

Abstract
The α -methyl-L-tryptophan (α -MTrp) method for the study of the brain serotonergic system is based on the fact that labelled α -MTrp is taken up by and, in part, retained in the brain, and this retention (trapping) is proportional to brain serotonin (5-HT) synthesis. A 3-compartment model is proposed in which the plasma, the precursor and irreversible pools are mathematically distinct compartments. The irreversible compartment is assumed to be the one in which the tracer is trapped. By definition, the tracer from the irreversible compartment does not exchange directly with the plasma compartment. The rate at which labelled α -MTrp is trapped is converted to the rate of 5-HT synthesis by dividing it by a conversion factor, called the lumped constant, and multiplying it by the plasma-free tryptophan concentration. Our results revealed that brain 5-HT synthesis can be influenced by the extraneuronal concentration of 5-HT and that, generally, the influence is not uniform throughout the brain. They also suggest that brain trapping of labelled α -MTrp relates to 5-HT synthesis. The proposed procedure for converting the rate at which labelled α -MTrp is trapped to brain 5-HT synthesis rates is based on measurements that suggest that plasma-free Trp relates to brain 5-HT synthesis. However, as with all biological models, there is likely room for improvement in our approach.
Medical subject headings: brain; models, biological; radioactive tracers; serotonin; serotonin uptake inhibitors; tomography, emission-computed; tryptophan; tryptophan hydroxylase
Abstract
La méthode à l'α -méthyl-L-tryptophane (α -MTrp) pour l'étude du système sérotoninergique du cerveau repose sur le fait que l'α -MTrp marqué est absorbé et retenu en partie dans le cerveau et que cette rétention (piégeage) est proportionnelle à la synthèse de la sérotonine (5-HT) dans le cerveau. On propose un modèle à trois compartiments où le plasma, le précurseur et les accumulations irréversibles sont des compartiments distincts sur le plan mathématique. On suppose que le compartiment irréversible est celui où le marqueur est piégé. Par définition, le marqueur n'a pas d'échange direct avec le compartiment du plasma. On convertit le taux de piégeage de l'α -MTrp marqué en taux de synthèse de la 5-HT en le divisant par un facteur de conversion, appelé constante localisée, et en le multipliant par la concentration de tryptophane sans plasma. Nos résultats révèlent que la synthèse de la 5-HT dans le cerveau peut être influencée par la concentration extraneuronale de 5-HT et que, en général, cette influence n'est pas uniforme à travers le cerveau. Ils suggèrent également que la rétention cérébrale d'α -MTrp marquée est liée à la synthèse de la 5-HT. La façon proposée pour convertir le taux de piégeage de l'α -MTrp marqué en taux de synthèse de la 5-HT dans le cerveau repose sur des mesures indiquant qu'il y a un lien entre le Trp sans plasma et la synthèse de la 5-HT dans le cerveau. Comme dans le cas de tous les modèles biologiques, toutefois, il est probablement possible d'améliorer notre approche.
Introduction
Serotonin (5-hydroxytryptamine [5-HT]), one of the monoamine neurotransmitters of the brain, is synthesized exclusively in serotonergic neurons from the essential amino acid L-tryptophan (Trp). The synthesis is catalyzed by 2 enzymes, tryptophan hydroxylase (TPH, E.C. 1.14.16.4) and aromatic amino acid decarboxylase (E.C. 4.1.1.28); the former has been accepted as the rate-limiting enzyme in the synthesis of 5-HT.1,2,3 Serotonergic neurons project diffusely throughout the brain and synapse with many different neurons. Because of this wide distribution, it is not surprising that many brain disorders have been associated with some kind of dysfunction in the brain serotonergic system.4 5-HT is partially deaminated by monoamine oxidase (MAO, E.C. 1.4.3.4) after synthesis, as well as after release into the synapse. 5-HT is the preferred substrate for MAO-A.5 However, MAO-B, the form found in serotonergic neurons, has a sufficiently high affinity for the deamination of 5-HT in serotonergic neurons if it is not protected by the 5-HT-binding protein6 or stored in vesicles. The deamination product 5-hydroxyindolacetic acid (5-HIAA) is removed by the acid transport system into the cerebrospinal fluid (CSF).7 There have been several attempts to relate the concentration of 5-HIAA in the CSF to the brain utilization of 5-HT, but the measurements, in addition to being rather invasive, can rarely be taken as a measure of the activity of the brain serotonergic system or of brain 5-HT synthesis.7,8 5-HIAA in the CSF might be, in some situations, an approximate index of 5-HT metabolism in the brain,9,10 but its direct relation to 5-HT synthesis has not been established to complete satisfaction.7,8,11,12 It has been shown, in humans, that a measurement bias can be introduced by the sampling method itself.11,12 Experiments with dogs suggest that there is very little mixing of the lumbar CSF with that found in the brain.12 Furthermore, it has been demonstrated in experiments with rats that the 5-HIAA concentration in CSF is more representative of the activity of MAO than the activity of the brain serotonergic system.7,8
There has been a need for imaging methods that will permit the study of the brain serotonergic system in vivo, especially in humans. Methods have recently been proposed to assess the functioning of the brain serotonergic system by measuring the density of 5-HT1A13 and 5-HT214,15 receptors, as well as 5-HT synthesis rates.16,17,18,19,20,21 These methods should permit a better understanding of dysfunctions in the brain serotonergic system and the influence of medications on them, as well as regional imbalances and the effects of drugs on these imbalances. My colleagues and I have been investigating the use of labelled α -methyl-L-tryptophan (α -MTrp) as a tracer for the imaging of the brain serotonergic system. In particular, our studies have focused on regional 5-HT synthesis and the influence of different drugs on this process.22,23,24,25,26,27 We have also investigated the effects of local brain lesions at serotonergic terminals on brain 5-HT synthesis in brain areas distant from the lesions.25,27,28,29
Characteristics of α -MTrp
α -MTrp, an artificial amino acid, is an analog of L-Trp. α -MTrp is a substrate for TPH and is converted under in vivo conditions to 5-hydroxy-α -methyl-L-tryptophan, which is also a substrate for aromatic amino acid decarboxylase.30 This therefore yields α -methyl-serotonin (α -M5-HT). α -M5-HT is not a substrate for MAO and, as such, remains in the brain for quite a long time after being formed from α -MTrp. In addition, α -MTrp is not incorporated into brain proteins.16,31 Studies from our laboratories have also shown that there is an excellent concordance between the localization of endogenous 5-HT and radioactively labelled α -MTrp, as well as TPH and radioactively labelled α -MTrp.32 Recently, we measured the brain trapping of α -[14C]MTrp in rats treated with cycloheximide, an inhibitor of the protein synthesis, and found that this treatment has no effect on the brain 5-HT synthesis calculated from the trapping of α -[14C]MTrp.33 These data with cycloheximide suggest that even after brain protein synthesis was inhibited, the brain trapping of α -[14C]MTrp was the same as that in the saline-treated rats. This indicates that α -[14C]MTrp is trapped by a process related to 5-HT synthesis because, despite the protein synthesis inhibition, the trapping of α -[14C]MTrp did not change.
On the basis of these findings, we have proposed a 3-compartment model in which the plasma and the precursor and irreversible pools are mathematically distinct compartments.16,17,34 It should be emphasized that these compartments, biologically, may not be distinct and easily identifiable, except for the plasma compartment. This is generally the case with most biological models — the compartments are described as mathematical creations that are often not biologically realistic.35 The compartmental structure for the α -MTrp model is shown in Fig. 1. The rate constants in the model are:
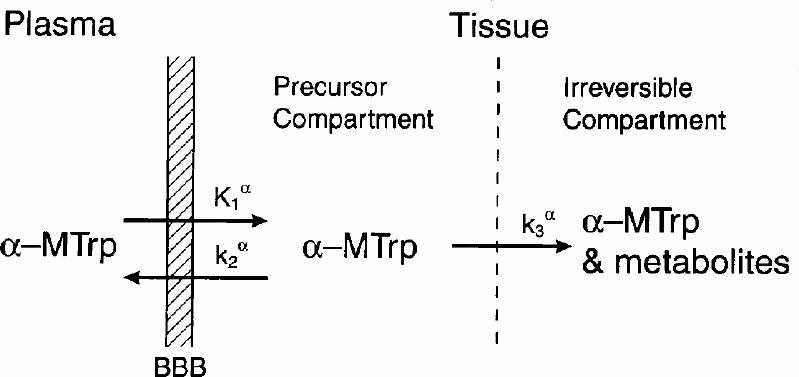
Fig. 1: Schematic representation of the brain compartmental structure used in the modelling of labelled α -methyl-L-tryptophan. The rate constants are assumed to be first-order rate constant and have units of min-1 except K1α (in mL/g per min), which is the product of the first-order rate constant k1α (per min) and the plasma volume (in mL/g) from which tracer is transferred into the brain precursor pool. The k2α is the constant for the movement of the tracer from the precursor pool to plasma, and k3α is the rate constant for the movement of the tracer from the precursor pool to the brain irreversible pool.
· K1α (mL/g per min) for the movement of the tracer from the plasma to the brain precursor pool,
· k2α (per min) from the brain precursor pool to the plasma, and
· k3α (per min) from the brain precursor pool to the brain irreversible pool.
By definition, K1α is equal to the product of the first-order rate constant k1α (per min) and the plasma volume (in mL/g) from which the tracer is exchanged with the precursor pool. An irreversible compartment is defined as one in which the tracer, its metabolite or both gets trapped, and it does not exchange directly with the plasma compartment. It is important to note that this model, as originally proposed,16,17 is not based on a metabolic conversion of labelled α -MTrp to α -M5-HT, as implied by some investigators.36,37 Rather, it is based on an irreversible trapping of labelled α -MTrp, its metabolite(s) or both. The issues related to the metabolic conversion have been discussed in more detail elsewhere.38,39 After solving the homogenous differential equations representing the movement of the tracer between compartments and expressing the tissue concentration of the tracer in the form of distribution volume (DV, in mL/g), one obtains (Equation 1):16,17,18
where Θ (min) is defined as the exposure time and is equal to the plasma integral divided by the tracer concentration at time T (Θ = ∫ 0TCp*(t) dt/Cp*(T)), and Kα (in mL/g per min) is the trapping (unidirectional net uptake) constant for α -MTrp.
In rat16,34,38 autoradiographic experiments using α -[14C]MTrp, and in dog17,40 and human38,41,42 experiments using positron emission tomography (PET) and α -[11C]MTrp, we investigated the presence of the tissue irreversible compartment. A comparison was done of the fit, by including k3α (3-compartment model) or excluding k3α (2-compartment model) in the fitting of the experimental data. As shown in Fig. 2 for the rat experiments and in Fig. 3 for the human experiment, there was a significantly better fit when k3α , the constant for an irreversible step (i.e., presence of on irreversible compartment) was included in the model structure. These results suggest that labelled α -MTrp is taken up into the brain irreversible compartment and also that an irreversible compartment for this tracer is present in the brain.38 This is contrary to the conclusion of Shoaf et al36 for their measurements in the brain of an anesthetized monkey; this point has been discussed in more detail elsewhere.38
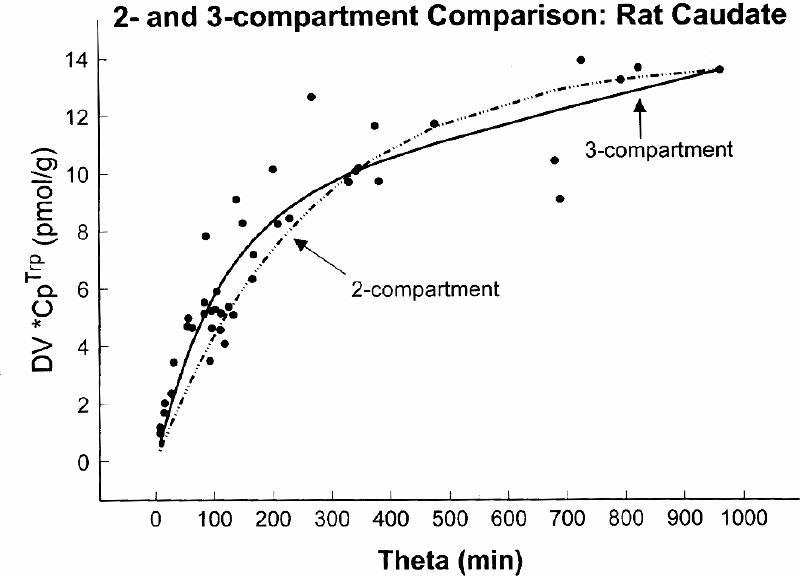
Fig. 2: A comparison between 2- and 3-compartment fits to the experimental data obtained in rats. The rats, each represented by one point on the graph, were killed at different times after tracer injection. The y axis represents distribution volume of the tracer (DV, in mL/g) obtained by dividing tissue tracer radioactivity with the plasma radioactivity measured at the end of experiment, and the x axis (theta, min) represents an integral of the plasma radioactivity divided with the plasma radioactivity at the end of experiment. A statistical evaluation of residues indicated that the 3-compartment model provided a significantly (F = 26, p < 10–4) better fit to the experimental data than the 2-compartment model.
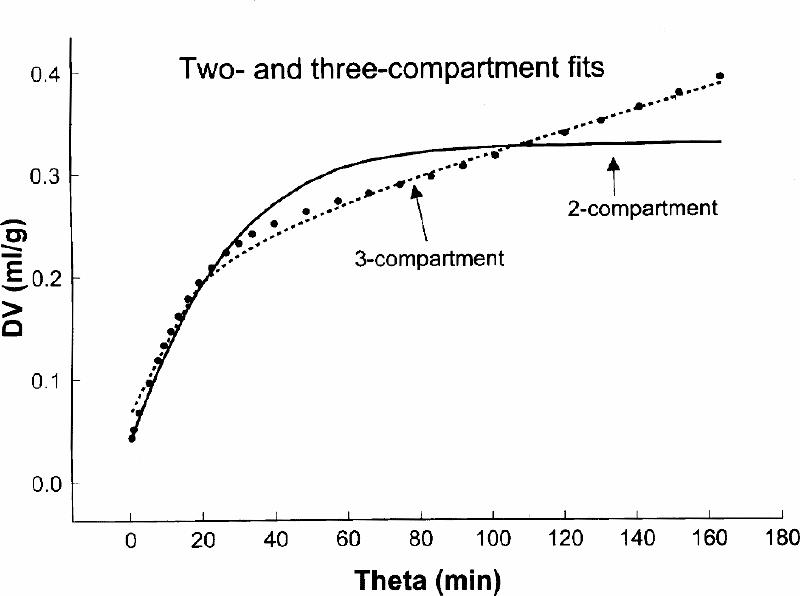
Fig. 3: A comparison between 2- and 3-compartment fits obtained in a human scanned for 90 min after tracer injection. The axes are defined as in Fig. 2. The 3-compartment fit was significantly better (F = 120, p < 10-6]. The fitting was done with the SAAM II program (SAAM Institute, University of Washington, Seattle, WA).
The 5-HT synthesis rate R (in pmol/g per min) is calculated from Kα by first converting it to the uptake constant for Trp metabolism via the 5-HT metabolic pathway (KT, in mL/g per min). This is accomplished by dividing Kα by the conversion factor named the “lumped constant” (LC).33,39,43 After obtaining KT, the 5-HT rates are calculated by multiplying KT with the plasma concentration of the free (non-albumin bound) Trp (CpTrp, in pmol/mL), such that:
R = (Kα /LC)(CpTrp)
This conversion was recently criticized by Chugani and Muzik44 on the grounds that there is uncertainty regarding which plasma Trp concentration should be used in the calculation. Indeed, many different views have been voiced on this subject.45,46,47,48 However, our use of the plasma-free Trp concentration in this calculation is supported by good experimental45,46,47 and theoretical48 evidence in the literature. Certainly, this does not imply that in all experimental settings or for all pathologic conditions, this concentration of Trp is the most appropriate to use. There should be no question that the product of KT and plasma Trp concentration represents brain 5-HT synthesis rate. Rather, the question should be which is, in a particular situation, the most appropriate concentration of the plasma Trp to be used in the calculation. In addition, the use of the plasma-free Trp concentration in experiments done with labelled Trp as a tracer for the measurement of 5-HT synthesis rates provides results similar to those obtained with other methods. The fact that plasma-free Trp concentration could vary should not negate its use in the calculation. It suggests that we must find a way to deal with this reality. The suggestion of Chugani and Muzik44 that Kα is not dependent on the changes in the plasma Trp is probably erroneous because Kα , by definition, includes in its formulation the free fraction of α -MTrp.16,17 This, in rats, correlates with the plasma-free fraction of Trp [unpublished data]. Gharib et al37 recently reported that the LC is not constant throughout the brain. This conclusion, however, was based on an erroneous data analysis, as discussed in our recent publication.39 As a matter of fact, their results obtained with labelled α -MTrp and a very different methodological approach accord very well with ours.39
Effect of regional lesions of serotonergic terminals on TPH activity
It has been shown in many electrophysiological experiments that the receptors found on the terminals of serotonergic neurons may control the release of 5-HT49,50,51 and possibly its synthesis.49,52 It is not surprising that the synthesis of 5-HT occurs in those terminals because TPH is present in the neuronal terminals. However, the exact control of the synthesis is not yet well understood. In male Wistar rats, we produced a local lesion with 5,7-dihydroxytryptamine (5,7-DHT),53 a specific toxin for serotonergic neurons.54 5,7-DHT (3 μg of free base in 250 nL of saline) was injected stereotaxically over 20 min into the dorsolateral hypothalamus (coordinates: –3.0 mm from bregma, 1.0 mm lateral and 8.1 mm below the brain surface).55 Five days later, the rats were injected with 1 mCi (1 Bq = 2.7 х 10-11 Ci approx) of α -[3H]MTrp (specific activity (SA) = 10 Ci/mmol) and were killed 24 h thereafter. The brains were sliced and TPH was stained with a specific antibody.32,56,57 The autoradiograms obtained for TPH and α -[3H]MTrp were quantified using appropriate standards with the aid of an image analysis system.53 The results suggest that there is a highly significant correlation between the concentration of TPH in the dorsal raphe and its activity. The activity of TPH was measured by the presence of labelled α -MTrp, its metabolite(s) or both. On the basis of the conversion rates of labelled α -MTrp into labelled α -M5-HT in the dorsal raphe16,37 24 h after an an injection of α -[3H]MTrp, the radioactivity came mainly from α -[3H]M5-HT. There was no significant correlation between the TPH concentration and its activity, measured as the presence of α -[3H]M5-HT, in the median forebrain bundle contralateral to the lesion.53 However, there was a highly significant correlation between TPH concentration and its activity, as measured from the presence of the radioactivity derived from α -[3H]MTrp (mainly α -[3H]M5-HT), in the median forebrain bundle ipsilateral to the lesion. These data suggest that TPH is transported along the medial forebrain bundle in a nonactivated state or that there is no access to TPH by the substrates (Trp and oxygen) or cofactors required for the 5-HT synthesis. However, after the terminals are damaged, TPH becomes active either because terminal feedback control has been lost or because the enzyme is more freely accessed with all the required substrates and cofactors. We also measured, in a separate group of rats with 5,7-dihydroxytryptamine (5,7-DHT) lesions in the dorsolateral hypothalamus, regional 5-HT synthesis using α -[14C]MTrp and autoradiography.28 There was a rather profound ipsilateral effect of this hypothalamic lesion, which resulted in an increase in 5-HT synthesis in many of the structures investigated. Furthermore, the lesion induced an increase in the TPH mRNA in the ipsilateral dorsal raphe,58 suggesting that with the loss of the serotonergic terminals, the cells lose control of 5-HT synthesis and, as a result, require more TPH for greater synthesis of 5-HT.
The influence of the terminal integrity after a freezing lesion in the rat parietal cortex on ipsi- and contralateral 5-HT synthesis has also been investigated with labelled α -MTrp.29 In these experiments, a general autoradiographic procedure was used on rats with freezing lesions produced by a cold probe (4 mm in diameter contacted for 5 s, –50°C) 72 h before 5-HT synthesis was measured. Control rats went through the same surgical procedure, but a room-temperature probe was used. The results suggest that there is an increase in 5-HT synthesis in many brain structures ipsilateral to the lesion.
It has been reported that an increase in the trapping of labelled α -MTrp can occur in pathologic conditions of the human brain in which there is an activation of the kynurenine pathway.19,20,21 It is possible that this pathway is activated in the rim around the lesion itself, because in that area, we have observed that the blood–brain barrier is compromised.29 This activation of the kynurenine pathway would be consistent with its activation in inflammatory diseases.57 But it is unlikely that this pathway would be activated in brain structures that are far from the lesion where there has been no alteration in blood–brain barrier permeability.27,28,29
Serotonergic drugs and 5-HT synthesis
We studied, using autoradiography and α -[14C]MTrp (about 30 μCi, SA 55 mCi/mmol) as a tracer, the influence of several drugs known to affect monoaminergic systems (e.g., reserpine, fenfluramine, fluoxetine, buspirone, ± 3,4-methylenedioxy-methamphetamine [MDMA]) in the rat brain.24 As well, we studied the effect of elevations of plasma Trp and oxygen in dogs17 and the reduction of plasma Trp in humans,42 using PET and α -[11C]MTrp (up to 10 mCi, SA about 100 Ci/mmol) as a tracer. Results of the studies revealed that brain 5-HT synthesis can be influenced by an extraneuronal concentration of 5-HT (studies with fluoxetine, fenfluramine, MDMA and reserpine), and that, generally, the influence is not uniform throughout the brain.22,23,27,28,29,59,60
A single dose of fluoxetine (10 mg/kg, intraperitoneally, 2 hours before tracer injection) produced a significant increase in 5-HT synthesis in terminal areas and, at the same time, a decrease in cell body structures.23 This treatment did not have any effect on plasma Trp, either total or free (non-albumin bound). The reduction in synthesis in the cell bodies is consistent with 5-HT synthesis control through somatodendritic 5-HT1A autoreceptors, which have been shown to control the firing of serotonergic neurons.61 After an 8-day treatment with fluoxetine, a general reduction in brain 5-HT synthesis was observed, but there were brain structures in which no effect was observed. This decrease in 5-HT synthesis with prolonged fluoxetine treatment would be expected because reuptake inhibitors make more 5-HT available in the extraneuronal space for neurotransmission. It is interesting to note that a higher dose fluoxetine (30 mg/kg, intraperitoneally, 2 h before tracer injection) produced a large increase in 5-HT synthesis across the brain. Thus, the effect of fluoxetine on brain 5-HT synthesis may also be influenced by the dose of the drug, and this may be relevant to clinical situations where the drug is abused. The rather large increase in the synthesis of 5-HT could have other behaviour effects not yet investigated.
We have also evaluated the effect of a single dose (5 mg/kg) of d-fenfluramine,62 a drug which simultaneously releases63,64 and prevents the reuptake65 of 5-HT, on brain 5-HT synthesis in the rat. Because the mode of action of d-fenfluramine is somewhat similar to that of fluoxetine, the influence on synthesis was also similar. When compared with the synthesis rates in the saline-treated group, d-fenfluramine treatment significantly decreased synthesis in the dorsal raphe and increased rates in some terminal areas.62 There was no significant influence on the plasma Trp concentration. In rats treated with 5 mg/kg once a day for 7 days, we observed a reduction in synthesis in the dorsal raphe, but there was an increase in several projection areas. The increase was particularly pronounced in the locus ceruleus (127% of that in control group), suggesting a possible influence from the loss of noradrenergic synapses. This would be in agreement with the reported neurotoxic effect of D-fenfluramine on brain serotonergic terminals.66 If de-enervation occurs, there could be an upregulation of TPH activity similar to that observed in the dorsal raphe, a brain region remote to the 5,7-DHT hypothalamic lesion.28
We found that treatment with reserpine (10 mg/kg, intraperitoneally, 30 min before tracer) profoundly reduced 5-HT synthesis in many discrete areas of the rat brain.22 This is probably related to the inhibitory action of released 5-HT, and possibly a response to other monoamines (i.e., dopamine, norepinephrine and histamine) known to be released by reserpine.67,68,69 The reduction could be a result of the direct effects of other neurotransmitters (e.g., dopamine) inhibiting the activity of TPH,70 or it might be related to the action of released 5-HT through the terminal and somatodendritic autoreceptors via feedback mechanisms.71
We have also shown that treatment with the aromatic amino acid decarboxylase inhibitor NSD-1015 has, by itself, a direct effect on brain 5-HT synthesis.22 This is because it increases the plasma and, most likely, the brain concentration of Trp. TPH is not saturated with the substrate Trp, so changes in brain Trp, at least to some extent, will influence brain 5-HT synthesis, as assessed by brain functions mediated by 5-HT.72 This effect has been reported in experiments with rats,73 in experiments with people in which plasma Trp depletion precipitated a negative mood state in healthy men74,41 and in patients with depression responding to antidepressant therapy.75 A more marked mood change in women than in men76 would accord with our observation of larger reductions in the brain 5-HT synthesis in women than men.42
The influence of MDMA (Ecstasy) on 5-HT synthesis was investigated in rats to determine the effects of giving the same total amount of drug on different dosing schedules.60 In addition, the influence of MDMA on brain 5-HT synthesis was studied in the dog at different times after a single dose of MDMA was administered.40 In rat experiments, we observed that MDMA given twice in 10 mg/kg doses every 12 hours or 4 times in 5 mg/kg doses every 12 hours (total dosage 20 mg/kg per day) had very different effects on 5-HT synthesis. In the rats treated with 2 doses, 5-HT synthesis was significantly reduced throughout the brain; however, 5-HT synthesis was increased in those administered the 4 doses over 2 days. In rats given MDMA in 8 doses of 5 mg/kg every 12 h for 4 days, there was a significant reduction in 5-HT synthesis, measured 14 days after the last dose, in many brain structures. These results suggest that the response of the serotonergic system to this drug may depend on the total amount of the drug injected at one time; 10 mg/kg was enough to profoundly reduce synthesis. This was most likely due to TPH inhibition via the autoreceptors, produced by released 5-HT. The reduction in synthesis measured 14 days after 8 injections could be related, at least in part, to a partial de-enervation of the neurons projecting from the dorsal raphe.77,78 Of course, it also may represent a downregulation of synthesis, as a result of the feedback action of released 5-HT. However, it should also be noted that there could be TPH activation in the neurons with damaged terminals, as was similarly observed in brains with 5,7-DHT hypothalamic lesions.28 If this were to happen, the reduction in some neurons could be substantially greater than that in others. Finally, there is also the possibility that dopamine released by MDMA79 has some influence on 5-HT synthesis and the apparent toxicity of MDMA.80,81
In the experiments with dogs,40 the influence of MDMA on brain 5-HT synthesis was evaluated using PET and α -[11C]MTrp. Brain 5-HT synthesis increased about 6-fold 1 hour after the MDMA injection (compared with baseline rates before the injection), whereas 5 hours after the injection, 5-HT synthesis was about half of baseline levels, or approximately 13 times lower than the rate 1 hour after the MDMA injection. These results again suggest that brain 5-HT synthesis can be controlled, to some extent, by the concentration of extraneuronal 5-HT. It should be noted that MDMA releases 5-HT from the neuronal storage and prevents the reuptake of released 5-HT into the vesicular storage.82,83 In addition, the data suggest that a large surge in 5-HT synthesis a relatively short time after the MDMA injection and the subsequent release of dopamine79 might contribute to the behavioural effects reported by human users.84,85 In addition, a large drop in synthesis some 5 hours after the injection could explain, at least in part, the recreational users' need for a “buster dose.”84,85
Effect of plasma tryptophan on brain 5-HT synthesis
The influence of plasma Trp on human behaviour has been studied for a long time.74,86,87 Studies indicate that a decrease in plasma Trp precipitates more depressive symptoms, as well as other symptoms associated with the brain serotonergic system, in female subjects than in males.75,76 Trp depletion studies75 in patients responding to antidepressant therapy suggest that, indeed, plasma Trp can modulate brain 5-HT synthesis levels. These observations are in accord with the hypothesis that major depression may be associated with a decrease (probably more accurately termed an imbalance) in 5-HT neurotransmission.84,88,89 However, the exact nature and biological basis remains ambiguous.
We investigated, using PET and α -[11C]MTrp directly in men and women, the effect of plasma Trp depletion on brain 5-HT synthesis,42 and found a substantially larger reduction in brain 5-HT synthesis (as measured by the uptake of α -[11C]MTrp) in women than in men; synthesis was reduced by approximately 40 times in women, compared with a 10-time reduction in men. Certainly, our sample was small, but our findings agree with behavioural measures that have been used to assess the effects of lowering plasma Trp in female and male subjects75 and with reports that women are particularly vulnerable to decreases in brain serotonergic function.88 We did find a difference in brain 5-HT synthesis between male and female subjects,42 but when these data were combined with another set (albeit obtained with a different scanner), we observed a bimodal distribution.89 There is obviously a need for further study on sex differences and 5-HT synthesis rates. The measurements of Kα reported by Chugani et al20 cannot be directly compared with our results42 because Kα , as measured by our method,16,17,18 includes another variable, the free fraction of α -[11C]MTrp, which could confound the comparison.
A study of plasma increases in Trp on brain 5-HT synthesis in the dog (calculated from trapping of α -[11C]MTrp using PET)17 suggests that there is an increase in 5-HT synthesis; one would expect because TPH is not normally saturated with Trp.73,90 The highest plasma concentration of Trp in that experiment (381 nmol/mL) would probably result in a brain concentration of about 84 nmol/g (assuming brain-to-plasma ratio between 0.1691,92 and 0.2893 [mean 0.22] reported for the rat), which would be just about twice the Km of TPH for Trp (Km was reported to be about 50 μmol/L94,95). This way, the estimated brain concentration of Trp would be about 107 μmol/L. Assuming a brain-to-plasma ratio for Trp in dogs of about 0.22, one should notice that the brain concentration of Trp at the baseline (plasma of 16 nmol/mL17) would be only about 4.7 μmol/L, which is far below the Km for TPH. It should be noted that all of the brain concentrations are estimated from data obtained in rats, which may not necessarily be valid for dogs. In addition, the Km of TPH in the dog brain under the conditions used in those experiments is unknown. Because the TPH in the brain could be almost saturated when plasma Trp concentration is high, there is a possibility that the LC conversion constant might change, and this would introduce a bias in the calculated synthesis rates. However, a general trend should still be valid. It should be noted that the brain-to-plasma ratio of Trp may increase with the increase of plasma Trp.96
Conclusion
The experimental results obtained so far suggest that labelled α -MTrp is a good tracer for studying the brain serotonergic system under the assumptions outlined in our publications. There is an irreversible trapping of the tracer in the brain at a baseline, and the trapping is sensitive to a variety of pharmacological manipulations. Imaging in laboratory animals could be very useful to obtain relevant information on TPH activation and the effects of local lesions of the brain serotonergic system. The study of TPH activity and its modulation with different drugs could yield valuable information on the differential control of synthesis in different brain structures (e.g., cell body v. projection areas). The advantages of the α -MTrp method include that it is minimally invasive, it does not require other drug treatments and it can be used in both laboratory animals and humans. Under certain conditions, a fraction of Trp exchanging with the brain might differ from the plasma-free Trp; in those cases, the 5-HT synthesis rates calculated with the plasma-free Trp may not be absolute. However, even in these cases, one should be able to get valid information about the change produced by the treatment. Indeed, regional changes produced by drugs are probably the most important factors responsible for their beneficial effects. If the changes are analyzed, as we recently proposed,89 the value, or a change therein, does not influence the comparisons.
Acknowledgments
My sincere thanks to the fellows and students who performed the actual experiments: Z. Cohen, M. Grdisa, A. Jevric-Causevic, V. Ljubic-Thibal, D. Mück-Seler, S. Nagahiro, S. Nishizawa, H. Okazawa, A. Takada, S. Takahashi, Y. Tohyama, K. Tsuiki, M. Vanier and F. Yamane. I would also like to express my thanks to the many collaborators with whom I have had many valuable discussions. I would also like to thank Ms. Valerie-Ann Cherneski for her editorial help.
Footnotes
Based on the Innovations in Neuropsychopharmacology Award Lecture presented at the 23rd Annual Meeting of the Canadian College of Neuropsychopharmacology in Marrakesh, Morocco, Apr. 10–12, 2000
The research reported here was supported, in part, by Medical Research Council of Canada (MT-13368) and US Public Health Service (R01-NS29629) grants.
Competing interests: None declared.
Correspondence to: Dr. Mirko Diksic, Montreal Neurological Institute, McGill University, 3801 University St., Montreal QC H3A 2B4; fax 514 398-8195; mirko@pet.mni.mcgill.ca
Submitted Oct. 16, 2000 Revised Jan. 17, 2001 Accepted Jan. 31, 2001
References
- 1.Carlsson A, Lindqvist M. The effect of L-tryptophan and some psychotropic drugs on the formation of 5-hydroxytryptophan in the mouse brain in vivo. J Neural Transm 1972;33:23-43. [DOI] [PubMed]
- 2.Lopez-Ibor, JJ Jr. The involvement of serotonin in psychiatric disorders and behaviour. Br J Psychiatry Suppl 1988;3:26-39. [PubMed]
- 3.Heninger G.R. Indoleamines: the role of serotonin in clinical disorders. In: Bloom FE, Kupfer DJ, editors. Psychopharmacology: the fourth generation of progress. New York: Raven Press; 1995. p. 471-82.
- 4.Azmitia EC, Segal M. An autoradiographic analysis of the differential ascending projections of the dorsal and median raphe nuclei in the rat. J Comp Neurol 1978;179:641-67. [DOI] [PubMed]
- 5.Fowler CJ, Tipton KF. Deamination of 5-hydroxytryptamine by both forms of monoamine axydase by the rat brain. J Neurochem 1982;38:733-6. [DOI] [PubMed]
- 6.Tamir H, Gershon MD. Storage of serotonin and serotonin binding protein in synaptic vesicles. J Neurochem 1979;33:35-44. [DOI] [PubMed]
- 7.Burns D, London J, Brunswick DJ, Pring M, Garfinkel D, Rabinowitz JL, et al. A kinetic analysis of 5-hydroxyindoleacetic acid extraction from rat brain and CSF. Biol Psychiatry 1976;11:125-57. [PubMed]
- 8.Wolf WA, Youdim MBH, Kuhn DM. Does brain 5-HIAA indicate serotonin release or monoamine oxidase activity? Eur J Pharmacol 1985;109:381-7. [DOI] [PubMed]
- 9.Garelis E, Young SN, Lal S, Sourkes TL. Monoamine metabolites in lumbar CSF: their sites of origin in relation to clinical studies. Brain Res 1974;79:1-8. [DOI] [PubMed]
- 10.Wood JH. Neurochemical analysis of cerebrospinal fluid. Neurology 1980;30:645-51. [DOI] [PubMed]
- 11.Jakupcevic M, Lackovic Z, Stefoski D, Bulat M. Nonhomogenous distribution of 5-hydroxyindoleacetic acid and homovanillic acid in the lumbar cerebrospinal fluid of man. J Neurol Sci 1977;31: 165-71. [DOI] [PubMed]
- 12.Bulat M. Cerebrospinal fluid and brain: basic controversies and new vistas. Period Biol 1996;98:11-5.
- 13.Gunn RN, Sargent PA, Bench CJ, Rabiner EA, Osman S, Pike VW, et al. Tracer kinetic modelling of the 5-HT1A receptor ligand [carbonyl-11C]WAY-100635 for PET. Neuroimage 1998;8: 426-40. [DOI] [PubMed]
- 14.Cho R, Kapur S, Du L, Hrdina P. Relationship between central and peripheral serotonin 5-HT2A receptors: a positron emission tomography study in healthy individuals. Neurosci Lett 1999; 261:139-42. [DOI] [PubMed]
- 15.Ito H, Nyberg S, Halldin C, Lundkvist C, Farde L. PET imaging of central 5-HT2A receptors with carbon-11-MDL 100,907. J Nucl Med 1998;39:208-14. [PubMed]
- 16.Diksic M, Nagahiro S, Sourkes TL, Yamamoto YL. A new method to measure brain serotonin synthesis in vivo: I — Theory and basic data for a biological model. J Cereb Blood Flow Metab 1990;10:1-12. [DOI] [PubMed]
- 17.Diksic M, Nagahiro S, Chaly T, Sourkes TL, Yamamoto YL, Feindel W. Serotonin synthesis rate measured in living dog brain by positron emission tomography. J Neurochem 1991;56:153-62. [DOI] [PubMed]
- 18.Muzik O, Chugani DC, Chakraborty P, Mangner T, Chugani HT. Analysis of [C-11]alpha-methyl-tryptophan kinetics for the estimation of serotonin synthesis rate in vivo. J Cereb Blood Flow Metab 1997;17:659-69. [DOI] [PubMed]
- 19.Chugani DC, Chugani HT, Muzik O, Shah JR, Shah AK, Canady A, et al. Imaging epileptic tubers in children with tuberous sclerosis complex using α -[11C]methyl-L-tryptophan positron emission tomography. Ann Neurol 1998;44:858-66. [DOI] [PubMed]
- 20.Chugani DC, Heyes MP, Kuhn DM, Chugani HT. Evidence α -[C-11]methyl-L-tryptophan metabolism via the kynurenine pathway in tuberous sclerosis complex [abstract]. Soc Neurosci Abstr 1998;24:1757.
- 21.Chugani DC, Muzik O, Chakraborty P, Mangner T, Chugani HT Human brain serotonin synthesis capacity measured in vivo with α -[C-11]methyl-L-tryptophan. Synapse 1998;28:33-43. [DOI] [PubMed]
- 22.Mück-Seler D, Diksic M. The acute effects of reserpine and NSD-1015 on the brain serotonin synthesis rate measured by an autoradiographic method. Neuropsychopharmacology 1995;12: 251-62. [DOI] [PubMed]
- 23.Mück-Seler D, Jevric-Causevic A, Diksic M. Influence of fluoxetine on regional serotonin synthesis in the rat brain. J Neurochem 1996;67:2434-42. [DOI] [PubMed]
- 24.Mück-Seler D, Takahashi S, Diksic M. The effect of MDMA on the 5-HT synthesis rate in the rat brain: an autoradiographic study. Brain Res 1998;810:76-86. [DOI] [PubMed]
- 25.Tsuiki K, Yamamoto YL, Diksic M. Effect of acute fluoxetine treatment on the brain serotonin synthesis as measured by the α -methyl-L-tryptophan autoradiographic method. J Neurochem 1995;65:250-6. [DOI] [PubMed]
- 26.Okazawa H, Yamane F, Blier P, Diksic M. Effects of acute and chronic administration of the serotonin1A agonist buspirone on serotonin synthesis in the rat brain. J Neurochem 1999;72: 2022-31. [DOI] [PubMed]
- 27.Tsuiki K, Yamamoto YL, Diksic M. Effect of acute fluoxetine treatment on the brain serotonin synthesis as measured by the alpha-methyl-L-tryptophan autoradiographic method. J Neurochem 1995;65:250-6. [DOI] [PubMed]
- 28.Tsuiki K, Mück-Seler D, Diksic M. Autoradiographic evaluation of the influence of hypothalamic 5,7-dihydroxytryptamine lesion on brain serotonin synthesis. Biochem Pharmacol 1995;49:633-42. [DOI] [PubMed]
- 29.Tsuiki K, Takada A, Nagahiro S, Grdisa M, Diksic M, Pappius HM. Synthesis of serotonin in traumatized rat brain. J Neurochem 1995;64:1319-25. [DOI] [PubMed]
- 30.Roberge AG, Missala K, Sourkes TL. Alpha-methyltryptophan: effects on synthesis and degradation of serotonin in the brain. Neuropharmacology 1972;11:197-209. [DOI] [PubMed]
- 31.Madras BK, Sourkes TL. Metabolism of α -methyltryptophan. Biochem Pharmacol 1965;14:1499-506. [DOI] [PubMed]
- 32.Cohen Z, Tsuiki K, Takada A, Beaudet A, Diksic M, Hamel E. In vivo-synthesized radioactively labelled a-methyl serotonin as a selective tracer for visualization of brain serotonin neurons. Synapse 1995;21:21-8. [DOI] [PubMed]
- 33.Diksic M, Tohyama Y, Firke Merid M. Cycloheximide, an inhibitor of protein synthesis, does not influence brain trapping of α -[14C]methyl-L-tryptophan [abstract 50]. Proceedings of the International Chemical Congress of Pacific Basin Societies; 2000 Dec 14–19; Honolulu (HA). Washington: American Chemical Society; 2001.
- 34.Diksic M, Grdisa M. α -Methyl-L-tryptophan as a tracer to study brain serotonergic system. Neurochem Res 1995;20:1353-60. [DOI] [PubMed]
- 35.Norwich KH. Molecular dynamics in biosynthesis: the kinetics of tracers in intact organisms. New York: Pergamon Press; 1977. p. 133.
- 36.Shoaf SE, Carson RE, Hommer D, Williams WA, Higley JD, Schmall B, et al. The suitability of [11C]-α -methyl-L-tryptophan as a tracer for serotonin synthesis: studies with dual administration of [11C] and [14C] labeled tracer. J Cereb Blood Flow Metab 2000;20:244-52. [DOI] [PubMed]
- 37.Gharib A, Balende C, Sarda N, Weissmann D, Plenevaux A, Luxen A, et al. Biochemical and autoradiographic measurements of brain serotonin synthesis rate in the freely moving rat: a reexamination of the α -methyl-L-tryptophan method. J Neurochem 1999;72:2593-600. [DOI] [PubMed]
- 38.Diksic M. Does labelled α -methyl-L-tryptophan image ONLY blood–brain barrier transport of tryptophan? [letter] J Cereb Blood Flow Metab 2000;20:1508-11. [DOI] [PubMed]
- 39.Diksic M, Leyton M, Benkelfat C. Is α -methyl-L-tryptophan a good tracer for brain serotonin synthesis measurements, and is the lumped constant different in different brain structures of the rat brain? J Neurochem 1999;73:2621-2. [DOI] [PubMed]
- 40.Nishisawa S, Mzengeza S, Diksic M. Acute effects of 3,4-methylene-dioxymethamphetamine on brain serotonin synthesis in the dog studied by positron emission tomography. Neurochem Int 1999;34:33-44. [DOI] [PubMed]
- 41.Benkelfat C, Seletti E, Mark A, Dean P, Palmour RM Young SN. Mood-lowering effects of tryptophan deplition: enhanced susceptibility in young man at genetic risk for major affective disorders. Arch Gen Psychiatry 1994;51:687-97. [DOI] [PubMed]
- 42.Nishizawa S, Benkelfat C, Young SN, Leyton M, Mzengeza S, de Montigny C. Differences between males and females in rates of serotonin synthesis in human brain. Proc Natl Acad Sci U S A 1997;94:5308-13. [DOI] [PMC free article] [PubMed]
- 43.Vanier M, Tsuiki K, Grdisa M, Worsley K, Diksic M. Determination of the lumped constant for the alpha-methyltryptophan method of estimating the rate of serotonin synthesis. J Neurochem 1995;64:624-35. [DOI] [PubMed]
- 44.Chugani DC, Muzik O. α -[C-11]Methyl-L-tryptophan PET maps brain serotonin synthesis and kynurenine pathway metabolism. J Cereb Blood Flow Metab 2000;20:2-9. [DOI] [PubMed]
- 45.Bloxam DL, Curzon G. A study of proposed determinants of brain tryptophan concentration in rats after portocaval anastomosis or sham operation. J Neurochem 1978;31:1255-63. [DOI] [PubMed]
- 46.Chaouloff F, Kennett GA, Serrurrier B, Merino D, Curzon G. Amino acid analysis demonstrates that increased plasma free-tryptophan causes the increase tryptophan during exercise in rat. J Neurochem 1986;46:1647-50. [DOI] [PubMed]
- 47.Takada A, Grdisa M, Diksic M, Gjedde A, Yamamoto YL. Rapid steady-state analysis of blood-brain transfer of L-Trp in rat, with special reference to the plasma protein binding. Neurochem Int 1993;23:351-9. [DOI] [PubMed]
- 48.Salter M, Knowles RG, Pogson CI. How does displacement of albumin-bound tryptophan cause sustained increases in the free tryptophan concentration in plasma and 5-hydroxytryptamine synthesis in brain? Biochem J 1989;262:365-8. [DOI] [PMC free article] [PubMed]
- 49.Sharp T, Bramwell SR, Grahame-Smith DG. 5-HT1 agonists reduce 5-hydroxytryptamine release in rat hippocampus in vivo as determined by brain microdialysis. Br J Pharmacol 1989;96: 283-90. [DOI] [PMC free article] [PubMed]
- 50.Haddjeri N, Blier P, de Montigny C. Long-term antidepressant treatments result in a tonic activation of forebrain 5-HT1A receptors. J Neurosci 1998;18:10150-6. [DOI] [PMC free article] [PubMed]
- 51.Pineyro G, Blier P. Autoregulation of serotonin neurons: role in antidepressant drug action. Pharmacol Rev 1999;51:533-91. [PubMed]
- 52.Hjorth S, Carlsson A. Buspirone: effects on central monoaminergic transmission — possible relevance to animal experimental and clinical findings. Eur J Pharmacol 1982;83:299-303. [DOI] [PubMed]
- 53.Ljubic-Thibal V, Diksic M, Hamel E, Raison S, Pujol JF, Weissman D. Ipsilateral alteration in tryptophan hydroxylase activity in rat brain after hypothalamic 5,7-dihydroxytryptamine lesion. Brain Res 1996;724:222-31. [DOI] [PubMed]
- 54.Björklund A, Nobin A, Stenevi R. The use of neurotoxic dihydroxytryptamines as tools for morphological studies and localized lesioning of central indolamine neurons. Z Zellforsch Mikrosk Anat 1973;145:479-501. [DOI] [PubMed]
- 55.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. New York: Academic Press; 1986.
- 56.Weissman D, Belin MF, Aguera M, Meunier C, MaÎtre M, Cash CD, et al. Immunohistochemistry of tryptophan hydroxylase in the rat brain. Neurosci 1987;23:291-304. [DOI] [PubMed]
- 57.Heyes MP, Saito K, Crowley JS, Davis LE, Demitrack MA, Der M, et al. Quinolinic acid and kynurenine pathway metabolism in inflammatory and non-inflammatory neurological disease. Brain 1992;115:1249-73. [DOI] [PubMed]
- 58.Ljubic-Thibal V, Morin A, Diksic M., Hamel E. Origin of the serotonergic innervation to the rat dorsolateral hypothalamus: retrograde transport of cholera toxin and upregulation of tryptophan hydroxylase mRNA expression following selective nerve terminals lesion. Synapse 1999;32:177-86. [DOI] [PubMed]
- 59.Mück-Seler D, Diksic M. Serotonin synthesis increased in terminals four days after reserpine treatment: an autoradiographic study in rat brain. Neurochem Res 1997;22:11-6. [DOI] [PubMed]
- 60.Mück-Seler D, Takahashi S, Diksic M. The effect of MDMA (3,4-methylenedioxymethamphetamine) on 5-HT synthesis rate in the rat brain: an autoradiographic study. Brain Res 1998;810:76-86. [DOI] [PubMed]
- 61.Blier P, de Montigny C. Modification of 5-HT neuron properties by sustained administration of the 5-HT1A agonist gepirone: electrophysiological studies in the rat brain. Synapse 1987;1:470-80. [DOI] [PubMed]
- 62.Yamane F, Tohyama Y, Diksic M. Acute and chronic D-fenfluramine treatments have different effects on serotonin synthesis rates in rat brain: an autoradiographic study. Neurochem Res 1999;24:1611-20. [DOI] [PubMed]
- 63.Mennini T, Garattini S, Cocia S. Anorectic effect of fenfluramine isomers and metabolites: relationship between brain levels and in vitro potencies on serotonergic mechanisms. Psychopharmacology 1985;85:111-4. [DOI] [PubMed]
- 64.Garattini S, Mennini T, Bendotti C, Invernizzi R, Samanin R. Neurochemical mechanism of action of drugs which modify feeding via the serotonergic system. Appetite 1986;7(Suppl):15-8. [DOI] [PubMed]
- 65.Knapp S, Mandel AJ. Coincidence of blockade of synaptosomal 5-hydroxytryptamine uptake and decrease in tryptophan hydroxylase activity;effects of fenfluramine. J Pharmacol Exp Ther 1976;198:123-32. [PubMed]
- 66.Molliver D, Molliver M. Anatomic evidence for a neurotoxic effect of D-fenfluramine upon serotonergic projections in the rat. Brain Res 1990;511:165-8. [DOI] [PubMed]
- 67.Oishi R, Suemaru K, Furuno K, Gomita Y, Saeki K. Possible explanation for the antagonism by nicotine agonist reserpine-induced deplition of monoamines in mouse brain. Naunyn Schmiedebergs Arch Pharmakol 1993;348:154-7. [DOI] [PubMed]
- 68.Ugedo L, Garro MA, Pineda J, Giralt MT, Miralles A, Olmos G. Acute and chronic effects of reserpine on biochemical and functional parameters of central and peripheral α 2-adrenoceptors. Eur J Pharmacol 1993;239:149-57. [DOI] [PubMed]
- 69.Russel WL, Henry DP, Phebus LA, Clemens JA. Release of histamine in rat hypothalamus and corpus striatum in vivo. Brain Res 1990;512:95-101. [DOI] [PubMed]
- 70.Johansen P, Wolf WA, Kuhn DM. Inhibition of tryptophan hydroxylase by benserazide and other catechols. Biochem Pharmacol 1991;41:625-8. [DOI] [PubMed]
- 71.Briley M, Moret C. Neurobiological mechanisms involved in antidepressant therapies. Clin Neuropharmacol 1993;16:387-400. [DOI] [PubMed]
- 72.Young SN. The clinical psychopharmacology of tryptophan. In: Wurtman RJ, Wurtman JJ, editors. Nutrition and the brain. Vol 7. Food constituents affecting normal and abnormal behaviors. New York: Raven Press; 1986. p. 49-88.
- 73.Fernstrom JD, Wurtman RJ. Brain serotonin content: physiological dependence on plasma tryptophan levels. Science 1971; 173:149-52. [DOI] [PubMed]
- 74.Young SN, Smith SE, Pihl RO, Ervin FR. Tryptophan depletion causes a rapid lowering of mood in normal males. Psychopharmacology 1985;87:173-7. [DOI] [PubMed]
- 75.Delgado PL. Depression: the case for a monoamine deficiency. J Clin Psychiatry 2000;61(Suppl 6):7-11. [PubMed]
- 76.Ellenbogen MA, Young SN, Dean P, Palmour RM, Benkelfat C. Mood response to acute tryptophan depletion in healthy volunteers: sex differences and temporal stability. Neuropsychopharmacology 1996;15:465-74. [DOI] [PubMed]
- 77.Ricaurte G, Bryan G, Strauss L, Seiden L Schuster C. Hallucinogenic amphetamine selectively destroys brain serotonin nerve terminals. Science 1985;229:986-8. [DOI] [PubMed]
- 78.Battaglia G, Yeh SY, O'Hearn E, Molliver ME, Kuhar MJ, DeSouza EB. 3,4-Methylene-dioxymethamphetamine and 3,4-methylenedioxyamphetamine destroy serotonin nerve terminals in rat brain: quantification of neurodegeneration by measurement of [3H]-paroxetine-labelled uptake sites. J Pharmacol Exp Ther 1987;242:911-6. [PubMed]
- 79.Stone DM, Hanson GR, Gibb JW. Differences in the central serotonergic effects of methylenedioxymethamphetamine (MDMA) in mice and rats. Neuropharmacology 1987;26:1657-61. [DOI] [PubMed]
- 80.Commins DL, Axt KJ, Vosmor G, Seiden LS. 5,6-Dihydroxytryptamine, a serotonergic neurotoxin, is formed endogenously in the rat brain. Brain Res1987;403:7-14. [DOI] [PubMed]
- 81.Seiden LS, Vosmerm G. Formation of 6-hydroxydopamine in candate nucleus of the rat brain after a single large dose of methamphetamine. Pharmacol Biochem Behav 1984;21:29-31. [DOI] [PubMed]
- 82.Rudnick G, Wall SC. The molecular mechanism of “ecstasy” [3,4-methylenedioxy-methamphetamine (MDMA)]. Serotonin transporters are targets for MDMA-induced serotonin release. Proc Natl Acad Sci U S A 1992;89:1817-21. [DOI] [PMC free article] [PubMed]
- 83.Schuldiner S, Sreiner-Mordoch S, Yelin R, Wall SC, Rudnick G. Amphetamine derivates interact with both plasma membrane and secretory vesicle biogenic amine transporters. Mol Pharmacol 1993;44:1227-31. [PubMed]
- 84.Downing J. The psychological and physiological effects of MDMA on normal volunteers. J Psychoactive Drugs 1986;18:335-40. [DOI] [PubMed]
- 85.Steele TD, McCann UD, Ricaurte GA. 3,4-Methylenedioxymethamphetamine (MDMA, “Ecstasy”): pharmacology and toxicology in animals and humans. Addiction 1994;89:539-51. [DOI] [PubMed]
- 86.Young SN, Sourkes TL. Antidepressant action of tryptophan. Lancet 1974;2(7885):897-8. [DOI] [PubMed]
- 87.Delgado PL, Charney DS. Neuroendocrine challenge studies in affective disorders. In: Horton RW, Katona C, editors. Biological aspects of affective disorders. London: Academic Press; 1991. p 146-90.
- 88.Smith KA, Fairburn CG, Cowen PJ. Relapse of depression after rapid depletion of tryptophan. Lancet 1997;349:915-9. [DOI] [PubMed]
- 89.Okazawa H, Leyton M, Benkelfat C, Mzengeza S, Diksic M. Gender-related regional differences in serotonin synthesis rate images detected by statistical mapping analysis. J Psychiatry Neurosci 2000;25:359-70. [PMC free article] [PubMed]
- 90.Delgado PL, Moreno FA. Role of norepinephrine in depression. J Clin Psychiatry 2000;61(Suppl 1):5-12. [PubMed]
- 91.Carlsson A. Measurements of monoamine synthesis and turnover with special reference to 5-hydroxytryptamine. Adv Biochem Psychopharmacol 1974;10:75-81. [PubMed]
- 92.Gal EM. Hydroxylation of tryptophan and its control in brain. Pavlov J Biol Sci 1975;10:145-60. [DOI] [PubMed]
- 93.Gessa GL, Tagliamonte A. Possible role of free serum tryptophan in the control of brain tryptophan level and serotonin synthesis. Adv Biochem Psychopharmacol 1974;11:119-31. [PubMed]
- 94.Gal EM, Christiansen PA, Yunger LM. Effect of p-chloroamphetamine on cerebral tryptophan-5-hydroxylase in vivo: reexamination. Neuropharmacology 1975;14:31-9. [DOI] [PubMed]
- 95.Mandell AJ, Knapp S. Regulation of serotonin biosynthesis in brain: role of the high affinity uptake of tryptophan into serotonergic neurons. Fed Proc 1977;36:2142-8. [PubMed]
- 96.Grahame-Smith DG. Studies in vivo on the relationship between brain tryptophan, brain 5-HT synthesis, and hyperactivity in rats treated with a monoamine oxidase inhibitor and L-tryptophan. J Neurochem 1971;18:1053-66. [DOI] [PubMed]