

Abstract
In allopolyploid species, fair meiosis could be challenged by homeologous chromosome pairing and is usually achieved by the action of homeologous pairing suppressor genes. Oilseed rape (Brassica napus) haploids (AC, n = 19) represent an attractive model for studying the mechanisms used by allopolyploids to ensure the diploid-like meiotic pairing pattern. In oilseed rape haploids, homeologous chromosome pairing at metaphase I was found to be genetically based and controlled by a major gene, PrBn, segregating in a background of polygenic variation. In this study, we have mapped PrBn within a 10-cM interval on the C genome linkage group DY15 and shown that PrBn displays incomplete penetrance or variable expressivity. We have identified three to six minor QTL/BTL that have slight additive effects on the amount of pairing at metaphase I but do not interact with PrBn. We have also detected a number of other loci that interact epistatically, notably with PrBn. Our results support the idea that, as in other polyploid species, metaphase I homeologous pairing in oilseed rape haploids is controlled by an integrated system of several genes, which function in a complex manner.
POLYPLOIDY plays an important role in the evolution and speciation of higher plants (Otto and Whitton 2000). It is estimated that >70% of angiosperms are polyploid, arising either by multiplication of a basic set of chromosomes (autopolyploidy) or as a result of combining related, but not completely homologous, genomes (allopolyploidy) (Comai 2005). Recent genomic analyses have revealed that even classic diploid species such as maize and Arabidopsis are actually ancient polyploids (Blanc and Wolfe 2004 and references therein). The success of polyploidy is now largely attributed to the fact that whole-genome duplication supplies genetic raw materials for DNA diversification through gene complementary effect, altered regulatory interactions, and genetic and epigenetic alterations (Liu and Wendel 2003; Osborn et al. 2003b; Comai 2005).
If the combination of two genomes offers a greater adaptability to organisms (Comai 2005), newly formed polyploid plants face an immediate challenge during meiosis: the two sets of homeologous chromosomes may be sufficiently similar to one another that pairing between homeologous chromosomes may occur, which would create complex meiotic configurations, leading to unbalanced gametes, aneuploid progenies (Ramsey and Schemske 2002), chromosome rearrangements (e.g., Benavente et al. 2001; Sánchez-Morán et al. 2001), and hence impaired fertility (Gillies 1989; Ramsey and Schemske 2002). Precise control of chromosome pairing (i.e., restriction of pairing to homologous chromosomes only) is therefore a prerequisite for meiotic and reproductive stability in polyploids. It is certainly not a matter of chance that most if not all allopolyploids exhibit a diploid-like meiotic behavior and disomic inheritance at meiosis with only homologous chromosomes being associated as bivalents at metaphase I. Current knowledge suggests that the meiotic diploidization, which has occurred upon hybridization in polyploid species, is probably achieved by two complementary mechanisms: (1) the structural divergence between homeologous chromosomes that may already exist at the diploid stage and be accentuated through sequence elimination upon allopolyploidization (Feldman et al. 1997; Salina et al. 2004) and (2) the involvement of genes that suppress homeologous pairing (reviewed in Jenczewski and Alix 2004). For instance, the Ph1 locus is required to prevent homeologous pairing in allopolyploid wheat (Riley and Chapman 1958; Sears and Okamoto 1958), as evidenced by the formation of numerous bivalents and multivalents at metaphase I of meiosis in nulli-5B haploids of wheat. Similar or related homeologous-pairing suppressors might also exist in oat, cotton, tobacco, fescue, goatgrass, and ryegrass (Jenczewski and Alix 2004). In most of these species, cytological diploidization is likely to be controlled by several genes that have effects on different processes throughout the premeiotic interphase and the meiotic prophase (Jenczewski and Alix 2004) and do not always interact in an additive way (e.g., Mello-Sampayo and Canas 1973). Although much of this understanding remains at the descriptive level, very recent cloning of the Ph1 locus (Griffiths et al. 2006) has provided, for the first time, an opportunity to characterize the function of a homeologous pairing regulator. However, the very peculiar nature of the Ph1 locus, which consists of a segment of subtelomeric heterochromatin from chromosome 3AL inserted into a cluster of cdc-2-related genes, also suggests that the mechanisms characterized in wheat may not be the same as in other polyploid species.
Oilseed rape (Brassica napus; AACC; 2n = 38) is an attractive model to provide further insights into the molecular mechanisms responsible for the cytological diploidization of allopolyploid species. Oilseed rape is a widely cultivated allopolyploid species, originating from multiple hybridizations between the ancestors of modern B. oleracea (CC; 2n = 18) and B. rapa (AA; 2n = 20). It is now largely accepted that the genomes of the diploid progenitors of B. napus are widely replicated although it is still not clear whether they have evolved from a hexaploid ancestor or via segmental duplication of one or two ancestral genomes (Truco et al. 1996; Parkin et al. 2003, 2005; Lukens et al. 2004; Lysak et al. 2005). B. napus exhibits a clear bivalent-pairing regime and a disomic inheritance, which demonstrate that homologs pair at meiosis at the expense of homeologous pairing. However, de novo nonreciprocal translocations resulting from recombination between homeologous chromosomes/regions were consistently reported at frequencies of 0.43–1.6% of total recombination events in several mapping populations of euploid B. napus (AACC, 2n = 38) (Parkin et al. 1995; Sharpe et al. 1995; Udall et al. 2005), with the highest frequencies being observed with resynthesized B. napus. Preexisting reciprocal (Lombard and Delourme 2001; Osborn et al. 2003a; Piquemal et al. 2005) and nonreciprocal translocations (Udall et al. 2005) have also been identified among different accessions of B. napus and shown to stimulate further rearrangements in their vicinity. Some of these de novo or preexisting homeologous exchanges were shown to increase the range of genetic variation observed for important ecological and agronomic traits like flowering time (Pires et al. 2004), seed yield (Osborn et al. 2003b), or pest resistance (Zhao et al. 2006).
The basis of the diploid-like meiotic behavior of B. napus has therefore been a subject of debate during the 20th century. Different authors have proposed that homeologous pairing is genetically regulated in oilseed rape (Attia and Robbelen 1986; Sharpe et al. 1995) and its close relatives (Prakash 1974; Hardberg 1976; Eber et al. 1994). A major contribution to this debate came from Renard and Dosba (1980) and Attia and Robbelen (1986), who observed that homeologous pairing was commonplace at metaphase I in B. napus haploids (AC; n = 19), and the number of bivalents in pollen mother cells varied with varieties, with high- and low-pairing varieties being clearly distinguished. Jenczewski et al. (2003) combined a segregation analysis with a maximum-likelihood approach to demonstrate that the distribution of the number of univalents (nonpaired chromosomes) among these haploids was consistent with the segregation of a diallelic major gene, named PrBn for Pairing regulator in B. napus, in a background of polygenic variation.
In this article we report the genetic mapping of PrBn and other complementary genetic factors that influence the amount of homeologous pairing at metaphase I in oilseed rape haploids. Bulked segregant analysis, involving thousands of previously mapped as well as anonymous molecular markers, was used first to find markers linked to PrBn. We then used and compared the results of conventional interval mapping and composite interval mapping, binary-trait-oriented as well as data-mining approaches to detect and localize the additive and epistatic loci contributing to the variation of homeologous pairing at metaphase I in oilseed rape haploids.
MATERIALS AND METHODS
Plant materials:
A population of 244 haploids was produced from F1 hybrids obtained by crossing a single plant of “Darmor-bzh” (high-pairing parent) with a single plant of “Yudal” (low-pairing parent). The details for obtaining the haploid plants were described previously (Jenczewski et al. 2003). The meiotic pairing behavior was observed on the pollen mother cells at metaphase I. On average, 20 pollen mother cells were examined for each haploid and the mean number of nonpaired chromosomes (univalents) was calculated (Jenczewski et al. 2003). A subset of 116 haploids (of the 244 phenotyped plants) with most contrasting phenotype (58 highest-pairing and 58 lowest-pairing plants) was selected to construct a full genetic linkage map and to perform quantitative trait loci (QTL) and binary trait loci (BTL) analyses (selective genotyping approach; Ayoub and Mather 2002). These 116 haploids were taken from series 2 (31 haploids), 3 (61 haploids), and 4 (24 haploids) as described in Jenczewski et al. (2003).
Molecular analyses:
Genomic DNA was extracted from the young leaves of all individual haploid plants according to the method of Doyle and Doyle (1990). DNA concentration was adjusted to 10, 50, and 1 ng/μl for random amplified polymorphic DNA (RAPD), amplified fragment length polymorphism (AFLP), and simple sequence repeat (SSR) assays, respectively.
Bulked segregant analysis:
Ten plants showing the lowest and 10 showing the highest number of univalents at metaphase I were selected and their genomic DNA was pooled to form the high-pairing and the low-pairing bulks, respectively (Michelmore et al. 1991).
RAPD markers:
Seventy-five RAPD primers (Operon Technologies, Alameda, CA) were selected from all linkage groups of the reference maps published by Foisset et al. (1996) and Lombard and Delourme (2001) and tested on the two bulks. Once a RAPD marker was found to be polymorphic between the two bulks, more markers from the same linkage group were tested. PCRs were performed using the same protocol as in Foisset et al. (1996). Amplified products were separated on 1.8% agarose gels and images acquired after staining by ethidium bromide. RAPD markers were named by a combination of primer code and the size of the DNA fragments amplified.
AFLP markers:
AFLP assays were conducted essentially as described by Vos et al. (1995). Briefly, DNA was digested either by EcoRI plus MseI or by PstI plus MseI. After ligation of double-stranded adaptors to the ends of the restriction fragments, preamplification was performed with EcoRI (or PstI) and MseI primer pairs with one selective nucleotide at their 3′-end. The selective amplification was performed with EcoRI (or PstI) and MseI primer pairs with three selective nucleotides at their 3′-end. The EcoRI+3 and PstI+3 primers were labeled by IRD700 or IRD800 fluorochrome (MWG Biotech). The amplified products were separated on 5.5% denaturing acrylamide gels and detected with an automated DNA sequencer (LI-COR Biosciences). Nine hundred randomly selected primer pairs (300 EcoRI+3–MseI+3 and 600 PstI+3–MseI+3) were tested on the two bulks. AFLP markers were named using the conventional code for each primer pair plus a letter based on fragment sizes. AFLP markers that revealed polymorphism between the two bulks were then checked on all 20 individuals making up the bulks. Those showing a cosegregation ratio (with the pairing type at metaphase I) ≥15/20 were retained and tested on the segregating population of 244 haploids.
SSR markers:
Forty-six SSR primers were obtained from the UK Cropnet Database (www.ukcrop.net/perl/ace/search/BrassicaDB) and other sources (Lowe et al. 2004; Piquemal et al. 2005). PCRs were performed in a total volume of 5 μl containing M13 universal fluorescent tag to label the amplified DNA fragments. The amplified products were electrophoresed through 6.5% denaturing polyacrylamide gels and detected by the same DNA sequencer as AFLP.
Construction of genetic linkage maps:
Three hundred fifty-eight AFLP markers, generated by 41 primer pairs, were used to establish a frame for each linkage group. Thirty-nine RAPD and 46 SSR markers were then selected from a recent reference map (R. Delourme, unpublished data) so that they can bridge the gaps between AFLP markers, enrich a region of interest, or make connections with linkage groups established by Lombard and Delourme (2001). Segregating markers were scored for each haploid plant and linkage analysis was performed with MAPMAKER/EXP version 3.0 (Lincoln et al. 1992). Linkage groups were established with a threshold LOD score of 5.0 and a maximum recombination frequency of 0.4. Kosambi function was used to evaluate the genetic distances (in centimorgans) between linked markers.
QTL and BTL analyses:
QTL analysis was performed using the number of univalents at metaphase I as a quantitative trait. As explained in Jenczewski et al. (2003), this variable was chosen because it can be reliably scored and it reflects the whole extent of pairing at metaphase I in a synthetic way. This variable did not follow a normal distribution, but rather a bimodal distribution in our segregating population, which could make standard statistical procedures potentially inaccurate to identify the location and effect of QTL (McIntyre et al. 2001 and references therein). It was therefore necessary to compare the results from traditional QTL mapping to those from other approaches that are robust against nonnormality of phenotypes.
QTL analysis:
We first explored single marker-trait associations using linear regression analysis. Composite interval mapping (Zeng 1994) was then used to map additive QTL affecting the number of univalents. This approach was implemented using QTL Cartographer 2.5 (Wang et al. 2005) with a conditioning window size of 10 cM and a walk speed of 2 cM. The number of markers to be used as cofactors was determined according to the results of single-marker regression analysis. Five cofactors were selected by forward selection, backward elimination, or forward selection–backward elimination (with Prob_into = Prob_out = 0.005) stepwise regressions. An experimentwide critical LOD score of 3.3 was determined by permutation test analyses (1000 permutations), as suggested by Churchill and Doerge (1994). Significant QTL peaks from the composite interval mapping analysis were used as the initial model for multiple interval mapping (Kao et al. 1999) with QTL Cartographer 2.5. Multiple interval mapping models searched for new QTL to add to the current model in an iterative way and tested their significance after each search cycle. The Bayesian information criterion was used to decide when no additional QTL could be added to the current model.
We finally used the regression interval mapping method of Haley and Knott (1992) and the nonparametric QTL mapping approach proposed by Kruglyak and Lander (1995), both shown to be robust against the nonnormality of the phenotypes (Rebai 1997). These analyses were implemented using R/QTL (Broman et al. 2003).
BTL analysis:
Given the fact that the number of univalents follows a bimodal distribution (Jenczewski et al. 2003), we have also considered the meiotic behavior of the segregating haploids as a binary trait. We used the generalized linear model framework proposed by Sen and Churchill (2001) as well as the probabilistic approach developed by Coffman et al. (2005) to detect and map the BTL associated with the high- and the low-pairing behaviors. Sen and Churchill's approach was implemented using R/QTL (Broman et al. 2003) while the proc BTL (SAS, Cary, NC) was used to identify the best single- (additive BTL) and two-marker models associated with the meiotic phenotype (default values were used). Akaike's information criterion (AIC) was used for model selection. The model with the smallest AIC was identified as the model best supported by the data.
QTL/BTL were named using the abbreviation uni (for number of univalents) followed by the number of the linkage group where the QTL/BTL was found and a terminal suffix, separated by a period, providing a unique identifier for multiple QTL/BTL on a single linkage group.
Epistatic interactions:
We searched for digenic epistasic interactions by conducting all possible two-way ANOVAs between every pair of markers and using the number of univalents as a quantitative variable. This approach was implemented using the GLM procedure of SAS. To survey the whole genome for digenic and epistatic effects, we evaluated 77,028 [n(n − 1)/2] possible interactions for a map of n = 393 markers. The threshold to claim a statistically significant interaction was set at P ≤ 0.0005 and R2 ≥ 5%. This threshold, which was based on the probability of getting one single false positive interaction and adjusted to roughly account for the linkage between the markers and therefore the number of actual independent tests, corresponds to a LOD score of 3.3.
Tree-based methods:
Classification and regression trees, proposed by Breiman et al (1984), form a data-mining alternative to parametric QTL and BTL approaches.
Regression tree is a tool for fitting the response variable, given a set of predictors that can be both categorical and numerical. Here the response variable is the number of univalents measured at metaphase I and the predictors are the markers. In the sequel we denote the set of all the markers by M and a single marker belonging to the set M by m. The algorithm splits recursively the data set into more homogeneous subsets with respect to the distribution of the number of univalents. The homogeneity measure is based on a sample variance of the number of univalents. The algorithm starts by looking for a marker m that defines the split of the data leading to the maximal gain in homogeneity. The initial data set is then divided into two subsets of plants according to the values of m and referred to as the tree nodes. The process of finding the best split is then repeated on each of the resulting nodes until a stopping rule is applied (see details below). Once the tree is built the mean number of univalents is calculated for each leaf node. The rule for predicting the number of univalents for a new plant with a set of markers M consists of finding an appropriate leaf node (the markers belonging to M must satisfy the conditions defining this node) and assigning to the plant a mean univalents number calculated for this node.
Bagging method:
One major problem with trees is their high variance: even a small change in data can result in a very different series of splits. To reduce this variance we applied a bootstrap aggregation method (bagging) proposed by Breiman (1996) for the trees. Instead of calculating the prediction for a single regression tree, bagging averages it over a collection of trees based on bootstrap samples drawn from the original data set, improving thereby the prediction accuracy. We calculated B=500 regression trees for the number of univalents. Every single tree was based on a training set of size 80, sampled with replacement from the original data. The following stopping rules were applied for a single-tree construction: the minimal size of a node to be split was fixed at 20 plants, the minimal size of a leaf node was fixed at 7 plants, and the maximal tree depth was fixed at 30 generations. The prediction of the number of univalents for a new plant was calculated by averaging the predictions of all the trees, leaving this plant out of the bootstrap sample. To assess the regression accuracy a mean square prediction error (PE) is calculated. One may finally acknowledge that the number of nodes for the trees constructed on different data sets can be different; in our case, for 500 bootstrap samples we obtained rather small trees, with the number of nodes in general less than five.
Variable importance:
We applied two criteria for measuring the marker importance. First, we considered the number of trees including a marker m over a total number of calculated trees. This measure, denoted by B(m), takes into account the markers participating frequently in tree construction, even if they do not improve notably the tree prediction. Conversely, some markers may participate rarely in a tree collection, but once they appear they improve greatly the prediction. The second importance measure I(m) takes into account these markers as well. For a marker m, we calculated a prediction error estimate restricted to the trees including m, PE(m). Then, we recalculated it after randomly permuting the values of m (we denote by  the marker after permutation). The idea behind this is that random permutations of important marker values will change the predicted values a lot and, consequently, will increase the prediction error. The importance measure of marker m is thus defined as a relative difference between
the marker after permutation). The idea behind this is that random permutations of important marker values will change the predicted values a lot and, consequently, will increase the prediction error. The importance measure of marker m is thus defined as a relative difference between 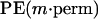 and PE(m):
and PE(m):
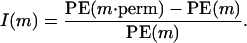 |
Note that it may happen that the prediction error decreases after permuting the values of m and so the importance I(m) may be negative. Note also that no threshold for the importance measure is available at the moment, so no inference for I(m) can be done.
The bagging algorithm and the computations of I(m) for the markers were handled using the rpart package of the R software (version 2.2).
RESULTS
Mapping of PrBn:
To cover the B. napus genome in the most exhaustive way, we used previously mapped and evenly distributed molecular markers (RAPD and AFLP), as well as ∼9900 anonymous AFLP markers to screen the high- and the low-pairing bulks.
Seventy-five RAPD primers and 900 random AFLP primers generated ∼10,000 markers showing polymorphism between the two parents of our segregating population; ∼120 of them have already been mapped and spanned over all the linkage groups of the reference map. Among all these polymorphic markers, only one RAPD marker (M13.1420) located on linkage group DY15 and 59 anonymous AFLP markers (24 EcoRI–MseI type and 35 PstI–MseI type) showed polymorphism between the two bulks. Further testing of 10 additional RAPD markers from linkage group DY15 allowed the identification of 6 more markers (AJ06.1100, O20.1360, J07.2960, N20.1320, T16.2010, and AG12.600) that also differentiated the bulks. These 7 RAPD markers as well as 34 AFLP markers (6 of EcoRI–MseI type and 28 PstI–MseI type selected from the 59 candidates) were further verified on all 20 individuals of the two bulks. None of them fully cosegregated with the phenotype (high- vs. low-pairing behavior at metaphase I) of the 20 haploid plants, but the 7 RAPD markers and 15 AFLP markers showed matching rates (genotypes vs. phenotypes) equal to or greater than the preset threshold (15/20), typically being 16–18/20 (example in Figure 1). These markers were used to genotype the entire segregating population of 244 haploids.
Figure 1.—

Partial cosegregation of E86M35a (arrows) with the pairing type of the 20 haploids of the two bulks.
All seven RAPD markers and seven of the AFLP markers formed one single linkage group of ∼70 cM, named DY15a, as it corresponded to the upper portion of DY15 on the reference map. The relative position and genetic distances between the seven RAPD markers were similar to those on the reference map. Among the eight remaining AFLP markers, two were linked to each other while the others were unlinked. We first considered the meiotic behavior (defined as high pairing or low pairing at metaphase I) of the segregating haploids as a genetic marker with two alleles and tried to map this marker, which should correspond to PrBn, on DY15a; our attempts resulted in an erratic position, 30–40 cM away from either end of the linkage group. The possibility of PrBn residing on other regions of DY15 was eliminated, as markers (RAPD and SSR) located on other parts of DY15 did not show any link with PrBn in bulked segregant analysis screening or when tested directly on the segregating population (data not shown). We therefore turned to map PrBn as a quantitative or binary trait locus.
A preliminary QTL analysis by interval mapping was then performed to map PrBn as a QTL on DY15a using the genotyping data of the 14 markers and the mean number of univalents as the quantitative trait value. A strong QTL was detected, which had a LOD score of ∼7 and explained 25% of the variation for the number of univalents (data not presented). We believe that this region contains PrBn, the main locus controlling homeologous pairing at metaphase I in oilseed rape haploids (see below for a more accurate estimate of PrBn position).
Construction of a full genetic map and identification of other QTL/BTL:
One hundred sixteen haploid plants were genotyped for 443 markers (358 AFLPs, 46 SSRs, and 39 RAPDs) to construct a full linkage map using MAPMAKER. Linkage groups were assigned and aligned to those on the reference map using common markers (at least 5 for each linkage group). Linkage group DY8 is represented by two subgroups still unlinked. All linkage groups add up to a total map length of ∼2400 cM, providing an estimated genome coverage of ∼90%. Average distance between two markers is ∼6 cM.
Mapping main-effect QTL:
Only two QTL, located on DY15 (PrBn) and on the very lower part of DY4 (uni4.1), met the criteria for statistical significance for all the analytical approaches we have implemented (Table 1; Figures 2 and 3). PrBn was identified as the main QTL, which explained twice as much of the variation for the number of univalents as uni4.1, the weaker QTL on DY4 (Table 1). We observed that the PrBn region showed a multiple-peak profile, which could be the indication of multiple-linked QTL (Figure 2); we discarded this hypothesis after reiterating composite interval mapping analyses with different cofactors on DY15 while keeping the other cofactors fixed (for a total of five cofactors). Regardless of the cofactor on DY15, only one QTL was detected on this linkage group. Considering that the model with the highest LOD score and the smallest AIC fitted the data best, the position of PrBn was set within an interval of 20 cM (one LOD support interval) centered by the marker E90M58h (Figure 2). Considering only the haploids that showed no recombination within this interval, we observed that 30 of the 116 haploids had a meiotic behavior that did not match their PrBn genotypes (thereafter named Meiotic Phenotype Unaccounted by PrBn, MPUP, haploids; supplemental Figure 1 at http://www.genetics.org/supplemental/). Extending this estimate to the entire population of haploids that had been phenotyped (Jenczewski et al. 2003), we observed that 28% (52 plants) of them were MPUP haploids (supplemental Figure 2 at http://www.genetics.org/supplemental/).
TABLE 1.
Characteristics of the QTL detected by composite interval mapping that contribute to the variation of the number of univalents
| QTLa | LGb | Peak markerc | Position (cM) | LOD score | R2 (%) | Additive effectd |
|---|---|---|---|---|---|---|
| PrBn | 15 | E36M65h | 34.9 | 7.7 | 24 | −3.2 univalents |
| uni4.1 | 4 | E35M64k | 135.67 | 3.3 | 11.5 | −2.0 univalents |
| (uni1a.1) | 1a | R10.2560 | 110.25 | 2.9 | 8.2 | +1.6 univalents |
| (uni13.1) | 13 | P91M34b | 43.4 | 2.5 | 7.9 | +1.6 univalents |
| Whole model | 12.32 | 40 |
QTL that meet the statistical significance criteria for only some but not all the analytical approaches implemented in this study are listed in parentheses.
Linkage group.
Peak markers define the position of the highest point on the probability curve (LOD plot).
Substitution effect of the Darmor-bzh allele by the Yudal allele.
Figure 2.—

Likelihood-ratio profile for linkage groups DY1a, DY4, DY13, and DY15 generated from composite interval mapping showing the position of the QTL affecting the mean number of univalents. LOD score threshold was determined using 1000 random permutations of the data and averaged 3.3. x-axis, map distances (centimorgans); y-axis, LOD score.
Figure 3.—

Genomic locations of the QTL, BTL, and epistatic loci affecting the number of univalents at metaphase I. These loci were detected by single-marker regression (stars), by composite interval mapping (solid bars), by the probabilistic BTL-mapping approach (circles, single-locus model; ovals, two-locus model), or by two-way ANOVAs (squares) in a haploid population, produced from F1 hybrids between Darmor-bzh (high-pairing parent) and Yudal (low-pairing parent). Map distances (centimorgans) are indicated on the left and the locus name on the right.
Two additional QTL, which had a LOD score just below the threshold (3.3), were also detected by composite interval mapping on DY1a (uni1a.1) and DY13 (uni13.1) (Table 1; Figures 2 and 3). These same two QTL were also detected by multiple interval mapping when considering the first round of QTL detection but were not identified by the nonparametric approach proposed by Kruglyak and Lander (1995) or by the regression interval mapping method of Haley and Knott (1992) (Figure 3). Instead these last two approaches provided evidence for a second QTL on DY1a (uni1a-2), which was also identified by single-marker regression analysis (supplemental Table 1 at http://www.genetics.org/supplemental/). As expected, most of the AFLP markers initially identified by bulked segregant analysis, but not mapped on DY15, were located within the minor QTL regions (Figure 3).
The genetic model composed of PrBn, uni4.1, uni1a.1, and uni13.1 accounted for 40% of the overall variation for the number of univalents.
Mapping BTL:
Six weak additive BTL were detected in addition to PrBn irrespective of the two procedures we used (Table 2). These BTL were generally consistent with the QTL reported above (Figure 3).
TABLE 2.
Characteristics of the BTL detected using a generalized linear model framework (Sen and Churchill 2001) and the probabilistic approach of Coffman et al. (2005)
| Generalized linear model (Sen and Churchill 2001): | Probabilistic approach (Coffman et al. 2005)
|
||||
|---|---|---|---|---|---|
| BTL | LGa | LOD score | LOD score | P > LOD | AICb |
| PrBn | 15 | 5.75 | 8.3 | <10−3 | 137.1 |
| uni4.1 | 4 | 1.85 | 1.65 | 0.022 | 167.6 |
| uni1a.1 | 1a | 1.81 | 1.9 | 0.013 | 166.5 |
| uni1a.2 | 1a | 1.78 | 1.75 | 0.020 | 167.1 |
| uni1b.1 | 1b | 1.67 | 1.7 | 0.005 | 165.4 |
| uni1b.2 | 1b | 1.70 | 2 | 0.009 | 165.8 |
| uni13.1 | 13 | 1.42 | 1.95 | 0.01 | 166.3 |
Linkage group.
Akaike's information criterion. The models with the smallest AIC are considered as the ones best supported by the data.
The first 18 best single-marker models identified by proc BTL were systematically obtained for markers targeting the PrBn region. The very best model was observed for marker E86M35a, which is only 6 cM apart from the peak marker identified by composite interval mapping (i.e., E90M58h; 6th best single-marker model). The same region produced the highest LOD score with Sen and Churchill's generalized linear model framework. Irrespective of the procedures we used, the one-LOD support interval for PrBn was set to cover a region of only 7 cM centered by the markers E86M35a/P76M37d.
Four significant or “just-below-the-threshold” QTL (uni1a.1, uni1a.2, uni4.1, and uni13.1) were also detected as significant BTL (Table 2). By contrast, the other two additional BTL, which are located at the opposing distal end of DY1b (uni1b.1 and uni1b.2), were not considered as significant by composite interval mapping even if they were identified by linear regression analysis (supplemental Table 1 at http://www.genetics.org/supplemental/; Figure 3).
The best two-marker models, selected by proc BTL, all contained PrBn but only one (uni13.1) among the five best markers detected by the single-marker model was included (Table 3). This observation suggests that several loci that had no intrinsic effect on the phenotype could interact with PrBn.
TABLE 3.
Five best two-locus models detected by proc BTL
| Closest markers to
|
||||
|---|---|---|---|---|
| BTL1 | BTL2 (LGa) | LOD score | P > LOD | AICb |
| E86M35a (PrBn) | N20-900 (14) | 13.2 | <10−3 | 128 |
| E86M35a (PrBn) | P91M34b (13) (uni13.1) | 12.4 | <10−3 | 129.3 |
| E86M35a (PrBn) | E38M59h (717) | 12.3 | <10−3 | 132.5 |
| E86M35a (PrBn) | E35M67e (3) | 11.3 | <10−3 | 134 |
| E86M35a (PrBn) | X02.500 (8a) | 11.95 | <10−3 | 135 |
Linkage group.
Akaike's information criterion. The models with the smallest AIC are considered as the ones best supported by the data.
Detection of epistatic effects:
Seventeen of the 77,028 possible interactions among any pair of markers were found to be significant by two-way ANOVAs (P ≤ 0.0005 and R2 ≥ 5%; Table 4) while only one was expected by chance. This indicates that real interactions exist in the genome. No significant interaction was detected among the main-effect QTL/BTL (including PrBn). The strongest interaction was found between markers Q05-800 (DY2) and F04-CD1 (DY13) (P = 2.35 × 10−5) as well as other nearby markers. A very strong interaction was also found between the PrBn region (N20.1320 and E86M40h) and the top part of DY14 (close to N20-900; P ≤ 0.0002 and 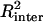 ≥ 10%), which is consistent with the best two-marker model selected by proc BTL (Table 3). The second region that interacted most with PrBn was found on DY717, centered by marker E86M41a (P ≤ 0.005 and R2 ≥ 5%). This region is slightly different from the one detected by proc BTL on DY717 (Figure 3) and it remains to be understood whether these markers point to a single or to two linked epistatic QTL.
≥ 10%), which is consistent with the best two-marker model selected by proc BTL (Table 3). The second region that interacted most with PrBn was found on DY717, centered by marker E86M41a (P ≤ 0.005 and R2 ≥ 5%). This region is slightly different from the one detected by proc BTL on DY717 (Figure 3) and it remains to be understood whether these markers point to a single or to two linked epistatic QTL.
TABLE 4.
Epistatic interactions detected by two-way ANOVAs
| Marker 1 | QTLa | LGb | Marker 2 | QTLa | LGb | P-value |
|---|---|---|---|---|---|---|
| E81M37k | uni1a.2 | 1a | E86M40q | 717 | 0.000226 | |
| E35M64a | 2 | E46M64i | 6 | 0.000407 | ||
| Q05.800 | 2 | F04.CD1 | 13 | 2.35E-05 | ||
| Q05.800 | 2 | E90M40d | 13 | 0.000186 | ||
| Q05.800 | 2 | E86M38o | 13 | 0.000425 | ||
| E35M64p | 3 | E46M66e | 6 | 0.000417 | ||
| E35M64p | 3 | E36M65i | 717 | 0.000382 | ||
| E33M64a | 5 | E86M40q | 717 | 0.000234 | ||
| E86M35b | 5 | E86M40q | 717 | 0.000409 | ||
| P87M78k | 717 | H12.1010 | 8b | 0.000339 | ||
| R10.360 | 717 | E90M40c | 13 | 0.000381 | ||
| E86M40q | 717 | P69M77a | 15 | 0.000421 | ||
| P91M33a | 9 | P91M33b | 15 | 0.000252 | ||
| P87M78f | 9 | P91M33b | 15 | 0.000331 | ||
| P87M78f | 9 | E33M50n | 19 | 0.000491 | ||
| N20.900 | 14 | E86M40h | PrBn | 15 | 0.000127 | |
| N20.900 | 14 | N20.1320 | PrBn | 15 | 0.000428 |
QTL and BTL identified by the conventional or binary-trait-oriented approaches.
Linkage group.
Using tree-based methods to uncover the most influential loci:
We then used tree-based methods as an exploratory technique to identify from the large number of mapped genetic markers (predictor variables) those that best explain the meiotic phenotype of haploids (response variable: number of univalents). These methods are useful when predictors are associated in some nonlinear fashion, as no implicit assumptions about the model for the response are made. Tree-based methods provided an opportunity to search for the presence of multiple, potentially interacting QTL (even with higher-order interactions) and to rank markers according to their impact on the number of univalents but irrespective of whether they have an effect on their own (additive QTL) or only through interactions (epistatic QTL). Tree-based methods are also robust against the nonnormality of the phenotype as no hypothesis is made on the distribution of the response variable.
Tree-based methods first confirmed the results from the different statistical approaches reported previously. As expected according to the results presented above, the markers located in the interval around PrBn provided the highest importance measures (Figure 4). The peak markers for uni1a.1, uni1a.2, uni1b.2, and uni4.1 as well as four of the loci found to interact with PrBn (N20-900, E86M41a, E35M67e, and E38M59h) often participated in tree construction although they displayed only a low importance measure. They therefore improved the prediction of the number of univalents slightly but almost systematically.
Figure 4.—

Tree-based identification of markers that best explain the meiotic phenotype of B. napus haploids (response variable: number of univalents). Every mapped marker m is plotted according to (i) B(m), the number of trees containing this marker (x-axis) and (ii) I(m), the relative increase in prediction error estimate obtained after permuting of the values of m (y-axis). The higher the increase in prediction error is, the more influential the marker (e.g., E86M35a).
Tree-based methods then provided additional insights into the genetic architecture of homeologous chromosome pairing in B. napus haploids. First, they showed that the two loci that were previously shown to interact with PrBn (N20-900, E86M41a) improved the prediction of the number of univalents as often as or even more often than those targeting the additive QTL (Figure 4). Second, partitioning the measure of marker importance into four additive components (that represent the contribution of haploids whose meiotic behaviors match or do not their genotypes in the PrBn region) allowed us to identify the loci that improved the prediction of the number of univalents in the MPUP haploids. This approach revealed that E38M59h, and to a lesser extent E35M67e, both found to interact with PrBn in previous analyses (best two-marker model; Table 3), as well as the peak marker for uni1b.2 (i.e., CB10081), contribute mainly to the meiotic phenotype of haploids that displayed a high number of univalents despite the presence of the permissive allele (i.e., Darmor-bzh) at PrBn (Table 5). Conversely, the two epistatic QTL that interacted with PrBn (N20-900 and E86M41a), the peak marker for uni4.1 (Bras002b), as well as two loci on linkage group DY19 (E33M50e, E43M66f) were found to be the most influential to explain the meiotic behavior of haploids that displayed a low number of univalents despite the presence of the strongest suppressive allele (i.e., Yudal) at PrBn (Table 5). But in no case did the segregation of these loci explain the shift in the meiotic behavior observed in all the haploids whose meiotic behavior is unaccounted for by PrBn.
TABLE 5.
Importance measure for markers contributing to the prediction of the meiotic behavior of the MPUP haploids
|
PrBn = Darmor
|
PrBn = Yudal
|
|||||
|---|---|---|---|---|---|---|
| Marker | LGa | QTLb | No. uni < 6 | No. uni > 7 (MPUP) | No. uni > 7 | No. uni < 6 (MPUP) |
| P69M39a | 1a | uni1a-2 | 0.005 | 0.007 | 0.045 | <0 |
| CB10572a | 1a | uni1a-1 | <0 | 0.010 | <0 | 0.015 |
| JLP015 | 1b | uni1b-1 | <0 | 0.012 | <0 | 0.007 |
| CB10081 | 1b | uni1b-2 | <0 | 0.043 | <0 | 0.017 |
| E35M67e | 3 | 0.002 | 0.034 | <0 | 0.003 | |
| Brass002b | 4 | uni4-1 | 0.001 | <0 | <0 | 0.058 |
| E38M59h | 717 | <0 | 0.086 | 0.002 | 0.000 | |
| E86M41a | 717 | <0 | 0.014 | 0.002 | 0.040 | |
| N20-900 | 14 | 0.000 | 0.002 | <0 | 0.079 | |
| E86M35a | 15 | PrBn | 0.343 | <0 | 0.290 | <0 |
| E86M40a | 15 | PrBn | 0.170 | <0 | 0.120 | <0 |
| E33M50e | 19 | 0.000 | 0.011 | <0 | 0.049 | |
| E43M66f | 19 | 0.023 | <0 | 0.041 | 0.060 | |
Linkage group.
QTL and BTL identified by the conventional or binary-trait-oriented approaches. Marker importance measure has been decomposed into four additive components that represent the contribution of the haploids whose meiotic behaviors match (PrBn = Darmor and no. uni < 6; PrBn = Yudal and no. uni > 7) or do not match (PrBn = Darmor and no. uni > 7; PrBn = Yudal and no. uni < 6) their genotypes in the PrBn region to the measure of marker importance. Note that the higher the importance measure is, the more influential the marker. uni, univalents.
DISCUSSION
Apart from wheat (Griffiths et al. 2006), little effort has been made to understand the mechanisms responsible for the meiotic diploidization of allopolyploid species (Jenczewski and Alix 2004). Jenczewski et al. (2003) recently obtained evidence for the genetic control of homeologous chromosome pairing in B. napus by examining the segregation pattern for pairing behavior at metaphase I in a population of haploids produced from F1 hybrids between two natural B. napus lines with high- and low-pairing behaviors at the haploid stage (at metaphase I). They observed that the parental metaphase I pairing patterns were inherited in a Mendelian fashion, supporting the presence of a single major gene that determines the homeologous chromosomal pairing in haploids. In this study, we provide new insights into the genetic architecture of this trait and demonstrate that the hereditary components of homeologous chromosome pairing are polygenic rather than monogenic. Notwithstanding this conclusion, our survey has pointed to a locus on DY15 that is far more influential than all the others.
Mapping PrBn, the most influential locus reducing homeologous pairing at metaphase I:
A systematic bulked segregant analysis screening using ∼10,000 previously mapped as well as anonymous molecular markers pointed to a single region of linkage group DY15 where the strongest QTL was subsequently mapped irrespective of the statistical procedures used. The segregation of this QTL explained up to 24% of the variance for the number of univalents (composite interval mapping estimate) and ∼75% of the haploids from the segregating population had a meiotic behavior matching their genotypes in this region. Taking into account (i) the number of AFLP markers used to screen the two bulks, (ii) the fact that most of the markers identified with bulked segregant analysis were located on DY15, (iii) that none of the remaining markers had higher cosegregation ratios with the phenotype, and (iv) that none of the 120 RAPD and AFLP markers previously mapped and distributed on all the other 18 linkage groups was polymorphic between the two bulks of contrasting haploids, the existence of a major gene on other linkage groups is very unlikely. Accordingly, we believe that PrBn, the main locus controlling homeologous pairing, is located within an interval of 10–20 cM on DY15 (C genome). Notwithstanding this conclusion, ∼28% of the haploids that showed no recombination within the PrBn interval had a meiotic behavior that did not match their PrBn genotypes (supplemental Figure 2 at http://www.genetics.org/supplemental/). Accordingly, part of the variation measured for the amount of homeologous chromosome pairing at metaphase I cannot be explained by the polymorphism at a single locus, and the effect of PrBn on the meiotic phenotype of oilseed rape haploid could be a collective property of a network of genes, rather than that of a gene alone. This prompts us to explore the oligogenic models in which multiple loci may modify the penetrance and/or expressivity of the traits.
Cumulative effects of weaker additive QTL/BTL:
Through the use and comparison of different statistical methods, we were able to detect consistently and map three to six QTL/BTL in addition to PrBn that contribute to the variation for homeologous pairing at metaphase I. Identification of additional QTL/BTL is consistent with the results of Jenczewski et al. (2003), who found that the distribution for the number of univalents among the haploids from the segregating population was not the mixture of the two parental distributions. Our results demonstrate that (i) a repressive allele(s) at at least one QTL is required in addition to PrBn (Yudal allele) to obtain the same low-pairing behavior in the haploids of the segregating population as in Yudal parental haploids and (ii) the cumulative effect of weak additive QTL is sufficient to explain why the mean number of univalents in the high-pairing subpopulation of the progeny was higher than that in the parental Darmor-bzh haploids. Progressive decrease in the number of univalents was reciprocally observed in the low-pairing subpopulation of the progeny due to the accumulation of permissive alleles. But in no case could the accumulation of the repressive/permissive alleles at weak additive QTL/BTL explain the shift in the meiotic behavior observed in the MPUP haploids.
Can environmental variation be responsible for the occurrence of the MPUP haploids?
Although variations in temperature have been shown to affect chromosome pairing and recombination (Riley et al. 1973), we are not convinced that environmental variation can explain the meiotic phenotype of the MPUP haploids. Indeed, Jenczewski et al. (2003) found that only a small proportion (7%) of the overall variance in the number of univalents could be attributed to the environment and that haploids produced from the parental Darmor-bzh and Yudal genotypes always had the expected (high-pairing and low-pairing at metaphase I) meiotic behavior. This observation has been confirmed in many independent experiments that took place over almost 10 years and encompassed a much wider range of environmental variations than those accounted for in this study. The MPUP haploids have been observed only in the segregating population produced using the high- and the low-pairing varieties as parents. Furthermore, the metaphase I meiotic pairing behavior of some MPUP haploids has been observed more than once in an interval of several weeks and almost constant metaphase I pairing levels were always obtained. All these observations indicate that environmental variation cannot explain the shift in the meiotic behavior observed in the MPUP haploids.
Do preexisting nonreciprocal translocations play a role?
Udall et al. (2005) proposed that preexisting homeologous nonreciprocal translocations (HNRTs) could contribute to the genetic variation for chromosome pairing behavior between the parental genotypes and segregate among the haploids of the offspring population, which we have examined in this work. This hypothesis is attractive, for it provides a mechanistic scenario for understanding why homeologous chromosomes with preexisting HNRTs (therefore carrying homologous segments) are able to pair and recombine even in the presence of a PrBn suppressive allele. According to this hypothesis, three pairs of homeologous chromosomes with preexisting HNRTs would be needed to shift the phenotype from low to high pairing and all the haploids with a high-pairing meiotic phenotype unaccounted for by the Yudal allele at PrBn would have the same genotype in these regions. These two expectations do not hold true in light of the data. First, about one-third of the haploids having the suppressive Yudal allele at PrBn displayed a high-pairing behavior at metaphase I whereas only 12.5% should have inherited the three translocated chromosomes (assuming an independent segregation). Second, tree-based methods clearly demonstrate that the meiotic phenotype of the MPUP haploids cannot be predicted by the segregation of only three regions. Accordingly, the shift in the meiotic behavior observed in the MPUP haploids is not due to the segregation of preexisting HNRTs. Nevertheless, we acknowledge that the BTL detected on the top part of DY1b (marker JLP015) could correspond to a preexisting HNRT. Indeed, JLP015 is a PCR-specific marker derived from a B. oleracea subfamily of SINE S1 retroelements but mapped onto a linkage group of the A genome (Prieto et al. 2005).
Epistatic interactions:
Epistasis (interactions among two or more genetic loci) is ubiquitous in the genetic control and evolution of most complex traits (Lynch and Walsh 1998) and broadly important in cancer and evolution (Wolf et al. 2000). Meiotic recombination is particularly prone to epistatic interactions as it involves (i) many successive and interdependent steps and (ii) protein complexes whose components act synergistically. It was therefore not surprising that we found epistatic QTL contributing to the control of homeologous pairing. We are aware that, given the sample size and the methodology used, it was not possible to give an accurate quantification of the number of interacting loci and consequently to measure the contribution of epistasis to the genetic variation for the amount of pairing at metaphase I in oilseed rape haploids.
A first concern comes from the fact that the detection of the interaction between any two loci may be confounded by the segregation of additional QTL, which might be different from one test to another. Several authors proposed to reanalyze the highly significant interactions detected in two-way ANOVAs by multiple regression with all QTL being fixed in the model (Li et al. 1997; Holland et al. 2002). If this procedure is efficient to remove the false positives, it is of no help when the number of nonadditive interactions has been underestimated during the two-way ANOVAs search and has led to overparameterization in our study. We confirmed the detection of two epistatic QTL using tree-based procedures that do not require any hypothesis to be made about the distribution of the response variable or about the form of underlying relationships between the predictor variables and the response. This approach, which is currently being tested as a screening procedure to identify risk-associated SNPs to complex disease models (Zhang and Bonney 2000; Lunetta et al. 2004), confirmed that the two loci that interacted with PrBn contributed more often to the prediction of the number of univalents than those targeting the additive QTL (Figure 4). A second concern is that two-way ANOVAs are capable of detecting interactions only between markers, by which epistasis between QTL can be identified but not reliably estimated (Wang et al. 1999). Several statistical models and methods have been regularly proposed to address these questions, but there is not yet a consensus regarding the most appropriate approach to detect QTL with no additive but only epistatic effects.
Of particular interest in our study are the two QTL located on DY14 and DY717, which interact directly with PrBn and appear to be as influential on the number of univalents as the additive QTL detected on DY4 and DY1a (Figure 4). Detailed analysis revealed that 90% of the haploids carrying a Yudal allele at QTL-DY14 had a meiotic behavior consistent with their PrBn genotype, whereas half the haploids having a Darmor-bzh allele at that locus were MPUP haploids (supplemental Figure 1 at http://www.genetics.org/supplemental/). The situation is somewhat different for QTL-DY717 because the Darmor-bzh allele at locus E86M41a (DY717) is associated with a higher proportion of MPUP haploids only when PrBn has a Yudal allele. These observations indicate that the Yudal alleles at QTL-DY14 and QTL-DY717 are required for PrBn to have a correct suppressive effect.
Similar interactions have already been described in wheat, in which homeologous chromosome pairing is controlled by a delicate balance between homeologous pairing promoters and suppressors, including Ph1 (see Jenczewski and Alix 2004 for a review). First, as allelic variation at the Ph1 locus is very unlikely (Griffiths et al. 2006), variation that exists between different wheat genotypes as to effectiveness of the diploidization mechanism (Driscoll and Quinn 1970; Martinez et al. 2001; Ozkan and Feldman 2001) is certainly the result of variation at other loci that interact with Ph1 in an additive or nonadditive way (Driscoll and Quinn 1970). This idea is reinforced by the detection of genotypes of Aegilops speltoides (Riley et al. 1961), Ae. mutica (Riley 1966), Ae. longissima (Mello-Sampayo 1972), and Secale cereale (Lelley 1976), where the suppression of the effect of Ph1 has been found. The inactivating effect on Ph1 observed on these genotypes evidently echoes the reduced PrBn effect encountered in the MPUP haploids in this study. Mapping of wheat Ph1 suppressors has very recently been achieved in Ae. speltoides and provided evidence for epistatic interactions among these regulators (Dvorak et al. 2006). Nonadditive interactions among wheat pairing regulators have also been highlighted by Mello-Sampayo and Canas (1973), who demonstrated that wheat–rye hybrids deprived of the short arms of both chromosomes 3A and 3D displayed higher amounts of homeologous pairing than the sum of the pairing values in the absence of either single chromosome arm. These authors concluded that the expression of any of these regulators depended upon the presence/absence of other regulators.
Concluding remarks:
Our results demonstrate that the control of homeologous pairing in B. napus haploids is far more complex than we previously thought even though PrBn still appears to be the main determinant in this process. Two scenarios can be proposed to reconcile the bimodal distribution of the mean number of univalents observed in the segregating population of haploids with the complex genetic architecture unraveled in this study. The first one is to assume that, as in other species, homeologous pairing in B. napus haploids is controlled by an integrated system of several genes that have effects on different processes throughout the premeiotic interphase and the meiotic prophase; these genes therefore do not interact in an additive way. According to this scenario, one may speculate that several loci that code for proteins also involved in the meiosis of oilseed rape haploids and positioned upstream of PrBn in the genetic pathway may partially inactivate PrBn and contribute to its reduced penetrance. The alternative scenario is to consider the ability of the homeologous chromosome to pair as a threshold character, each locus contributing a cumulative effect underlying the dichotomy of the phenotype. According to this second scenario, a threshold dose should be achieved for the meiotic phenotype to switch from low to high pairing and vice versa; the dosage effects of replicated genes could play a prime role in this process. An in-depth understanding of the epistatic interactions is therefore important to clarify the influence of genetic background on the activity of PrBn and to determine the extent to which PrBn activity is dosage dependent. Jenczewski et al. (2003) have already formulated the hypothesis that the allele present in genotypes with a high-pairing behavior at the haploid stage could be haplo-insufficient to explain why all B. napus accessions display regular bivalent associations and disomic inheritance irrespective of the amount of chromosome pairing in their haploid forms. One may finally note that lengthy research has been necessary to clone the Ph1 locus that was identified nearly 50 years ago (Griffiths et al. 2006). Nothing suggests that the task will be easier for oilseed rape, a multiple-round allopolyploid species.
Acknowledgments
We thank J. C. Letanneur for technical assistance and Antoine Lostanlen and OUEST-genopole for providing us the genotyping facilities. We also thank C. Mezard and O. Loudet (INRA Versailles, France), L. Moreau (UMR de Génétique Végétale du Moulon, France), as well as two anonymous reviewers for their help and fruitful comments on a previous version of this manuscript. Z. Liu was supported by a postdoctoral fellowship from Institut National de la Recherche Agronomique (INRA)-GAP. Research support was provided by INRA-DSPPV and INRA-Génétique et Amélioration des Plantes as well as by a grant from the Agence Nationale pour la Recherche.
References
- Attia, T., and G. Robbelen, 1986. Meiotic pairing in haploids and amphihaploids of spontaneous versus synthetic origin in rape, Brassica napus L. Can. J. Genet. Cytol. 28: 330–334. [Google Scholar]
- Ayoub, M., and D. E. Mather, 2002. Effectiveness of selective genotyping for detection of quantitative trait loci: an analysis of grain and malt quality traits in three barley populations. Genome 45: 1116–1124. [DOI] [PubMed] [Google Scholar]
- Benavente, E., K. Alix, J. C. Dusautoir, J. Orellana and J. L. David, 2001. Early evolution of the chromosomal structure of Triticum turgidum-Aegilops ovata amphiploids carrying and lacking the Ph1 gene. Theor. Appl. Genet. 103: 1123–1128. [Google Scholar]
- Blanc, G., and K. H. Wolfe, 2004. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell 16: 1667–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman, L., 1996. Bagging predictors. Mach. Learning 26: 123–140. [Google Scholar]
- Breiman, L., J. Friedman, R. Olshen and C. Stone, 1984. Classification and Regression Trees. Wadsworth, Belmont, CA.
- Broman, K. W., H. Wu, S. Sen and G. A. Churchill, 2003. R/qtl: QTL mapping in experimental crosses. Bioinformatics 19: 889–890. [DOI] [PubMed] [Google Scholar]
- Churchill, G. A., and R. W. Doerge, 1994. Empirical threshold values for quantitative trait mapping. Genetics 138: 963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffman, C. J., R. W. Doerge, K. L. Simonsen, K. M. Nichols, C. K. Duarte et al., 2005. Model selection in binary trait locus mapping. Genetics 170: 1281–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comai, L., 2005. Advantages and disadvantages of polyploidy. Nat. Rev. Genet. 6: 836–846. [DOI] [PubMed] [Google Scholar]
- Doyle, J. J., and J. L. Doyle, 1990. Isolation of plant DNA from fresh tissues. Focus 12: 13–15. [Google Scholar]
- Driscoll, C. J., and C. J. Quinn, 1970. Genetic variation in Triticum affecting the level of chromosome pairing in intergeneric hybrids. Can. J. Genet. Cytol. 12: 278–282. [Google Scholar]
- Dvorak, J., K. R. Deal and M.-C. Luo, 2006. Discovery and mapping of wheat Ph1 suppressors. Genetics 174: 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eber, F., A. M. Chèvre, A. Baranger, P. Vallée, X. Tanguy et al., 1994. Spontaneous hybridization between a male-sterile oilseed rape and two weeds. Theor. Appl. Genet. 88: 362–368. [DOI] [PubMed] [Google Scholar]
- Feldman, M., B. Liu, G. Segal, S. Abbo, A. A. Levy et al., 1997. Rapid elimination of low-copy DNA sequences in polyploidy wheat: a possible mechanism for differentiation of homeologous chromosomes. Genetics 147: 1381–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foisset, N., R. Delourme, P. Barrer, N. Hubert, B. S. Landry et al., 1996. Molecular-mapping analysis in Brassica napus using isozyme, RAPD and RFLP markers on a doubled-haploid progeny. Theor. Appl. Genet. 93: 1017–1025. [DOI] [PubMed] [Google Scholar]
- Gillies, C. B. (Editor), 1989. Chromosome pairing and fertility in polyploids, pp. 137–176 in Fertility and Chromosome Pairing: Recent Studies in Plants and Animals. CRC Press, Boca Raton, FL.
- Griffiths, S., R. Sharp, T. N. Foote, I. Bertin, M. Wanous et al., 2006. Molecular characterization of Ph1 as a major chromosome pairing locus in polyploid wheat. Nature 439: 749–752. [DOI] [PubMed] [Google Scholar]
- Hardberg, D. J., 1976. Cytotaxonomic studies of Brassica and related genera, pp. 47–68 in The Biology and Chemistry of the Crucifereae, edited by J. G. Vaughan, A. J. MacLeod and B. M. Jones. Academic Press, London.
- Haley, C. S., and S. A. Knott, 1992. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity 69: 315–324. [DOI] [PubMed] [Google Scholar]
- Holland, J. B., V. A. Portyanko, D. L. Hoffman and M. Lee, 2002. Genomic regions controlling vernalization and photoperiod responses in oat. Theor. Appl. Genet. 105: 113–126. [DOI] [PubMed] [Google Scholar]
- Jenczewski, E., and K. Alix, 2004. From diploids to allopolyploids: the emergence of efficient pairing control genes in plants. Crit. Rev. Plant Sci. 23: 21–25. [Google Scholar]
- Jenczewski, E., F. Eber, A. Grimaud, S. Huet, M.-O. Lucas et al., 2003. PrBn, a major gene controlling homeologous pairing in oilseed rape (Brassica napus) haploids. Genetics 164: 645–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao, C.-H., Z.-B. Zeng and R. D. Teasdale, 1999. Multiple interval mapping for quantitative trait loci. Genetics 152: 1203–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruglyak, L., and E. S. Lander, 1995. A nonparametric approach for mapping quantitative trait loci. Genetics 139: 1421–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelley, T., 1976. Induction of homeologous pairing in wheat by genes of rye suppressing chromosome 5B effect. Can. J. Genet. Cytol. 18: 485–489. [Google Scholar]
- Li, Z., S. R. M. Pinson, W. D. Park, A. H. Paterson and J. W. Stansel, 1997. Epistasis for three grain yield components in rice (Oryza sativa L.). Genetics 145: 453–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoln, S. E., M. J. Daly and E. S. Lander, 1992. Constructing Genetic Linkage Maps With MAPMAKER/EXP 3.0: A Tutorial and Reference Manual. Whitehead Institute Technical Report, Ed. 3, Whitehead Institute, Cambridge, MA.
- Liu, B., and J. F. Wendel, 2003. Epigenetic phenomena and the evolution of plant allopolyploids. Mol. Phylogenet. Evol. 29: 365–379. [DOI] [PubMed] [Google Scholar]
- Lombard, V., and R. Delourme, 2001. A consensus linkage map for rapeseed (Brassica napus L.): construction and integration of three individual maps from DH populations. Theor. Appl. Genet. 103: 491–507. [Google Scholar]
- Lowe, A. J., C. Moule, M. Trick and K. J. Edwards, 2004. Efficient large-scale development of microsatellites for marker and mapping applications in Brassica crop species. Theor. Appl. Genet. 108: 1103–1112. [DOI] [PubMed] [Google Scholar]
- Lukens, L. N., P. A. Quijada, J. Udall, J. C. Pires, M. E. Schranz et al., 2004. Genome redundancy and plasticity within ancient and recent Brassica crop species. Biol. J. Linn. Soc. 82: 665–674. [Google Scholar]
- Lunetta, K. L., L. B. Hayward, J. Segal and P. van Eerdewegh, 2004. Screening large-scale association study data: exploiting interactions using random forests. BMC Genet. 5: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M., and B. Walsh, 1998. Genetics and Analysis of Quantitative Traits. Sinauer, Sunderland, MA.
- Lysak, M. A., M. A. Koch, A. Pecinka and I. Schubert, 2005. Chromosome triplication found across the tribe Brassiceae. Genome Res. 15: 516–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez, M., T. Naranjo, C. Cuadrado and C. Romero, 2001. The synaptic behaviour of the wild forms of Triticum turgidum and T. timopheevi. Genome 44: 517–522. [DOI] [PubMed] [Google Scholar]
- McIntyre, L. M., C. Coffman and R. W. Doerge, 2001. Detection and location of a single binary trait locus in experimental populations. Genet. Res. 78: 79–92. [DOI] [PubMed] [Google Scholar]
- Mello-Sampayo, T., 1972. Promotion of homeologous pairing in hybrids of Triticum aestivum x Aegilops longissima. Genet. Iber. 23: 1–9. [Google Scholar]
- Mello-Sampayo, T., and A. P. Canas, 1973. Suppressors of meiotic chromosome pairing in common wheat, pp. 709–713 in 4th International Wheat Genetics Symposium, edited by E. R. Sears and L. M. S. Sears. Agricultural Experiment Station, College of Agriculture and University of Missouri, Columbia, MO.
- Michelmore, R. W., I. Paran and R. V. Kesseli, 1991. Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl. Acad. Sci. USA 88: 9828–9832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn, T. C., D. V. Butruille, A. G. Sharpe, K. J. Pickering, I. A. P. Parkin et al., 2003. a Detection and effects of a homeologous reciprocal transposition in Brassica napus. Genetics 165: 1569–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn, T. C., J. C. Pires, J. A. Birchler, D. L. Auger, Z. J. Chen et al., 2003. b Understanding mechanisms of novel gene expression in polyploids. Trends Genet. 19: 141–147. [DOI] [PubMed] [Google Scholar]
- Otto, S. P., and J. Whitton, 2000. Polyploid incidence and evolution. Annu. Rev. Genet. 34: 401–437. [DOI] [PubMed] [Google Scholar]
- Ozkan, H., and M. Feldman, 2001. Genotypic variation in tetraploid wheat affecting homeologous pairing in hybrids with Aegilops peregrina. Genome 44: 1000–1006. [PubMed] [Google Scholar]
- Parkin, I. A. P., A. G. Sharpe, D. J. Keith and D. J. Lydiate, 1995. Identification of the A and C genomes of amphidiploid Brassica napus (oilseed rape). Genome 38: 1122–1131. [DOI] [PubMed] [Google Scholar]
- Parkin, I. A. P., A. G. Sharpe and D. J. Lydiate, 2003. Patterns of genome duplication within the Brassica napus genome. Genome 46: 291–303. [DOI] [PubMed] [Google Scholar]
- Parkin, I. A. P., S. M. Gulden, A. G. Sharpe, L. Lukens, M. Trick et al., 2005. Segmental structure of the Brassica napus genome based on comparative analysis with Arabidopsis thaliana. Genetics 171: 765–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piquemal, J., E. Cinquin, F. Couton, C. Rondeau, E. Seignoret et al., 2005. Construction of an oilseed rape (Brassica napus L.) genetic map with SSR markers. Theor. Appl. Genet. 111: 1514–1523. [DOI] [PubMed] [Google Scholar]
- Pires, J. C., J. W. Zhao, M. E. Schranz, E. J. Leon, P. A. Quijada et al., 2004. Flowering time divergence and genomic rearrangements in resynthesized Brassica polyploids (Brassicaceae). Biol. J. Linn. Soc. 82: 675–688. [Google Scholar]
- Prakash, S., 1974. Probable basis of diploidization of Brassica juncea Coss. Can. J. Genet. Cytol. 16: 232–234. [Google Scholar]
- Prieto, J. L., N. Pouilly, E. Jenczewski, J. M. Deragon and A. M. Chèvre, 2005. Development of crop-specific transposable element (SINE) markers to study gene flow from oilseed rape to wild radish. Theor. Appl. Genet. 111: 446–455. [DOI] [PubMed] [Google Scholar]
- Ramsey, J., and D. W. Schemske, 2002. Neopolyploidy in flowering plants. Annu. Rev. Ecol. Syst. 33: 589–639. [Google Scholar]
- Rebai, A., 1997. Comparison of methods for regression interval mapping in QTL analysis with non-normal traits. Genet. Res. 69: 69–74. [Google Scholar]
- Renard, M., and F. Dosba, 1980. Etude de l'haploidie chez le colza (Brassica napus L. Var oleifera Metzger). Ann. Amél. Pl. 30: 191–209. [Google Scholar]
- Riley, R., 1966. The genetic regulation of meiotic behaviour in wheat and its relatives. Proceedings of the 2nd International Wheat Syposium (Lund 1963). Hereditas 2(Suppl.): 395–408. [Google Scholar]
- Riley, R., and V. Chapman, 1958. Genetic control of the cytologically diploid behaviour of hexaploid wheat. Nature 13: 713–715. [Google Scholar]
- Riley, R., G. Kimber and V. Chapman, 1961. Origin of genetic control of diploid-like behavior of polyploid wheat. J. Hered. 52: 22–25. [Google Scholar]
- Riley, R., V. Chapman and T. E. Miller, 1973. The determination of meiotic chromosome pairing, pp. 731–738 in 4th International Wheat Genetics Symposium, edited by E. R. Sears and L. M. S. Sears. Agricultural Experiment Station, College of Agriculture and University of Missouri, Columbia, MO.
- Salina, E. A., O. M. Numerova, H. Ozkan and M. Feldman, 2004. Alterations in subtelomeric tandem repeats during early stages of allopolyploidy in wheat. Genome 47: 860–867. [DOI] [PubMed] [Google Scholar]
- Sánchez-Morán, E., E. Benavente and J. Orellana, 2001. Analysis of karyotypic stability of homoeologous-pairing (ph) mutants in allopolyploid wheat. Chromosoma 110: 371–377. [DOI] [PubMed] [Google Scholar]
- Sears, E. R., and M. Okamoto, 1958. Intergenomic chromosome relationships in hexaploid wheat. 10th Int. Congr. Genet. 2: 258–259. [Google Scholar]
- Sen, S., and G. A. Churchill, 2001. A statistical framework for quantitative trait mapping. Genetics 159: 371–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe, A. G., I. A. P. Parkin, D. J. Keith and D. J. Lydiate, 1995. Frequent nonreciprocal translocations in the amphidiploid genome of oilseed rape (Brassica napus). Genome 38: 1112–1121. [DOI] [PubMed] [Google Scholar]
- Truco, M. J., J. Hu, J. Sadowski and C. F. Quiros, 1996. Inter- and intra-genomic homology of the Brassica genomes: implications for their origin and evolution. Theor. Appl. Genet. 93: 1225–1233. [DOI] [PubMed] [Google Scholar]
- Udall, J. A., P. A. Quijada and T. C. Osborn, 2005. Detection of chromosomal rearrangements derived from homeologous recombination in four mapping populations of Brassica napus L. Genetics 169: 967–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos, P., M. Hogers, M. Bleeker, M. Reijans, T. van de Lee et al, 1995. AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res. 23: 4407–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, D. L., J. Zhu, Z. K. Li and A. H. Paterson, 1999. Mapping QTLs with epistatic effects and QTL × environment interactions by mixed linear model approaches. Theor. Appl. Genet. 99: 1255–1264. [Google Scholar]
- Wang, S., C. J. Basten and Z.-B. Zeng, 2005. Windows QTL Cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh, NC (http://statgen.ncsu.edu/qtlcart/WQTLCart.htm).
- Wolf, J. B., E. D. I. I. I. Brodie and M. J. Wade, 2000. Epistasis and the Evolutionary Process. Oxford University Press, New York
- Zeng, Z.-B., 1994. Precision mapping of quantitative trait loci. Genetics 136: 1457–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H., and G. Bonney, 2000. Use of classification trees for association studies. Genet. Epidemiol. 19: 323–332. [DOI] [PubMed] [Google Scholar]
- Zhao, J. W., J. A. Udall, P. A. Quijada, C. R. Grau, J. Meng et al., 2006. Quantitative trait loci for resistance to Sclerotinia sclerotiorum and its association with a homeologous non-reciprocal transposition in Brassica napus L. Theor. Appl. Genet. 112: 509–516. [DOI] [PubMed] [Google Scholar]