

Abstract
We present a genome-wide view of the male gametophytic transcriptome in Arabidopsis based on microarray analysis. In comparison with the transcriptome of the sporophyte throughout development, the pollen transcriptome showed reduced complexity and a unique composition. We identified 992 pollen-expressed mRNAs, nearly 40% of which were detected specifically in pollen. Analysis of the functional composition of the pollen transcriptome revealed the over-representation of mRNAs encoding proteins involved in cell wall metabolism, cytoskeleton, and signaling and under-representation of mRNAs involved in transcription and protein synthesis. For several gene families, we observed a common pattern of mutually exclusive gene expression between pollen and sporophytic tissues for different gene family members. Our results provide a 50-fold increase in the knowledge of genes expressed in Arabidopsis pollen. Moreover, we also detail the extensive overlap (61%) of the pollen transcriptome with that of the sporophyte, which provides ample potential to influence sporophytic fitness through gametophytic selection.
The functional specialization of the haploid male gametophyte and the closed carpel in particular are thought to be key factors in the evolutionary success of flowering plants. Competition between male gametophytes for fertilization of a limited number of egg cells is common, and pronounced differences in pollen tube growth rate reflect genetic differences between individual microgametophytes (Mulcahy, 1979; Mulcahy et al., 1996). Gametophytic selection provides a barrier against poorly functioning haploid genomes, serving to reduce the influence of random events and to promote the rigorous selection of superior haploid genotypes. The supporting argument is based on the criterion that sexual reproduction would provide a long-term beneficial effect only if population sizes were at least 10 times the reciprocal of rates at which favorable mutations occur (Maynard Smith, 1971). Unlike sporophytes, with a typical spontaneous mutation frequency of 10–6 per locus, populations containing 107 individuals are common for the microgametophyte. These facts argue for the rapid evolution of gametophytically expressed genes that encode specialized functions that contribute to improve the fitness of the male gametophyte. On the contrary, gametophytic selection can influence the fitness of the sporophyte only if there is significant overlap between genes expressed in both gametophyte and sporophyte generations (Mulcahy, 1979; Mulcahy et al., 1996).
It was isozyme studies that first suggested widespread overlap of gametophytic and sporophytic gene expression. In various species, 60% to 72% of isozymes analyzed were expressed in both gametophytic and sporophytic tissues (Tanksley et al., 1981; Sari-Gorla et al., 1986; Pedersen et al., 1987). Studies of the kinetics of [3H]cDNA to poly(A+) RNA hybridization confirmed this overlap and quantified the complexity of haploid gene expression. These estimates suggested that Tradescantia paludosa and maize (Zea mays) pollen contain approximately 20,000 to 24,000 different mRNA sequences (Willing and Mascarenhas, 1984; Willing et al., 1988). The complexity of pollen mRNA populations were significantly lower when compared with roots, in both T. paludosa and maize, where it was estimated that 30,000 genes were expressed. The overlap between pollen and root mRNAs in these species was estimated to be 65%.
Hybridization studies of pollen cDNA libraries also suggested that the majority of pollen-expressed mRNAs showed gametophytic-sporophytic overlap. In fact, only 10% of pollen-expressed mRNAs were considered to be pollen specific (Stinson et al., 1987; Mascarenhas, 1990). However, these estimations did not account for cross-hybridization between closely related gene family members. Subsequent studies have shown that a number of pollen-specific genes have closely related counterparts expressed in sporophytic tissues (Belostotsky and Meagher, 1993, 1996; Brander and Kuhlemeier, 1995; Lopez et al., 1996), but the extent to which this characterizes the male transcriptome is unknown.
The assembly of gene-specific data concerning the male gametophytic transcriptome has revealed approximately 150 different genes, assigned to 16 distinct function groups, with strong evidence for pollen-specific expression in about 30 (Twell, 2002). Thus, compared with the estimated 20,000 pollen-expressed mRNAs and 2,000 to 7,000 pollen-specific mRNAs (Willing and Mascarenhas, 1984; Willing et al., 1988), current knowledge of the pollen transcriptome is strongly deficient. Microarray technology now provides the opportunity to reveal this unknown fraction and to compile comprehensive data concerning the extent of overlap between male gametophytic and sporophytic gene expression.
In addition to its vital role in sexual reproduction, pollen provides a microcosm of cellular development, which is an attractive system in which to dissect the fundamental processes of cell growth and division, cellular differentiation, and intercellular communication (Bedinger, 1992; Twell, 1994, 2002). An essential step toward the detailed understanding of these processes is to define the transcriptome at the cellular level. In plants, the male gametophyte is a uniquely accessible cell type for such studies, enabling RNA isolation from a pure cell population and transcriptome analysis of what is essentially a single cell type poised for an explosive growth phase.
Here, we report the first genome-wide view of the pollen transcriptome for the model species Arabidopsis. We demonstrate the unique composition of the pollen transcriptome based on comparative analysis with sporophytic tissues. The identities of 992 pollen-expressed mRNAs were identified, nearly 40% of which were detected specifically in pollen. We also analyze the functional composition of the pollen transcriptome and demonstrate common patterns of mutually exclusive gene expression between pollen and sporophytic tissues for different gene family members.
RESULTS
Reduced Complexity of the Pollen Transcriptome
Transcriptome analysis of the male gametophyte required the development of a procedure for the isolation of viable mature pollen grains from Arabidopsis (see “Materials and Methods”). This allowed us to isolate homogenous populations of mature pollen grains. Microscopic examination of such populations revealed that the proportion of aborted pollen grains was less than 2%, with no contaminating sporophytic cells and little or no other cellular debris (data not shown).
Affymetrix Arabidopsis 8K GeneChip arrays were used to explore the pollen transcriptome in comparison with that of the developing sporophyte at various stages of plant development. Microarrays were hybridized with cRNA probes made from total RNA isolated from mature pollen. Microarray hybridization data for pollen RNA prepared from two independently grown plant populations were compared. Only genes with a positive hybridization signal accompanied by a detection call value of 1 in both experiments were scored as pollen expressed. Sporophytic Affymetrix 8K GeneChip data were downloaded from the GARNet Web site (http://www.Arabidopsis.info). This provided transcriptome data for Arabidopsis plants at open cotyledon stage (stage 1.00; Boyes et al., 2001), four rosette leaf stage (1.08), first visible flower bud stage (5.10), and first open flower stage (6.00). Sporophytically expressed genes were identified using the same algorithm used for pollen microarray data.
The Affymetrix 8K GeneChip arrays contain oligonucleotide probes representing 7,792 annotated genes with Arabidopsis Genome Initiative (AGI) numbers. This represents approximately 28% of the total of 27,117 annotated protein-coding genes in Arabidopsis (The Institute for Genomic Research, 2002). In pollen, 992 genes consistently gave a positive hybridization signal, representing nearly 13% of the unigene targets present on the micorarray (Fig. 1). Complete microarray data are publicly available at the Nottingham Arabidopsis Stock Centre (NASC) microarray database (http://Arabidopsis.info/prototype). The fully annotated list of pollen-expressed mRNAs is available as a supplemental information (Supplemental Fig. 1; supplemental data can be viewed at http://www.plantphysiol.org). Given that the contribution of sperm cells to the detectable pollen transcriptome is likely to be negligible (Xu et al., 2002), these data essentially represent the transcriptome of the vegetative cell present in mature pollen.
Figure 1.

Quantitative evaluation of gametophytic and sporophytic gene expression. A, Relative proportion of stage-specific mRNAs in mature pollen and in four sporophytic stages: open cotyledon (S 1.00), four leaf rosette (S 1.08), first visible flower bud (S 5.10), and first open flower (S 6.00) stages. The number of stage-expressed and stage-specific genes are indicated. B, Distribution of mRNAs between three relative abundance classes. Three mRNA abundance classes were defined: high (≤10-fold less than the maximum signal), medium (10- to 100-fold less), and low (≥100-fold less). The number of stage-expressed genes is indicated.
The number of diverse mRNAs expressed in pollen calculated from microarray analysis was significantly lower by 30% to 60% (Fig. 1A) than that of other sporophytic samples. This may not be surprising given that all sporophytic RNA samples represented more complex tissues. However, a striking feature was that 39% of pollen-expressed mRNAs were detected exclusively in pollen. In all other sporophytic samples, the percentage of stage-specific mRNAs was less than 8%, reflecting the broadly similar tissue compositions and cellular functions throughout sporophytic development. Although less pronounced than between sporophytic stages, there was a significant proportion (61%) of pollen-expressed mRNAs that were also expressed in one or more sporophytic stages.
Analysis of the distribution of mRNAs among three mRNA abundance classes; high (up to 10-fold less than the maximum signal), medium (10- to 100-fold less), and low (more than 100-fold less; Fig. 1C) revealed that the proportion of mRNAs forming the high-abundance class in pollen (17%) was significantly above that in sporophytic samples (5.2%–8.2%). There was a tendency toward reduction of the medium-abundance class and expansion of the low-abundance class during sporophytic development. Fifty-five percent of pollen-expressed mRNAs forming the highly abundant class were only detected in pollen. Moreover, the proportion of pollen-specific genes (55%) was much higher than in the two lower abundance classes (36%). The high proportion of the pollen transcriptome contributed by pollen-specific mRNAs highlights the potential significance and regulatory specialization of this abundant subset of the pollen transcriptome.
Transcriptomic Data Reflect in Vivo Gene Expression
Expression data obtained from the 8K GeneChip array were verified using two approaches: first by reverse transcriptase (RT)-PCR analysis of RNA isolated from different tissues, and second by comparison with the expression profiles of previously characterized genes present on the microarray. RT-PCR primers were designed for a subset of genes with the highest and lowest hybridization signals that were also scored as pollen-specific based on 8K GeneChip data. Genes were considered pollen-specific if detected in both pollen microarrays experiments, but not in any sporophytic experiments. Primers were designed for nine of 11 genes with the highest signals and six of 17 genes with the lowest signals. All 15 putative pollen-specific mRNAs tested by RT-PCR were confirmed to be expressed in pollen but were not detected in roots, stems, and leaves (Fig. 2).
Figure 2.

Verification of 8K GeneChip expression data by RT-PCR. Expression of four genes considered pollen-specific according to 8K GeneChip data in roots (R), stems (S), leaves (L), flowers (F), and pollen (P) were verified by RT-PCR. These are compared with the expression of a constitutively expressed gene (At3g20770) and Rubisco small subunit 1b (At5g38430) as a negative control.
The purity of pollen populations and isolated RNA was confirmed by the lack of expression of the Rubisco small subunit 1b gene At5g38430 by RT-PCR (Fig. 2). Moreover, all six genes encoding light-harvesting chlorophyll a/b-binding proteins (LHCB2, At3g27690; LHCB3, At5g54270; LHCB4, At5g01530; LHCB4.3, At2g40100; LHCB5, At4g10340; and LHCB6, At1g15820) present on the microarray were abundantly expressed in the sporophyte and were among the 50 most abundantly expressed genes. However, none of these genes gave positive signals in pollen (data not shown).
The recent compilation of approximately 150 pollen-expressed genes from various species included 23 genes found in Arabidopsis (Twell, 2002). A positive microarray hybridization signal in pollen was verified for 10 of 11 of these genes that were present on the 8K GeneChip. The only exception was the lack of a microarray signal for AtSUC1 gene (At1g71880) in mature pollen. However, AtSUC1 expression was not examined in mature dehiscent pollen (Stadler et al., 1999). With regard to Arabidopsis homologs of published pollen-expressed or -specific genes, two Arabidopsis homologs (At1g55560 and At1g55570) of the abundantly expressed tobacco (Nicotiana tabacum) pollen-specific gene ntp303 (Weterings et al., 1992) were detected exclusively in pollen and were among the 25 most abundantly expressed genes. The same was true for At1g14420 (At59 in Kulikauskas and McCormick, 1997), a homolog of the tomato (Lycopersicon esculentum) late anther-specific gene lat59 encoding a pectate lyase protein (Wing et al., 1989), whose expression ranked slightly lower at 31 of 992. Similarly, several members of the polygalacturonase gene family (At2g23900, At3g07820, At3g07850, and At4g33440) were detected exclusively in pollen, in accord with data previously published for maize and tobacco (Allen and Lonsdale, 1993). Moreover, four of five actin genes expressed in reproductive tissues (see McDowell et al., 1996) were present on the 8K GeneChip. All four belonged into the high-abundance class in pollen including two that were pollen-specific (ACT4 [At5g59370] rank 8 and ACT12 [At3g46520] rank 172), and the remaining two were highly pollen-enriched (ACT1 [At2g37620] rank 53 and ACT11 [At3g12110] rank 74). Of two profilin genes present on the 8K GeneChip, PRF4 (At4g29340) was expressed exclusively in pollen and was ranked 84, whereas PRF1 (At2g19760) showed low-level constitutive expression. Again, these data are in accord with previously published data (Huang et al., 1996). Data presented in this paragraph can be found in Supplemental Figure 1.
Pollen and Sporophytic Transcriptomes Differ Significantly
To analyze in more detail the extent of gametophytic-sporophytic overlap, including the relationship between relative expression levels, we used scatter-plot analysis. The expression levels of individual genes was normalized using a scale of 0 to 100, and genes co-expressed in pairs of data sets were plotted. Scatter-plots are shown for three pairs of data sets (Fig. 3). Figure 3A exemplifies a typical scatter plot of pollen versus sporophytic expression data that illustrates a lack of correlation between pollen-expressed and other stage-expressed genes. Shared genes typically belonged to the lowest abundance classes. Pair wise comparison of sporophytic samples illustrated a tendency toward unification of transcriptomes through consecutive developmental stages (Fig. 3, B and C). For example, the relationship between cotyledon stage and rosette stage was weaker (Fig. 3B), and points were more evenly spread along a diagonal in rosette and first flower stages, reflecting very similar expression levels (Fig. 3C).
Figure 3.

Scatter plots comparing gene expression levels in pollen and at different sporophytic developmental stages. The expression levels of individual genes were normalized using a scale of 0 to 100. Genes co-expressed in pairs of transcriptome data sets were plotted. A, Pollen versus four leaf rosette stage, 2,906 genes compared; B, open cotyledon versus four leaf rosette stage, 2,617 genes compared; C, four leaf rosette versus first open flower stage, 2,629 genes compared.
We also performed quantitative analysis of the extent of gametophytic-sporophytic overlap that resolved all 31 possible intersections between the five independent transcriptome data sets. The outcome is displayed using a Venn diagram (Fig. 4; Ruskey, 1997), directly outlining numbers of genes comprising each of the 31 categories. For quantitative analyses, gene expression levels were not taken into account, each gene was treated only as expressed or not. Of 3,282 genes expressed in at least one sample, only 287 (8.7%) were shared by all five. Pollen expressed the highest percentage (39%) of specific genes compared with all sporophytic samples. In contrast, 830 genes not expressed in pollen, representing 29% of the total number of sporophytically expressed genes, were co-expressed in all four sporophytic data sets. The percentage of expressed genes shared by the three post-seedling data sets reached 65% of the total number of sporophytically expressed genes. Pollen and seedlings co-expressed 312 genes, representing the minimum overlap between all data sets. Moreover, 92% of these represented constitutive genes expressed in all five data sets. Taken together, Venn and scatter plot analyses highlight the unique composition of the pollen transcriptome in comparison with the developing sporophyte.
Figure 4.

Quantification of gametophytic-sporophytic and sporophytic-sporophytic overlaps of transcriptome profiles. Venn diagram illustrating all 31 possible intersections between five independent transcriptome data sets including the number of genes composing each of these overlapping categories. Transcriptome data sets were as follows: mature pollen (pollen), open cotyledon stage (S 1.00), four leaf rosette stage (S 1.08), first visible flower bud stage (S 5.10), and first open flower stage (S 6.00). Numbers in parentheses indicate the total number of genes expressed in each data set.
Preferential Expression of Genes Encoding Cell Wall and Cytoskeleton-Related Proteins in Pollen
To evaluate the functional specialization of the pollen transcriptome in comparison with the sporophyte, we analyzed the distribution of pollen-expressed mRNAs between gene function categories. All genes annotated on the 8K GeneChip were organized into 12 functional categories through the combination of categories automatically derived by Munich Information Center for Protein Sequences Arabidopsis thaliana Database (MAtDB) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases and using gene families available at MAtDB and The Arabidopsis Information Resource (TAIR). The gene function distribution for the 8K GeneChip is shown in Figure 5A. The distribution of pollen-expressed genes among functional categories was similar to that of the complete 8K microarray (Fig. 5D). The only exception was the under-representation of annotated transcription factors by 4%. More striking differences were found when the number of different genes expressed was replaced with their relative expression levels (Fig. 5E). Such data treatment led to the expansion of five functional categories and a reduction in five others. The most dramatic expansion from 3% to 15% was observed for cell wall proteins. In this regard, five of the six most abundant pollen-expressed mRNAs encoded proteins involved in cell wall metabolism, such as polygalacturonases and pectinesterases. Moreover, four of these mRNAs were pollen specific, and the remaining one was highly pollen preferential. Another major functional category contained genes encoding cytoskeletal proteins. This category contained 2% of pollen-expressed genes accounting for 5% of the total relative hybridization signal. For example, the previously mentioned pollen-specific actin 4 gene and two transcripts encoding tubulin β-4 and β-9 chains were highly pollen enriched; both belonged among the 25 most abundantly expressed genes. On the contrary, genes involved in transcription and protein synthesis were expressed at lower levels (Fig. 5, D and E).
Figure 5.

Distribution of expressed mRNAs among gene function categories. Pie charts display each gene function category according to the number of expressed genes (B, D, F, and H) or their relative signal intensity (C, E, G, and I). The number of expressed mRNAs present in each data set are indicated. Percentage data for each pie are provided as supplemental information (Supplemental Fig. 1). A, Distribution of genes embedded on the 8K GeneChip among 12 functional categories. B through I, Functional distribution of mRNAs expressed at four leaf rosette stage (B and C); total pollen-expressed mRNAs (D and E); pollen-specific mRNAs (F and G); and pollen-expressed mRNAs showing gametophytic-sporophytic overlap (H and I).
Sporophytic tissues, represented here by the four rosette leaf stage 1.08 (Fig. 5, B and C), displayed transcriptome profiles that were broadly similar to each other, but distinct from pollen. Compared with the distribution of genes on 8K GeneChip, the most significant increase was observed in the number of genes involved in basic and energy metabolism, transport, and protein synthesis. Unlike pollen, genes involved in energy metabolism, transport, protein synthesis, and stress were preferentially expressed. The dramatic increase in expression of genes involved in energy metabolism from 2% to 11% was due to the extraordinary expression of genes involved in the light phase of photosynthesis. Apart from cell wall proteins, these categories account for the major differences between gametophytic and sporophytic transcriptomes.
The preferential expression of genes belonging to particular functional categories became even more apparent when only genes detected specifically in pollen were considered (Fig. 5, F and G). Narrowing of both transcription and protein synthesis categories was accompanied by a dramatic overrepresentation of genes involved in cell wall metabolism, signaling, and the cytoskeleton. In particular, the sum of the hybridization signals for genes in these last three classes reached 45% of the total signal intensity for all pollen-specific genes. On the contrary, the complementary set of pollen-expressed genes showing gametophytic-sporophytic overlap resembled the distribution of functional categories in the sporophyte (Fig. 5, H and I). Apparent under-representation was found for only two categories, energy metabolism and protein synthesis, reflecting their significant under-representation in the complete mature pollen transcriptome. Conversely, mRNAs encoding cytoskeletonrelated proteins were over-represented among subsets showing pollen-specific or gametophytic-sporophytic overlap.
Pollen Expresses Discrete Subsets of Analyzed Gene Families
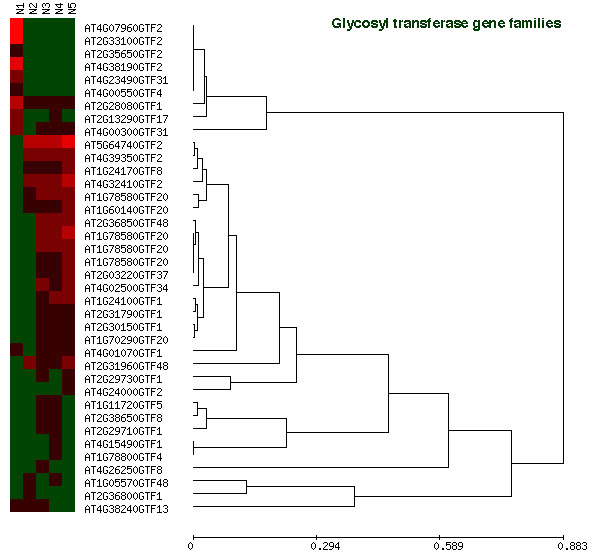
To further investigate the relationship between pollen and sporophytically expressed genes, we used hierarchical clustering analysis (Expression Profile data CLUSTering and analysis [EPCLUST] and Self Organizing Tree Algorithm [SOTA]) of pollen and sporophytic data sets. Because the complete pollen transcriptome data set was considered too large for comprehensive analysis, six well-characterized gene families were selected from MAtDB and TAIR. Criteria for selection included a reasonable size (>30 members) and appropriate representation (>25% of members) on the 8K GeneChip (Tab. I). The gene families and superfamilies chosen were: receptor-like kinases (RLKs; Shiu and Bleecker, 2001), glycoside hydrolases, glycosyltransferases, carbohydrate esterases (Carbohydrate-Active enZYmes server; http://afmb.cnrs-mrs.fr/CAZY/ind), expansins (Lee et al., 2001; Expansins Central homepage; http://www.bio.psu.edu/expansins/), and translation initiation factors (Factors in Arabidopsis translation database; http://www.cm.utexas.edu/browning/db/). Hierarchical clustering of the first two families is shown in Figures 6 and 7. Results for the remaining four families are available separately (Supplemental Figs. 2, 3, 4, 5). Application of both EPCLUST and SOTA algorithms gave very similar results, so only EPCLUST-generated trees are presented.
Table I.
Gene families selected for the hierarchical clustering analysis (EPCLUST)
| No. of Genes
|
|||||
|---|---|---|---|---|---|
| Gene Family | Total Family Members | Present on the 8K GeneChip | Expressed in One or More Dataset(s) | Pollen-Expressed | Pollen-Specific |
| RLKa | 612 | 226 (37%) | 62 (27%) | 23 | 21 |
| GHFb | 386 | 154 (40%) | 51 (33%) | 14 | 13 |
| GTFc | 359 | 132 (37%) | 38 (29%) | 11 | 6 |
| CESd | 98 | 26 (27%) | 13 (50%) | 3 | 3 |
| EXPe | 35 | 13 (37%) | 7 (54%) | 1 | 0 |
| TIFf | 101 | 42 (42%) | 31 (74%) | 12 | 4 |
Receptor-like kinases. b Glycoside hydrolases. c Glycosyl transferases. d Carbohydrate esterases. e Expansins. f Translation initiation factors.
Figure 6.

Expression profiles of the glycoside hydrolase gene superfamily. Hierarchical cluster analysis of 51 members of the glycoside hydrolase superfamily in mature pollen (1) and in four sporophytic data sets: S 1.00 (2), S 1.08 (3), S 5.10 (4), and S 6.00 (5). For each expression profile, the AGI number of the corresponding gene and gene family are indicated.
Figure 7.

Expression profiles of the RLK gene family. Hierarchical cluster analysis of 62 members of RLK gene family in mature pollen (1) and in four sporophytic data sets (2–5) according to legend of Figure 6. For each expression profile, the AGI number of the corresponding gene and gene subfamily are indicated.
Four selected families and superfamilies contained proteins involved in cell wall metabolism. Within three superfamilies of carbohydrate-modifying enzymes and the family of expansins, large and unique subsets of genes were expressed in pollen (Fig. 6; Supplemental Figs. 2, 3, 4). Approximately 40% of genes composing glycoside hydrolase families were present on 8K GeneChip, one-third of which were expressed at least at one sample. Four members of glycoside hydrolase family GHF28 (polygalacturonases) were only detected in pollen. These polygalacturonase genes were highly expressed within high (At3g07820, At3g07850, and At2g23900) and medium (At4g33440) mRNA abundance classes. Within five pollen-expressed glycoside hydrolase families, GHF1 (β-glycosidases), GHF16 (xyloglucan endotransglycosylases), GHF28 (polygalacturonases), GHF35 (β-galactosidases), and GHF9 (glucanases/cellulases), there was no overlap between gene family members expressed in pollen and the sporophyte.
Among glycosyltransferase gene families, we identified 11 pollen-expressed genes, six of which were highly expressed and pollen specific (Table I; Supplemental Fig. 2). Four of these belonged to the GTF2 family (glucosyltransferases), including two encoding cellulose synthases (At2g33100 and At4g38190). Therefore pollen expresses specific subsets of enzymes involved in cellulose synthesis as well as degradation (At4g11050 from GHF9). We observed the same general pattern for a gene family encoding other abundant cell wall proteins, pectinesterases (Supplemental Fig. 3), but not for expansins (Supplemental Fig. 4).
The RLK superfamily was well represented on the 8K GeneChip, with 226 RLK genes or 37% of the total number of RLKs in the Arabidopsis genome. Sixty-two RLK genes distributed among 25 subfamilies were expressed in at least one sample (Fig. 7). The large majority (21 of 23) of pollen-expressed RLKs were detected exclusively in pollen. Furthermore, specificity for pollen versus sporophytic expression was observed not only for individual RLK family members, but also at the level of RLK subfamilies. Of a total of nine pollen-expressed RLK subfamilies, four did not show sporophytic expression. These were Pro-rich extensin-like receptor kinases, the receptor-like cytoplasmic kinase subfamily IX, Crinkly4-like, and the Leu-rich repeat subfamily VI. Five pollen-expressed RLK subfamilies were coexpressed in sporophyte, and the remaining 16 subfamilies were expressed only in the sporophyte.
Genes encoding translation initiation factors more frequently exhibited overlap between pollen and sporophytic expression compared with the other gene families analyzed (Supplemental Fig. 5). This overlap was relatively high (two-thirds of pollen-expressed genes) with the exception of poly(A)binding proteins (PAB). Two PABs were pollen specific (PAB-like [At2g36660] and PAB6 [At3g16380]), one gene was expressed in the sporophyte, but not in pollen (PAB1 [At2g23350]), and one was not detectably expressed (PAB3 [At1g22760]).
DISCUSSION
To identify, on a genome-wide scale, genes involved in pollen functions and those with overlapping expression between the male gametophyte and the sporophyte, we analyzed the transcriptome of mature Arabidopsis pollen using microarrays. This represents the first attempt to characterize the transcriptome of gametophytic cell types on a genomewide scale. Gene-by-gene approaches have previously identified only 20 different genes expressed in Arabidopsis pollen (Twell, 2002), such that the data sets generated here provide a 50-fold increase in knowledge of the number, identity, and relative expression levels of pollen-expressed genes in Arabidopsis. In the sporophyte, transcriptome profiling at the single-cell level has recently been reported for leaf epidermal and mesophyll cells in Arabidopsis using expressed sequence tag filter arrays (Brandt et al., 2002). Of 16,000 expressed sequence tags, 680 showed expression in one or both cells types, with 3% to 14% of sequences uniquely expressed in epidermis or mesophyll, respectively.
The pollen transcriptome was analyzed using Affymetrix Arabidopsis 8K GeneChip arrays harboring probe sets for 7,792 annotated genes (Institute for Genomic Research, 2002). Of these, 992 genes gave positive hybridization signals in pollen. Taking into account that the proportion of genes embedded on the 8K microarray is approximately 28%, we estimate the total number of pollen-expressed genes in Arabidopsis to be over 3,500. According to the criteria given, 39% of pollen-expressed genes were considered pollen specific. Although there were minor differences in the growth conditions of plants used for gametophytic and sporophytic samples, given the wide variety of developmental stages and the timing of sample collection, their comparison with pollen is unlikely to account for much of the variation of gene expression between pollen and sporophytic tissues. Therefore, we estimate the total number of genes preferentially or specifically expressed in Arabidopsis to be more than 1,400. On the contrary, a significantly lower percentage (3% and 14%, respectively) of mRNAs uniquely expressed in epidermal and mesophyll cells was reported (Brandt et al., 2002). This degree of cell-specific expression is likely to be severely overestimated because it does not take into account a number of other cell types that make up the sporophyte.
The fact that 1 in 20 Arabidopsis genes is preferentially or specifically active in pollen highlights the evolution and potential importance of transcriptional regulatory mechanisms that operate to specify gametophytic gene expression. Previous functional studies have defined multiple cis-regulatory elements involved in such transcriptional regulation in several species (for review, see Twell, 2002). Microarray data in a single species now provide the opportunity to use bioinformatic approaches to accelerate the identification of conserved regulatory elements that regulate pollen-specific expression.
We propose that 1,400 pollen-specific genes represent an ample pool for selection for pollen fitness at microgametophytic level as suggested by Mulcahy et al. (1996). On the contrary, the observed 61% gametophytic-sporophytic overlap provides the potential to improve the fitness of the sporophytic generation through gametophytic competition and selection. Our results strongly support the extent of gametophytic-sporophytic overlap previously estimated from isozyme and hybridization kinetic studies. There was 72% overlap for isozyme profiles in maize (Sari-Gorla et al., 1986) and 60% in barley (Pedersen et al., 1987) and tomato (Tanksley et al., 1981). Similarly, hybridization kinetic studies in T. paludosa (Willing and Mascarenhas, 1984) and maize (Willing et al., 1988) estimated that 65% of pollen-expressed transcripts were also present in roots. Here, the identity and relative expression levels of 605 genes that show gametophytic-sporophytic overlap are reported.
There were significant differences between estimates for the number of pollen-expressed genes in Arabidopsis based on microarray data and previous estimates based on mRNA reassociation kinetics in T. paludosa and maize (Willing and Mascarenhas, 1984). Although our data are likely to be minimum estimates that would lead to the underestimation of very low-abundance transcripts, we suggest that 20,000 pollen-expressed genes may be a significant overestimation. Despite a large difference in genome size, the total number of genes in rice is estimated to be only 1.2- to 1.9-fold larger than in Arabidopsis (Goff et al., 2002). We suggest that it is unlikely that T. paludosa and maize pollen will express 5- to 6-fold more genes than Arabidopsis.
Our results demonstrated reduced complexity of the pollen transcriptome compared with the sporophyte that was associated with the expression of a greater number of abundant mRNAs. This is in accord with previous findings in T. paludosa and maize (Willing and Mascarenhas, 1984). These observations are consistent with the limited, but highly specialized activities of mature pollen grains that are prepared for rapid germination and pollen tube growth (see Mascarenhas, 1990; Taylor and Hepler, 1997). Mature pollen is charged with mRNA and proteins in preparation for extensive cell wall synthesis and the establishment of a prominent actin-rich cytoskeleton. Although comparable transcriptome data are not available for other differentiated plant cell types, there could be similar requirements for abundant mRNAs sets in rapidly growing cells such as root hairs.
We demonstrated the pollen-specific expression of discrete subsets of genes within several gene families. The pollen-specific expression of specific members of gene families, such as those encoding cytoskeletal proteins and proteins involved in carbohydrate metabolism, could influence pollen fitness through regulatory and/or functional specialization.
For the RLK gene family, microarray data demonstrated pollen-specific expression not only of individual genes, but also for several RLK subfamilies. In total, 21 of 23 pollen-expressed RLK genes were only detected in pollen, suggesting multiple and specialized roles in pollen functions. Our description of 23 pollen-expressed receptor-like protein kinases extends data recently published on this gene family (Kim et al., 2002) and confirms the reported pollen-specific expression of AtPRKc (At07040) and AtPRKd (At5g35390). For two other pollen RLKs named in this article, AtPRKg (At2g26730) was not detected and AtPRKh (At1g48480) was detected only in sporophytic tissues. The significance of RLKs for pollen function was recently highlighted in a study showing interaction of the essential tomato pollen-specific LAT52 protein with the extracellular domain of the pollen-specific receptor kinase LePRK2 (Tang et al., 2002).
The majority of pollen-expressed genes showing gametophytic-sporophytic overlap belonged to the two lowest mRNA abundance classes. In contrast, 55% of genes forming the highly abundant pollen-expressed class were specifically expressed in pollen. This division of abundantly expressed pollen-specific transcripts and poorly expressed transcripts showing gametophytic-sporophytic overlap may reflect different evolutionary constraints in gametophyte and sporophyte. We speculate that the evolution of highly expressed pollen-specific genes depends on initial gene redundancy caused by gene duplication, which allows rapid gametophytic selection accompanied by mutations leading to the loss of the sporophytic component of expression.
The singularity of the pollen transcriptome became evident after allocation of pollen-expressed genes into function groups in comparison with their sporophytic equivalents. Pollen-tube walls are the product of gametophytic expression and possess a unique composition distinct from that of most sporophytic cell types (Li et al., 1999; Hepler et al., 2001). Moreover, the specialized tip growth mechanism of the pollen tube is associated with extensive cell wall synthesis, dynamic changes in cell wall structure, and continuous cell-cell interactions in the pistil (Hepler et al., 2001). Our results revealed a significant investment in the gametophytic expression of genes involved in cell wall metabolism, cytoskeleton, and signaling pathways, consistent with such functional requirements. With regard to the gametophytic synthesis of cell wall polysaccharides, there is limited information concerning the pollen phenotypes of functional knockouts of polysaccharide synthetic enzymes. A cellulose-synthase-like glycosyltransferase, CslA7, has recently been shown to be required for efficient pollen tube growth in Arabidopsis and may synthesize a β-linked polymer (Goubet et al., 2003). The corresponding gene, At2g35650, was expressed in pollen and sporophytic tissues according to RTPCR (Goubet et al., 2003), which was confirmed in our microarray analysis. Pollen microarray data will help to target further knockouts to discover the significance of pollen-expressed polysaccharide synthetic enzymes.
Genes involved in transcription and protein synthesis were generally expressed at lower levels in pollen than in the sporophyte. These data are consistent with the fact that pollen germination and early tube growth in many species are largely independent of transcription, but vitally dependent on translation (Hoekstra and Bruinsma, 1979). mRNA accumulates during pollen maturation and is stored for use in protein synthesis during pollen germination (Schrauwen et al., 1990; Honys et al., 2000). Moreover, in physiologically advanced species, such as T. paludosa, rRNA gene transcription is repressed during pollen maturation and preformed rRNA is stored for use in protein synthesis during pollen germination (see Mascarenhas, 1989). Our data support the concept that Arabidopsis pollen is charged with stored mRNA and preformed translational apparatus enabling rapid activation upon hydration and germination.
CONCLUSIONS
Genome-wide analysis of the pollen transcriptome unequivocally demonstrates the unique state of differentiation that distinguishes the mature male gametophyte from the sporophyte. This study revealed the identities and relative expression levels of genes comprising approximately 30% of the pollen transcriptome, including genes expressed specifically in pollen and those showing gametophytic-sporophytic overlap. The functional specialization of the mature male gametophyte for recognition of target tissues and rapid directional growth was highlighted by the overrepresentation of genes expected to be vital for fulfilling these tasks. These included genes involved in cell wall metabolism, the cytoskeleton, and cell signaling. Our data also highlight the diminished role for transcription and the important role of mRNA storage in pollen function. The main impact of this work is that it provides the first genomic and complex view of the male gametophyte. This knowledge now provides a significant opportunity to construct new genome-targeted questions concerning plant cellular functions and the regulation of male gametophyte development and evolution.
MATERIALS AND METHODS
Plant Materials and Pollen Isolation
Arabidopsis ecotype Landsberg erecta plants used for pollen isolation were grown in controlled-environment cabinets at 21°C under illumination of 100 μmol m–2 s–1 with a 16-h photoperiod. Pollen for microarray experiments was harvested from two independently grown populations. Roots were grown from plants in liquid cultures. Five surface-sterilized seeds were placed in a 250-mL Erlenmeyer flask with 50 mL of 0.5× Murashige and Skoog media (Sigma-Aldrich, St. Louis) supplemented with 1% (w/v) Suc. The roots were grown in the dark at 22°C with constant shaking for 6 weeks.
For the isolation of mature pollen, inflorescences from over 500 plants were harvested in a large Erlenmeyer flask, 300 mL of ice-cold 0.3 m mannitol was added, and the flask was vigorously shaken for 1 min. The pollen suspension was sequentially filtered through 100- and 53-μm nylon mesh. Pollen grains were concentrated by repeated centrifugation steps (50-mL Falcon tubes, 450 g, 5 min, 4°C), and the final compact pollen pellet was stored at –80°C. The purity of isolated pollen was determined by light microscopy and 4′,6-diamino-phenylindole-staining according to Park et al. (1998). Arabidopsis ecotype Landsberg erecta plants used in sporophytic microarray analyses at NASC were grown in controlled-environment cabinets at 21°C to 22°C under constant light.
RNA Extraction, Probe Preparation, and GeneChip Hybridization
Total RNA was extracted from 50 mg of isolated pollen using the RNeasy Plant Kit according to the manufacturers instructions (Qiagen, Valencia, CA). The yield and RNA purity were determined spectrophotometrically, and their integrity was checked using an Agilent 2100 Bioanalyser (Agilent Technologies, Boblingen, Germany) at NASC.
Biotinylated target RNA was prepared from 20 μg of total RNA as described in the Affymetrix GeneChip Technical Analysis Manual (Affymetrix, Santa Clara, CA). Double-stranded cDNA was synthesized using SuperScript Choice System (Invitrogen, Carlsbad, CA) with oligo(dT)24 primer fused to T7 RNA polymerase promoter. Biotin-labeled target complementary RNA (cRNA) was prepared by cDNA in vitro transcription using the BioArray High-Yield RNA Transcript Labeling kit (Enzo Biochem, New York) in the presence of biotinylated UTP and CTP.
The Arabidopsis 8K GeneChip array was hybridized with 15 μg of labeled target cRNA for 16 h at 45°C as described in the Affymetrix Technical Analysis Manual. GeneChips were stained with Streptavidin-Phycoerythrin solution and scanned with an Agilent 2500A GeneArray Scanner (Agilent Technologies).
RT-PCR
Samples of 1 μg total RNA isolated from roots, stem, leaves, flowers, and pollen were reverse transcribed in a 20-μL reaction using the ImProm-II Reverse Transcription System (Promega, Madison, WI) following the manufacturer's instructions with the exception that the oligo(dT)15 primer was replaced with a custom-synthesized 3′-RACE primer (5′-AAGCAGTGGTAACAACGCAGAGTAC(T)30VN-3′). Pollen, stem, leaf, and flower samples were isolated from plants grown as described. Pollen RNA used for RT-PCR verification was obtained from plants that were grown independently from those used to isolate RNA for microarray analysis.
For PCR amplification, 1 μL of 50× diluted RT mix was used. The PCR reaction was carried out in 25 μL with 0.5 unit of BioTaq DNA polymerase (Bioline, London), 1.2 mm MgCl2, and 20 pmol of each primer. The PCR program was as follows: 2 min at 95°C, 33 cycles of 15 s at 94°C, 15 s at the optimal annealing temperature (63°C to 67°C), and 30 s at 72°C, followed by 10 min at 72°C. As a reverse primer, NESTED primer (5′-AAGCAGTGGTAACAACGCAGAGT-3′) overlapping the 3′-RACE primer was used to eliminate genomic DNA amplification. The following gene-specific forward primers were designed using Primer3 software (http://www-genome.wi.mit.edu/cgi-bin/primer/primer3_www.cgi): At3g57690, 5′-TCCCTTGTCTCCCTCTTCAGCTACT-3′; At5g14380, 5′-CTAAGTTCTCTGTTGTCGGCACAGTC-3′; At1g02790, 5′-AACTTAGTTTCCCTATGTGTCCCAAA-3′; At4g27110, 5′-TGCAGTCGGTTATCTCGTTTAAGAAG-3′; At4g19440, 5′-TTGATGGATATGGTAAATTGGGTCAG-3′; At3g20770, 5′-ACATCGAGACAGTCGCTTACCGTAT-3′; and Rubisco, 5′-ATCTTACCTCCCTGACCTTACTGACGT-3′.
Data Analysis
Affymetrix MAS 5.0 Standard Image Analysis was performed using the Affymetrix Microarray Analysis Suite 5.0. Sporophytic data from public baseline GeneChip experiments used for comparison with the pollen transcriptome were downloaded from the GARNet Web site (http://www.Arabidopsis.info). To make data from all used GeneChips comparable, the output from all microarrays were scaled such that the top 2% and bottom 2% of signal intensities were excluded and the trimmed mean calculated as described by Welle et al. (2002). All signal values for each individual GeneChip were multiplied by a microarray-specific scaling factor obtained such that the trimmed mean was normalized to 5,000.
Microsoft Excel (Microsoft, Redmont, WA) was used to manage and filter the microarray data. For annotation of genes present on the 8K GeneChip, the Arabidopsis Genome Annotation Release 3.0 published by Institute for Genomic Research (2002) was used. Genes were sorted into functional categories created according to data mined from MAtDB (http://mips.gsf.de/proj/thal/db/index.html), KEGG (http://www.genome.ad.jp/kegg/), and TAIR (http://www.Arabidopsis.org/) databases. Hierarchical clustering of gene families was performed using expression profile data clustering and analysis software EPCLUST (http://ep.ebi.ac.uk/EP/EPCLUS), with correlation measure-based distance and average linkage clustering methods. SOTA analysis was performed using SOTA software with default parameters (Herrero et al., 2001; http://ep.ebi.ac.uk/EP/SOTA/).
Supplementary Material
Acknowledgments
We gratefully acknowledge the GARNet transcriptomic center at NASC for performing pollen microarray hybridizations and for providing public baseline 8K GeneChip data for sporophytic samples.
Article, publication date, and citation information can be found at www.plantphysiol.org/cgi/doi/10.1104/pp.103.020925.
This work was supported by a Royal Society/NATO Postdoctoral Fellowship (to D.H.), by the Ministry of Education of the Czech Republic (project no. CE:JI3/98:113100003 to D.H.), and by the Biotechnology and Biological Sciences Research Council under the Investigating Gene Function Initiative (to D.T.).
The online version of this article contains Web-only data. The supplemental material is available at http://www.plantphysiol.org.
References
- Allen RL, Lonsdale DM (1993) Molecular characterisation of one of the maize polygalacturonase gene family members which are expressed during late pollen development. Plant J 3: 261–271 [DOI] [PubMed] [Google Scholar]
- Bedinger P (1992) The remarkable biology of pollen. Plant Cell 4: 879–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belostotsky DA, Meagher RB (1993) Differential organ-specific expression of three poly(A) binding protein genes from Arabidopsis. Proc Natl Acad Sci USA 90: 6686–6690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belostotsky DA, Meagher RB (1996) A pollen-, ovule-, and early embryo-specific poly(A) binding protein from Arabidopsis complements essential functions in yeast. Plant Cell 8: 1261–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyes DC, Zayed AM, Ascenzi R, McCaskill AJ, Hoffman NE, Davis KR, Gorlach J (2001) Growth stage-based phenotypic analysis of Arabidopsis: a model for high throughput functional genomics in plants. Plant Cell 13: 1499–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brander KA, Kuhlemeier C (1995) A pollen-specific DEAD-box protein related to translation initiation factor eIF-4A from tobacco. Plant Mol Biol 27: 637–649 [DOI] [PubMed] [Google Scholar]
- Brandt S, Kloska S, Altmann T, Kehr J (2002) Using array hybridisation to monitor gene expression at the single cell level. J Exp Bot 53: 2315–2323 [DOI] [PubMed] [Google Scholar]
- Goff SA, Goff SA, Ricke D, Lan T-H, Presting G, Wang R, Dunn M, Glazebrook J, Sessions A, Oeller P, Varma H et al. (2002) A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296: 92–100 [DOI] [PubMed] [Google Scholar]
- Goubet F, Misrahi A, Park S-K, Zhang Z, Twell D, Dupree P (2003) AtCSLA7, a cellulose-synthase-like putative glycosyltransferase, is important for pollen tube growth and embryogenesis in Arabidopsis. Plant Physiol 131: 547–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepler PK, Vidali L, Cheung AY (2001) Polarized cell growth in higher plants. Annu Rev Cell Dev Biol 17: 159–187 [DOI] [PubMed] [Google Scholar]
- Herrero J, Valencia A, Dopazo J (2001) A hierarchical unsupervised growing neural network for clustering gene expression patterns. Bioinformatics 17: 126–136 [DOI] [PubMed] [Google Scholar]
- Hoekstra FA, Bruinsma J (1979) Protein synthesis of binucleate and trinucleate pollen and its relationship to tube emergence and growth. Planta 146: 559–566 [DOI] [PubMed] [Google Scholar]
- Honys D, Combe J, Twell D, Capkova V (2000) The translationally repressed pollen-specific ntp303 mRNA is stored in non-polysomal mRNPs during pollen maturation. Sex Plant Reprod 13: 135–144 [Google Scholar]
- Huang S, McDowell JM, Weise MJ, Meagher RB (1996) The Arabidopsis profilin gene family: evidence for an ancient split between constitutive and pollen-specific profilin genes. Plant Physiol 111: 115–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Institute for Genomic Research (2002) Arabidopsis Genome Annotation Release 3.0. ftp://ftp.tigr.org/pub/data/a_thaliana/ath1/
- Kim HU, Cotter R, Johnson S, Senda M, Dodds P, Kulikauskas R, Tang W, Ezcurra I, Herzmark P, McCormick S (2002) New pollen-specific receptor kinases identified in tomato, maize and Arabidopsis: The tomato kinases show overlapping but distinct localization patterns in pollen tubes. Plant Mol Biol 50: 1–16 [DOI] [PubMed] [Google Scholar]
- Kulikauskas R, McCormick S (1997) Identification of the tobacco and Arabidopsis homologues of the pollen-expressed LAT59 gene of tomato. Plant Mol Biol 34: 809–814 [DOI] [PubMed] [Google Scholar]
- Lee Y, Choi D, Kende H (2001) Expansins: ever-expanding numbers and functions. Curr Opin Plant Biol 4: 527–532 [DOI] [PubMed] [Google Scholar]
- Li H, Bacic A, Read SM (1999) Role of a callose synthase zymogen in regulating wall deposition in pollen tubes of Nicotiana alata Link et Otto. Planta 208: 528–538 [Google Scholar]
- Lopez I, Anthony RG, Maciver SK, Jiang CJ, Khan S, Weeds AG, Hussey PJ (1996) Pollen specific expression of maize genes encoding actin depolymerizing factor-like proteins. Proc Natl Acad Sci USA 93: 7415–7420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascarenhas JP (1989) The male gametophyte of flowering plants. Plant Cell 1: 657–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascarenhas JP (1990) Gene activity during pollen development. Annu Rev Plant Physiol Plant Mol Biol 41: 317–338 [Google Scholar]
- Maynard Smith J (1971) What use is sex? J Theor Biol 30: 319–335 [DOI] [PubMed] [Google Scholar]
- McDowell JM, Huang S, McKinney EC, An Y-Q, Meagher RB (1996) Structure and evolution of the actin gene family in Arabidopsis thaliana. Genetics 142: 587–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulcahy DL (1979) The rise of the angiosperms: a genecological factor. Science 206: 20–23 [DOI] [PubMed] [Google Scholar]
- Mulcahy DL, Sari-Gorla M, Bergamini Mulcahy G (1996) Pollen selection: past, present and future. Sex Plant Reprod 9: 353–356 [Google Scholar]
- Park S-K, Howden R, Twell D (1998) The Arabidopsis thaliana gametophytic mutation pollen1 disrupts microspore polarity, division asymmetry and pollen cell fate. Development 125: 3789–3799 [DOI] [PubMed] [Google Scholar]
- Pedersen S, Simonsen V, Loeschcke V (1987) Overlap of gametophytic and sporophytic gene expression in barley. Theor Appl Genet 75: 200–206 [Google Scholar]
- Ruskey F (1997) A survey of Venn diagrams. Electronic Journal of Combinatorics 4: DS5 [Google Scholar]
- Sari-Gorla M, Frova C, Binelli G, Ottaviano E (1986) The extent of gametophytic-sporophytic gene expression in maize. Theor Appl Genet 72: 42–47 [DOI] [PubMed] [Google Scholar]
- Schrauwen J, de Groot P, van Herpen M, van Lee T, Reynen W, Weterings K, Wullems G (1990) Stage-related expression of mRNAs during pollen development in lily and tobacco. Planta 182: 298–304 [DOI] [PubMed] [Google Scholar]
- Shiu S, Bleecker AB (2001) Receptor-like kinases from Arabidopsis form a monophyletic gene family related to animal receptor kinases. Proc Natl Acad Sci USA 98: 10763–10768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler R, Truernit E, Gahrtz M, Sauer N (1999) The AtSUC1 sucrose carrier may represent the osmotic driving force for anther dehiscence and pollen tube growth in Arabidopsis. Plant J 19: 269–278 [DOI] [PubMed] [Google Scholar]
- Stinson JR, Eisenberg AJ, Willing RP, Pe MP, Hanson DD, Mascarenhas JP (1987) Genes expressed in the male gametophyte of flowering plants and their isolation. Plant Physiol 83: 442–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Ezcurra I, Muschietti J, McCormick S (2002) A cysteine-rich extracellular protein, LAT52, interacts with the extracellular domain of the pollen receptor kinase LePRK2. Plant Cell 14: 2277–2287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanksley SD, Zamir D, Rick CM (1981) Evidence for extensive overlap of sporophytic and gametophytic gene expression in Lycopersicon esculentum. Science 213: 454–455 [DOI] [PubMed] [Google Scholar]
- Taylor LP, Hepler PK (1997) Pollen germination and tube growth. Annu Rev Plant Physiol Plant Mol Biol 48: 461–491 [DOI] [PubMed] [Google Scholar]
- Twell D (1994) The diversity and regulation of gene expression in the pathway of male gametophyte development. In RJ Scott, AD Stead, eds, Molecular and Cellular Aspects of Plant Reproduction. Cambridge University Press, Cambridge, UK, pp 83–135
- Twell D (2002) Pollen developmental biology. In SD O'Neill, JA Roberts, eds, Plant Reproduction. Annual Plant Reviews, Vol 6. Sheffield Academic Press, Sheffield, UK, pp 86–153 [Google Scholar]
- Welle S, Brooks AI, Thornton CA (2002) Computational method for reducing variance with Affymetrix microarrays. BMC Bioinformatics 3: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weterings K, Reijnen W, van Aarssen R, Kortsee A, Spijkers J, van Herpen M, Schrauwen J, Wullems G (1992) Characterisation of a pollen-specific cDNA clone from Nicotiana tabacum expressed during microgametogenesis and germination. Plant Mol Biol 18: 1101–1111 [DOI] [PubMed] [Google Scholar]
- Willing RP, Bashe D, Mascarenhas JP (1988) An analysis of the quantity and diversity of messenger RNAs from pollen and shoots of Zea mays. Theor Appl Genet 75: 751–753 [Google Scholar]
- Willing RP, Mascarenhas JP (1984) Analysis of the complexity and diversity of mRNAs from pollen and shoots of Tradescantia. Plant Physiol 75: 865–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing RA, Yamaguchi J, Larabell SK, Ursin VM, McCormick S (1989) Molecular and genetic characterization of two pollen-expressed genes that have sequence similarity to pectate lyases of the plant pathogen Erwinia. Plant Mol Biol 14: 17–28 [DOI] [PubMed] [Google Scholar]
- Xu H, Weterings K, Vriezen W, Feron R, Xue Y, Derksen J, Mariani C (2002) Isolation and characterisation of male-germ-cell transcripts in Nicotiana tabacum. Sex Plant Reprod 14: 339–346 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}