci. Genetics 135: 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, C., H. Lan, A. D. Attie, G. A. Churchill, D. Bulutuglo et al., 2004. Selective phenotyping for increased efficiency in genetic mapping studies. Genetics 168: 2285–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. H., S. Sen, C. S. Avery, E. Simpson, P. Chandler et al., 2001. Genetic analysis of a new mouse model for non-insulin-dependent diabetes. Genomics 74: 273–286. [DOI] [PubMed] [Google Scholar]
- Lander, E., and L. Kruglyak, 1995. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat. Genet. 11: 241–247. [DOI] [PubMed] [Google Scholar]
- Lehtovirta, M., J. Kaprio, C. Forsblom, J. Eriksson, J. Tuomilehto et al., 2000. Insulin sensitivity and insulin secretion in monozygotic and dizygotic twins. Diabetologia 43: 285–293. [DOI] [PubMed] [Google Scholar]
- Lin, J. M., H. Ortsater, H. Fakhrai-Rad, J. Galli, H. Luthman et al., 2001. Phenotyping of individual pancreatic islets locates genetic defects in stimulus secretion coupling to Niddm1i within the major diabetes locus in GK rats. Diabetes 50: 2737–2743. [DOI] [PubMed] [Google Scholar]
- Lynch, M., and B. Walsh, 1998. Genetics and Analysis of Quantitative Traits. Sinauer Associates, Sundrland, MA.
- Medici, F., M. Hawa, A. Ianari, D. A. Pyke and R. D. Leslie, 1999. Concordance rate for type II diabetes mellitus in monozygotic twins: actuarial analysis. Diabetologia 42: 146–150. [DOI] [PubMed] [Google Scholar]
- Paivarinne, H., and H. Kainulainen, 2001. DAPIT, a novel protein down-regulated in insulin-sensitive tissues in streptozotocin-induced diabetes. Acta Diabetol. 38: 83–86. [DOI] [PubMed] [Google Scholar]
- Permutt, M. A., J. Wasson and N. Cox, 2005. Genetic epidemiology of diabetes. J. Clin. Invest. 115: 1431–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piepho, H. P., 2001. A quick method for computing approximate thresholds for quantitative trait loci detection. Genetics 157: 425–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen, P., K. O. Kyvik, A. Vaag and H. Beck-Nielsen, 1999. Heritability of type II (non-insulin-dependent) diabetes mellitus and abnormal glucose tolerance—a population-based twin study. Diabetologia 42: 139–145. [DOI] [PubMed] [Google Scholar]
- Poulsen, P., K. Levin, I. Petersen, K. Christensen, H. Beck-Nielsen et al., 2005. Heritability of insulin secretion, peripheral and hepatic insulin action, and intracellular glucose partitioning in young and old Danish twins. Diabetes 54: 275–283. [DOI] [PubMed] [Google Scholar]
- Reynisdottir, I., G. Thorleifsson, R. Benediktsson, G. Sigurdsson, V. Emilsson et al., 2003. Localization of a susceptibility gene for type 2 diabetes to chromosome 5q34-q35.2. Am. J. Hum. Genet. 73: 323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronin, Y., A. Korol, M. Shtemberg, E. Nevo and M. Soller, 2003. High-resolution mapping of quantitative trait loci by selective recombinant genotyping. Genetics 164: 1657–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selfors, L. M., J. L. Schutzman, C. Z. Borland and M. J. Stern, 1998. soc-2 encodes a leucine-rich repeat protein implicated in fibroblast growth factor receptor signaling. Proc. Natl. Acad. Sci. USA 95: 6903–6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen, S., and G. A. Churchill, 2001. A statistical framework for quantitative trait mapping. Genetics 159: 371–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoehr, J. P., S. T. Nadler, K. L. Schueler, M. E. Rabaglia, B. S. Yandell et al., 2000. Genetic obesity unmasks nonlinear interactions between murine type 2 diabetes susceptibility loci. Diabetes 49: 1946–1954. [DOI] [PubMed] [Google Scholar]
- Watanabe, T. K., S. Okuno, K. Oga, A. Mizoguchi-Miyakita, A. Tsuji et al., 1999. Genetic dissection of “OLETF,” a rat model for non-insulin-dependent diabetes mellitus: quantitative trait locus analysis of (OLETF × BN) × OLETF. Genomics 58: 233–239. [DOI] [PubMed] [Google Scholar]
- Visscher, P., J. Whittaker and R. Jansen, 2000. Mapping multiple QTL of different effects: comparison of a simple sequential testing strategy and multiple QTL mapping. Mol. Breed. 6: 11–24. [Google Scholar]
- Xu, Z., F. Zou and T. J. Vision, 2005. Improving quantitative trait loci mapping resolution in experimental crosses by the use of genotypically selected samples. Genetics 170: 401–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Z. B., 1993. Theoretical basis for separation of multiple linked gene effects in mapping quantitative trait loci. Proc. Natl. Acad. Sci. USA 90: 10972–10976. [DOI] [PMC free article] [PubMed] [Google Scholar]