

Abstract
We have studied the receptor-specific function of four linker-insertion mutants of herpes simplex virus type 1 glycoprotein D (gD) representing each of the functional regions of gD. We used biosensor analysis to measure binding of the gD mutants to the receptors HVEM (HveA) and nectin-1 (HveC). One of the mutants, gD(∇34t), failed to bind HVEMt but showed essentially wild-type (WT) affinity for nectin-1t. The receptor-binding kinetics and affinities of the other three gD mutants varied over a 1,000-fold range, but each mutant had the same affinity for both receptors. All of the mutants were functionally impaired in virus entry and cell fusion, and the levels of activity were strikingly similar in these two assays. gD(∇34)-containing virus was defective on HVEM-expressing cells but did enter nectin-1-expressing cells to about 60% of WT levels. This showed that the defect of this form of gD on HVEM-expressing cells was primarily one of binding and that this was separable from its later function in virus entry. gD(∇243t) showed WT binding affinity for both receptors, but virus containing this form of gD had a markedly reduced rate of entry, suggesting that gD(∇243) is impaired in a postbinding step in the entry process. There was no correlation between gD mutant activity in fusion or virus entry and receptor-binding affinity. We conclude that gD functions in virus entry and cell fusion regardless of its receptor-binding kinetics and that as long as binding to a functional receptor occurs, entry will progress.
Entry of herpes simplex virus type 1 (HSV-1) into cells involves the coordinated actions of at least five viral glycoproteins: glycoprotein B (gB), gC, gD, gH, and gL (50). All but gC are indispensable for entry (1, 13, 27, 43), and gB, gD, gH, and gL are sufficient for cell fusion (55). With the exception of gD, the individual functions served by the essential glycoproteins are unknown. gD is a type I virion membrane glycoprotein of 369 amino acids in its mature form. The core of gD (residues 56 to 184) has a V-like immunoglobulin (Ig) fold. This is encircled by extensions at the N and C termini which comprise the principal functional domains of the protein (3). gD homologues are present only in the alphaherpesviruses (although not in varicella-zoster virus). In these viruses, gD plays a conserved and pivotal role during virus entry, binding to one of several specific receptors (2, 5, 17, 36, 51, 56). Two of these receptors, herpesvirus entry mediator (HVEM; also known as HveA) and nectin-1 (CD111; previously known as HveC), have been extensively studied because they are used by all wild-type (WT) strains of HSV-1 (17, 36).
HVEM is a member of the tumor necrosis factor receptor family (36). It has at least two natural ligands, LIGHT and lymphotoxin-α, as well as gD (31, 47). Nectin-1 is a cell surface protein comprising one V-like and two C-like Ig domains (28). The N-terminal V domain contains the gD-binding region (23, 30, 32). Nectin-1 functions as an adhesion molecule and is found at adherens junctions, where it can engage in a homotypic trans-interaction with nectin-1 on an apposed cell membrane (24, 52, 53, 63). Mutational analysis and studies of gD-specific antibodies that block receptor binding suggest that the two receptors bind to similar but distinct sites on gD. Interestingly, although nectin-1 and HVEM are structurally unrelated, they bind to gD with the same affinity and kinetics (26, 61).
Prior to the identification of gD receptors, a panel of gD linker-insertion mutants were tested for entry activity in a complementation assay and for structural integrity by reactivity with gD-specific monoclonal antibodies (4). The rationale was that linker-insertions that gave rise to antigenically intact but functionally impaired proteins might be located in functionally significant domains of gD. On this basis, four linear functional regions (FR) were defined (4). Their locations within the recently solved structure of the gD-HVEM complex (3) are shown in Fig. 1.
FIG. 1.

Structure of gD in complex with HVEM, showing the locations of FR and of linker-insertions. The structure shown (3) contains HVEM residues 4 to 105 (shown in blue) and gD residues 1 to 259. Residues 1 and 259 of gD are numbered and prefixed with an asterisk. FRI (residues 27 to 43), FRII (residues 125 to 161), and FRIII (residues 222 to 246) are shown in red, yellow, and green, respectively. FRIV (residues 277 to 310) was not resolved in the structure. The Ig-like core of gD, with the exception of the C", D, and E β-strands that are part of FRII, is shown in grey, as is the short stretch of amino acids downstream of FRIII. The curved grey arrow adjacent to gD residue 259 indicates that the C-terminal portion of gD (including FRIV) is thought to fold back on the Ig-like core. Arrows point to the sites of linker insertions for the FRI, FRII, and FRIII mutants gD(∇34), gD(∇126), and gD(∇243), respectively. The pink arrow shows the approximate viewpoint used in Fig. 8. (This figure was prepared using the SwissPdb Viewer [18] from PDB file 1JMA.)
FRI, FRII, and FRIII are close to each other in the structure. FRI is located entirely in the N-terminal extension and is supported by FRII and FRIII. FRII comprises the C", D, and E beta-strands of the Ig fold, and FRIII includes an alpha-helix adjacent to FRI. These three FR seem to form a discontinuous functional domain, and it has been suggested that this might have existed as the receptor binding site before acquisition of the Ig-like domain (3). FRIV was not resolved in the structure, but monoclonal antibody-binding studies (4, 35) suggest that the C-terminal extension folds back toward the main body of gD, bringing FRIV close to the other three FRs. The HVEM binding site on gD is unusual in comprising contiguous residues rather than being assembled from different parts of gD (3). All of the HVEM contact residues are contained within two segments of the N-terminal region of gD: residues 7 to 15 and 24 to 32.
In this study we reevaluated the biological function of representative FR linker-insertion mutants in the light of recent advances in our understanding of the interaction of gD with its receptors and of gD structure. In studying these mutants, for which accurate receptor-binding kinetics are available, we set out to answer two questions. First, does the function of gD mutants in cell fusion and virus entry correlate with receptor binding? Second, what effects, if any, do changes in receptor-binding kinetics have on the function of gD in cell fusion and virus entry? We show that when binding to both receptors is detectable, the FR mutants are affected similarly for both receptors. Additionally, at least regarding gD function, the cell fusion assay is a good model of virus entry. gD can function in virus entry and cell fusion regardless of its receptor binding kinetics; therefore, as long as binding to a functional receptor occurs, entry will progress.
MATERIALS AND METHODS
Cells and viruses.
A summary of all the cell types used in this study is provided in Table 1. VD60 cells (derived from Vero) express gD inducibly after HSV infection (27). Murine L cells, B78H1 cells, and B78H1-derived cell lines, as well as VD60, were maintained in Dulbecco's minimal essential medium supplemented with 5% (for VD60 cells) or 10% (for L and B78H1 cells) fetal calf serum, 100 U of penicillin per ml, and 100 μg of streptomycin per ml. For the B78H1-derived lines C10 (24, 33) and B78A (S. A. Connolly, G. H. Cohen, and R. J. Eisenberg, unpublished data), the medium was supplemented with 500 μg of G418 per ml. Cell lines B78C10-gD (expressing nectin-1) and B78A-gD (expressing HVEM) contain the HSV-1 gD gene under the control of its own promoter and support plaque formation by gD-negative virus. To construct these cells, B78C10 and B78A cells were transfected with plasmid pRM465, which contains the gD gene and its flanking regions (see below). Cells were selected through two rounds of limiting-dilution cloning in the presence of 125 μg of hygromycin per ml and 500 μg of G418 per ml and also maintained in this medium. CHOK1 cells were maintained in Ham's F12 medium supplemented with 10% fetal calf serum, 100 U of penicillin per ml, and 100 μg of streptomycin per ml. For the CHOK1-derived cell lines R3A (R. Xu, G. H. Cohen, and R. J. Eisenberg, unpublished data), which stably expresses nectin-1, and HVEM12 (36), which stably expresses HVEM, this was supplemented with 250 μg of G418 per ml.
TABLE 1.
Summary of the cell types used in this study
| Cell line | Parental cell type | Complements gD-negative virus | Nectin-1 and/or HVEM expression |
|---|---|---|---|
| L | NAa | No | Unknown |
| Vero | NA | No | Both |
| B78 H1 | NA | No | Neither |
| B78C10 | B78 H1 | No | Nectin-1 |
| B78A | B78 H1 | No | HVEM |
| B78A-gD | B78A | Yes | HVEM |
| B78C10-gD | B78C10 | Yes | Nectin-1 |
| VD60 | Vero | Yes | Both |
| CHO K1 | N/A | No | Neither |
| R3A | CHO K1 | No | Nectin-1 |
| HVEM 12 | CHO K1 | No | HVEM |
NA, not applicable.
HSV-1 KOSgDβ (9) contains a β-galactosidase expression cassette in place of the gD gene. It was propagated on VD60 cells (27).
Plasmids.
For complementation analysis, the following plasmids were used for HSV-1 gD expression: for WT gD-1, pRE4 (6); for gD-1 FRI mutant gD(∇34), plasmid D1-N29; for gD-1 FRII mutant gD(∇126), plasmid D1-H98; for gD-1 FRIII mutant gD(∇243), plasmid D1-H24; and for gD-1 FRIV mutant gD(Δ290-299), plasmid pHC240. Each FR mutant plasmid contains a linker insertion in the gD coding sequence. In these descriptions, the symbol ∇ indicates a linker-insertion after the named residue and the symbol Δ indicates a linker-insertion in place of the named residues. The construction of these plasmids is described in reference 4.
For the cell fusion assay, each of the FR mutant gD genes was subcloned from the above plasmids into pCAGGS/MCS (40). The resulting plasmids were termed pSH445 [containing gD(∇34)], pSH446 [containing gD(∇126)], pSH447 [containing gD(∇243)], and pRM461 [containing gD(Δ290-299)].
The wild-type HSV-1 glycoprotein expression vectors were as follows: for gB, pPEP98; for gD, pPEP99; for gH, pPEP100; and for gL, pPEP101 (42). Plasmid pCAGT7 contains the T7 RNA polymerase gene under control of the CAG promoter (41), and plasmid pT7EMCLuc contains the firefly luciferase gene under control of the T7 promoter. All of these plasmids were kindly provided by P. G. Spear.
The human nectin-1 expression plasmid pBG38 (17) was also a kind gift of P. G. Spear. The nectin-1 coding region from Vero cells was amplified by reverse transcription-PCR using primers pHveC5.1 and pHveC3.2 and cloned into pCDNA3.1+ exactly as described for porcine nectin-1 (34). The resulting plasmid was designated pRM500.
pRM465 contains the gD gene under its own promoter, flanking regions from HSV strain 17, and a hygromycin resistance gene. To construct pRM465, pCDNA3.1+(hygro) was digested with BclI and BglII to give two fragments. The 3,507-bp fragment, containing the hygromycin resistance gene and the origin, was used as the vector. A 6,457-bp BamHI fragment (corresponding to bases 136309 to 142766 of the strain 17 genome) containing the gD gene was inserted into this, resulting in the 9,961-bp construct pRM465, in which gD was in the sense orientation with respect to the original numbering of pCDNA3.1+(hygro).
Complementation assay.
L cells plated in 12-well dishes (2 × 105 cells/well) were transfected with gD expression vectors or empty vector using 2 μg of DNA per well and 10 μl of Geneporter per well in 1 ml of OptiMEM serum-free medium as specified in the manfacturer's protocol (Gene Therapy Systems). At 20 h posttransfection, the cells were infected at an input multiplicity of 20 PFU/cell with phenotypically complemented KOSgDβ (that is, KOSgDβ that had been grown on VD60 cells so that it contained gD in its envelope). Residual inoculum was inactivated after 2 h by a 1-min wash with acid-citrate buffer (10 mM KCl, 135 mM NaCl, 40 mM citric acid [pH 3.0]), and the cells were refed with 1 ml of medium per well. Twenty-four hours later, supernatants were harvested, cleared by low-speed centrifugation (1,500 rpm in a Beckman GPR bench-top centrifuge), and titrated on VD60, B78C10-gD, and B78A-gD cells.
Proteins.
All of the proteins used in biosensor experiments were expressed from recombinant baculovirus vectors in Sf9 cells and purified to homogeneity by published methods (49, 60). Throughout this paper, soluble truncated forms of proteins are described with the suffix “t.” The WT HSV-1 gDt, previously termed gD-1(306t), is truncated at residue 306, just upstream of the transmembrane domain, and has a 6-histidine tag at the C terminus (49). Analogous truncations of the four FR mutants, gD-1(∇34t), gD-1(∇126t), gD-1(∇243t) and gD-1(Δ290-299t), were constructed in the gDt background (39). Soluble forms of nectin-1 and HVEM were also produced by truncation just upstream of the transmembrane domain and also have 6-histidine tags. The nomenclature of both receptors has changed since the description of these soluble forms. Nectin-1t was called HveC(346t) and is described in reference 25. HVEMt was called HveA(200t) and is described in reference 59.
Optical biosensor analysis.
Binding of truncated soluble forms of gD and gD FR mutants to nectin-1t and HVEMt was analyzed in real time using a BIACORE X optical biosensor (BIAcore AB) at 25°C essentially as described previously (26, 45, 61). Sensorgrams were analyzed using BIAEvaluation software (version 3.0), and it was assumed that all forms of gDt were dimeric. Model curve fitting used global fitting with a 1:1 Langmuir binding model with drifting baseline. This represents the simplest model for a receptor-ligand interaction, where A + B = AB. Binding affinity (KD) was calculated from the rate of association (Kon) and the rate of dissociation (Koff) of the complex, using the formula 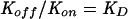 .
.
Fusion assays.
Fusion assays for characterization of the FR mutant forms of gD were carried out by a previously published method (42), except that the cells were lysed using the reporter lysis buffer (Promega) and luminosity readings were made using a Thermo Labsystems Luminoscan Ascent luminometer. Luminosity was expressed as relative light units (RLU). Additionally, equal expression of gD mutants was checked by cell enzyme-linked immunosorbent assay (CELISA) (see below). For fusion assays comparing human and Vero nectin-1, the assay was modified to use transient receptor expression. To generate target cells, CHOK1 cells (4 × 106 per 75-cm2 flask) were transfected in serum plus antibiotic-free OptiMEM, using Lipofectamine 2000 (3 μl per μg of DNA) with 2 μg of receptor expression plasmid (pBG38 for human nectin-1, and pRM500 for Vero nectin-1), 8 μg of pCAGGS/MCS (or other carrier DNA), and 10 μg of T7EMCluc. The cells were refed with normal medium after 3.5 h at 37°C. Effector cells were produced by transfecting CHOK1 as above with 3.2 μg each of pPEP98, pPEP100, pPEP101, pCAGT7, and one of the plasmids pPEP99, pSH445, pSH446, pSH447, or pRM461, or with the vector pCAGGS/MCS. At 12 h after transfection of effector cells and 24 h after transfection of targets, the cells were mixed in 24-well plates with 2 × 105 cells/well in a 1:1 effector-to-target ratio. The cells were lysed after 22 h of cocultivation at 37°C.
CELISA.
gD expression on the surface of transfected cells was detected by CELISA (16, 34) using the polyclonal gD-specific rabbit serum R7 (22). The same method was used to detect receptor expression on complementing cell lines using monoclonal antibody CK41 (23) for nectin-1 and the rabbit polyclonal antiserum R140 for HVEM (54).
Rate of virus entry.
Confluent monolayers of VD60 cells in 12-well plates were chilled to 4°C and inoculated with 100 PFU of KOSgDβ complemented with WT or FR mutant gD per well. After 90 min at 4°C for virus adsorption, the plates were transferred to 37°C and refed with warm (37°C) medium. At intervals after the temperature shift, the wells were washed with acid-citrate buffer (pH 3.0) to inactivate extracellular virus. At the end of the time course, a semisolid overlay (normal medium containing 1% carboxymethyl cellulose) was added to each well, and the cells were incubated until plaques formed. The mean plaque number (usually from triplicate wells) at each time point was then expressed as a percentage of the mean plaque number obtained without acid treatment to give an estimate of the percent virus entry.
RESULTS
In an earlier study, a panel of gD linker-insertion mutants was generated and tested for entry activity in a complementation assay (4). Four functional regions (FR) were defined in gD on the basis of this study: FRI, residues 27 to 43; FRII, residues 125 to 161; FRIII, residues 222 to 246; and FRIV, residues 277 to 310. Subsequently, a representative mutant from each FR was expressed as a truncated soluble protein for further analysis (39). These mutant forms of gD were as follows: for FRI, gD(∇34); for FRII, gD(∇126); for FRIII, gD(∇243); and for FRIV, gD(Δ290-299) (where ∇ indicates the residue after which the linker was inserted and Δ indicates residues replaced by the linker). The locations of these linker-insertions, as well as of FRI, FRII, and FRIII, are shown in Fig. 1 which depicts the structure of gD in complex with HVEM (3).
The gD receptors HVEM and nectin-1 had not been identified at the time of the construction of the FR mutants. We have reevaluated the FR mutants, testing receptor binding by using biosensor analysis and receptor-specific biological function in complementation and cell fusion assays.
Receptor binding by gD FR mutants.
Binding of gD to both HVEM and nectin-1 has been extensively studied using soluble forms of the proteins (8, 26, 34, 45, 57, 58, 61). We have previously used optical biosensor technology to study the receptor-binding kinetics of numerous truncated soluble forms of gD. This has revealed that FRIV is important for modulating receptor binding. Specifically, C-terminal truncations in gD up to residue 275 result in 100-fold higher affinity for receptors (45). We do not know the significance of these kinetic and affinity measurements to the function of gD in virions. To address this issue, we took advantage of the FR mutants which are available as soluble forms, suitable for binding assays, and as full-length forms, suitable for functional assays.
Table 2 shows the binding kinetic and affinity values of the four FR mutant forms of gDt for the two receptors HVEMt and nectin-1t. The HVEMt data were published previously (61) and are reproduced here for comparison.
TABLE 2.
Kinetic and affinity values for the binding of gD FR mutants to HVEM and nectin-1a
| gD | kon (103 s−1 M−1) | koff (10−2 s−1) | KD (10−6 M)b |
|---|---|---|---|
| HVEMtc | |||
| gD(306t) | 6.1 | 2.0 | 3.2 |
| gD(∇34t) | Does not bind | Does not bind | Does not bind |
| gD(∇126t) | 1.2 | 2.6 | 22.0 |
| gD(∇243t) | 11.0 | 1.5 | 1.3 |
| gD(Δ290-299t) | 240 | 0.8 | 0.03 |
| Nectin-1t | |||
| gD(306t) | 2.6 | 0.7 | 2.7 |
| gD(∇34t) | Not 1:1d | Not 1:1d | 4.0e |
| gD(∇126t) | 0.7 | 1.0 | 14.0 |
| gD(∇243t) | 8.1 | 1.8 | 2.2 |
| gD(Δ290-299t) | 150 | 0.76 | 0.05 |
Data are from a representative experiment.
KD = koff/kon; χ2 values for the global fits range from 0.7 to 4.0.
HVEM binding data are reproduced from reference 61.
Data did not fit the 1:1 Langmuir model used for analysis.
From Scatchard analysis.
The most striking receptor-binding phenotype was seen with gD(∇34t), which failed to bind HVEMt. This form of gD did bind to nectin-1t, but the kinetics for this interaction did not fit the 1:1 binding model used in the data analysis. Scatchard analysis of these data suggested that binding to nectin-1t was at approximately WT affinity, and this is consistent with published ELISA studies (25). These data suggest that gD(∇34t) is able to bind nectin-1t but that this binding differs in some way from WT gDt binding.
The affinity measurements of the other three mutants, for both receptors, varied over a large range from 100-fold higher than that of WT gDt, for gD(Δ290-299t), to 10-fold lower, for gD(∇126). Of interest, gD(∇243t) showed WT affinity (albeit with slightly different kinetics) in spite of its impaired function (4). We showed previously, using a series of gD C-terminal truncations, that for a given form of gD, the affinity and kinetics for HVEMt and nectin-1t were the same (26, 45, 61). The data presented here extend that work in that although their receptor-binding affinities varied over a 1,000-fold range, each of the FRII, FRIII, and FRIV mutants bound with essentially the same kinetics and affinity to HVEMt as to nectin-1t. Notably, and similar to data reported for gD truncation mutants, the on-rates were the most variable factor, largely accounting for the affinity differences. The off-rates for each of the mutants, with both receptors, were comparable to WT rates.
An obvious question is how these receptor-binding data relate to the ability of each gD FR mutant to use the two receptors in virus entry. To address this, we asked whether the function of gD in virus entry correlates with receptor-binding kinetics.
gD FR mutants in virus entry. (i) VD60 cells.
We tested whether transiently expressed gD FR mutants could complement the infectivity of a gD-negative virus, KOSgDβ (9). Initially, we repeated the complementation assay that was used to define the four FR on gD (4). L cells were transfected with gD expression plasmids and then superinfected with phenotypically complemented KOSgDβ. Progeny virus was titrated on the complementing cell line VD60 (27), with plaque formation providing a measure of the entry activity of the mutated forms of gD tested.
Figure 2 shows the complementation analysis of WT gD and the four FR mutants on VD60 cells. Consistent with previous data (4), all of the gD FR mutant-complemented virions showed impaired entry into VD60 cells. Entry activity was generally somewhat higher than in the original study. We attribute this to improvements in transfection technology. gD(∇34) was the most highly impaired in this assay, with entry activity almost at background levels. This was interesting in that the soluble form of this mutant was unable to bind HVEMt but bound nectin-1t with near-WT affinity (Table 2). The other three FR mutants, gD(∇126), gD(∇243), and gD(Δ290-299), each retained some entry activity, but these low levels did not correlate with binding affinity for either receptor.
FIG. 2.

Complementation analysis of FR mutants on VD60 cells. Titers of FR mutant gD-complemented virus are shown as percentages of the titer obtained with WT gD-complemented virus. Results are the mean of four experiments, except for the results for gD(∇34), which are the mean of six. Error bars represent ±1 standard deviation about the mean.
VD60 cells (27) are derived from Vero cells which express nectin-1 (34) and HVEM (14) homologues. Based on the lack of binding of gD(∇34t) to human HVEM (Table 2), it seemed likely that gD(∇34)-complemented virus would be unable to use primate HVEM. Clearly, this mutant was also unable to use primate nectin-1 to enter VD60 cells. However, we do not know which receptor(s) is used by WT HSV for entry into VD60 cells. We therefore next asked whether the observed entry activities reflected preferential usage of one of the known HSV receptors, HVEM and nectin-1.
(ii) Receptor-defined cells.
To test receptor-specific entry, we constructed receptor-defined complementing cell lines. This was necessary because KOSgDβ cannot form plaques unless gD is provided in trans. We took advantage of the murine melanoma cell line B78H1, which does not express endogenous HSV gD receptors and so does not support HSV entry. B78C10 and B78A cells, which stably express nectin-1 and HVEM, respectively, support HSV entry and, importantly for this study, plaque formation.
We transfected B78C10 and B78A with plasmid pRM465 containing the HSV-1 gD gene and its flanking regions so that expression of gD would be induced by HSV infection, analogous to VD60 cells. Selected stable clones, termed B78C10-gD and B78A-gD, expressed receptor at essentially the same level as their respective parental lines (Fig. 3) and supported plaque formation by KOSgDβ (Table 3). While KOS replicated to comparable levels on both parental (B78C10 and B78A) and complementing (B78C10-gD and B78A-gD) cell lines, KOSgDβ, replicated effectively only on B78C10-gD and B78A-gD, where gD was provided in trans.
FIG. 3.

Expression of HVEM (A) and nectin-1 (B) on receptor-defined complementing cell lines and parental lines. Cells were incubated with dilutions of antibodies at 4°C for 2 h and then fixed and processed as a CELISA. OD 405nm, optical density at 405 nm.
TABLE 3.
Plaque formation by gD-negative virus on receptor-defined complementing cell lines
| Cell line | Viral titer (PFU/ml)a
|
|
|---|---|---|
| KOS | KOSgDβ | |
| B78A | 4.5 × 106 | 0 |
| B78A-gD | 1.2 × 107 | 4.9 × 105 |
| B78C10 | 2.6 × 106 | 1.8 × 102 |
| B78C10-gD | 4.7 × 106 | 2.7 × 105 |
Titers on VD60 cells: KOS, 2.25 × 107 PFU/ml; KOSgDβ, 2.75 × 106 PFU/ml.
We next used these cell lines to ask whether the four FR mutants showed receptor-specific entry activity. The results are presented in Fig. 4. gD(∇34)-complemented virus was completely defective on HVEM-expressing B78A-gD cells but was able to enter nectin-1-expressing B78C10-gD cells to about 60% of WT levels (Fig. 4). The failure of gD(∇34)-complemented virus to enter B78A-gD cells was consistent with the binding data (Table 2) and also with the inability of gD(∇34) to mediate entry to VD60 cells (Fig. 2). The entry activity of gD(∇34)-complemented virus on B78C10-gD cells was interesting because it suggests that the functional defect of gD(∇34) on HVEM-expressing cells is predominantly one of binding and that gD(∇34) is not intrinsically nonfunctional. Second, it suggests that the impaired entry is not due to reduced levels of incorporation of gD into the virion envelope.
FIG. 4.

Complementation analysis of FR mutants on B78A-gD (HVEM) (A) and B78C10-gD (nectin-1) (B) cells. Titers of FR mutant gD-complemented virus are shown as percentages of the titer obtained with WT gD-complemented virus. Data are from a representative experiment.
The FRII, FRIII, and FRIV mutants showed generally higher entry activity on the nectin-1 cells than on the HVEM cells. Of the two cell lines, the pattern of activity on HVEM cells more closely resembled that obtained with VD60 cells.
gD FR mutants in cell fusion.
To confirm the receptor-specific activity shown by complementation and to provide an alternative assessment of function, we tested the FR mutants in a cell fusion assay (42). This assay reconstitutes the virus fusion machinery and provides a test of gD function that is independent of other aspects of virus entry. Additionally, the use of a different cell type from the complementation assay argues against any cell-type-specific effects.
The four essential HSV glycoproteins, gB, gD, gH and gL, are expressed in CHOK1 effector cells along with T7 RNA polymerase. These cells are cocultivated with CHOK1-derived target cells that express a receptor and are transfected with a plasmid containing the luciferase gene under the control of the T7 promoter. Fusion between effector and target cells allows T7 polymerase-driven expression of luciferase, which can be detected in a standard assay. Equal expression of each form of gD was confirmed in each assay by CELISA on effector cells plated at the start of cocultivation (a representative result is shown in Fig. 5A).
FIG. 5.

Activity of gD mutants in cell fusion. Effector CHOK1 cells expressing WT gB, gH, gL, and WT or mutant gD were mixed with receptor-expressing target cells. (A) Equal expression of each form of gD was checked by CELISA on effector cells with gD polyclonal serum R7. (B and C) RLU obtained with effector cells expressing each FR mutant form of gD fusing with HVEM target cells (B) or nectin-1 target cells (C) are plotted as percentages of RLU obtained with effector cells expressing WT glycoproteins. Data in panel A are from a representative experiment. Data in panels B and C are the mean of three experiments, each carried out in quadruplicate. Error bars represent ±1 standard deviation about the mean. OD 405nm, optical density at 405 nm.
Results obtained with the panel of FR mutants in fusion assays are shown in Fig. 5B (for HVEM-expressing target cells) and Fig. 5C (for nectin-1-expressing target cells). The results for nectin-1 target cells are strikingly similar to those obtained in the complementation assays using B78C10-gD cells. The gD(∇126), gD(∇243) and gD(Δ290-299) mutants showed somewhat higher percent activities on HVEM cells than were seen in complementation with B78A-gD cells, although the relative levels were similar in the two assays. Consistent with the complementation data, effector cells expressing gD(∇34) did not fuse with HVEM target cells yet fused with nectin-1 target cells at levels approaching those obtained with effectors expressing WT gD.
Two points arise from the results. The similarity of fusion and entry data argues that the entry defects seen with FR mutant-complemented virus are intrinsic properties of the mutant proteins and are not due to some virion-specific aspect of gD function or to uneven virion incorporation of gD. Secondly, it is striking that gD(∇34) is functional for both fusion and entry on nectin-1 expressing cells, consistent with binding data obtained by ELISA and biosensor methods. Nonetheless, virions complemented with gD(∇34) fail to enter VD60 cells.
Function of Vero nectin-1.
The results of binding, entry, and fusion assays suggested that gD(∇34) could not use nectin-1 to enter Vero-derived VD60 cells even though it binds to and uses human nectin-1 for entry and fusion. We showed previously that the nectin-1 homologue in Vero (African green monkey) cells contains a 3-amino-acid insertion in the V domain, adjacent to the gD-binding region, but we did not determine whether this affected its function as an HSV receptor (34). Accordingly, we set out to test the possibility that the impaired entry of gD(∇34)-complemented virus into VD60 cells reflected a species-specific effect.
Fluorescence-activated cell sorter analysis showed that VD60 cells express similar levels of nectin-1 and HVEM to those of their parental Vero cells (C. Krummenacher, G. H. Cohen, and R. J. Eisenberg, unpublished data), ruling out the lack of nectin-1 expression as an explanation for the lack of entry activity seen with gD(∇34)-complemented virus. Additionally, Vero nectin-1 can function as a receptor for WT virus when expressed transiently in B78H1 cells (Fig. 6A) and WT virus entry into Vero cells can be inhibited by some anti-nectin-1 monoclonal antibodies (data not shown).
FIG. 6.

Virus entry (A) and cell fusion (B) mediated by human and Vero nectin-1. (A) HSV-1 KOStk12 was titrated on cells transiently expressing human or Vero nectin-1 or control cells transfected with vector. Entry activity is shown as the mean slope of β-galactosidase activity over 50 min. (B) Target CHOK1 cells transiently expressing human or Vero nectin-1 were used in a fusion assay. Data shown are from a representative experiment, carried out in quadruplicate. Error bars in panel B represent ±1 standard deviation about the mean RLU.
We have been unable to test the function of gD(∇34) on Vero cells directly because the cells do not complement KOSgDβ and function poorly, if at all, as targets in the cell fusion assay. Here we tested whether Vero nectin-1 was able to function as a receptor for gD(∇34) by transiently expressing either human or Vero nectin-1 in CHOK1 cells and using these cells as targets in the fusion assay. Figure 6B shows a typical result. For all four FR mutants and WT gD, Vero nectin-1 supported fusion at levels indistinguishable from those obtained with human nectin-1 target cells.
We conclude that Vero nectin-1 is a functional receptor for WT gD and the FR mutants, but that at physiological expression levels in the context of the VD60 cell surface it is somehow prevented from mediating the entry of gD(∇34)-complemented virus.
Comparisons of the rate of entry of FR mutant-complemented virus.
The gD(∇243) mutant was interesting because it showed essentially WT binding to both nectin-1t and HVEMt but showed impaired entry and cell fusion. We have proposed that the functional defect in gD(∇243) defines a second, post-receptor-binding, function of gD (61). Such a defect might be expected to affect the rate of entry, and so we asked whether virus complemented with gD(∇243) was affected in this way. We also wanted to test whether the rate of entry assay revealed a correlation between receptor binding affinity and entry for gD(∇126) and gD(Δ290-299). The complementation and fusion assays had not revealed any convincing association; however, since neither assay is rate limited (20), functional differences between mutants that are dependent on variations in affinity might be missed.
Figure 7 shows the rate of entry of KOSgDβ complemented with WT gD or with gD(∇243). The rate of entry seen with WT gD-complemented virus (t1/2 = 34 min) is typical for L-cell-derived HSV and somewhat lower than that seen with Vero-derived virus. The lag time after shifting the temperature to 37°C is a characteristic of virus produced on L cells (C. G. Handler, G. H. Cohen, and R. J. Eisenberg, unpublished data) and is not seen with Vero-derived virus. We do not know the explanation for this phenomenon.
FIG. 7.

Rate of entry of FR mutant-complemented virus into VD60 cells. Equal numbers of PFU of each virus preparation were added to cells on ice. After a 90-min adsorption, the temperature was shifted to 37°C to allow entry to progress. Remaining extracellular virus was inactivated by acid treatment at the times indicated. The plaque number at each time point was expressed as a percentage of the plaque number obtained with no acid. Curves were generated using the curve-fitting option of the Delta Graph program. Data shown are the mean of three experiments. Error bars represent ±1 standard deviation about the mean.
Virus containing gD(∇243) showed a markedly reduced rate of entry; indeed, at the last time point tested in the assay, the plaque number had not reached 50% of the input PFU. Significantly, this reduction in rate occurs in spite of its WT receptor-binding characteristics. KOSgDβ complemented with gD(∇126) or gD(Δ290-299) showed marginally reduced rates of entry compared to WT gD-containing virus (data not shown). The similar rates of entry seen with these two virus preparations was interesting because the measured affinity of soluble forms of these two gD mutants for both nectin-1 and HVEM was 1,000-fold different (Table 2).
Thus, our data indicate that there is no correlation between the affinity of these forms of gD for receptors and their ability to function in place of WT gD. Additionally, they support the suggestion that gD(∇243) is impaired in a postbinding step in the entry process.
DISCUSSION
In this paper, we have evaluated the biological characteristics of four gD FR mutants in the light of measurements of receptor-binding kinetics and affinity obtained from biosensor analysis.
Kinetics and affinities of mutant forms of gD for receptors.
The most striking observation was that for each of the FRII, FRIII, and FRIV mutants, the binding kinetics and affinity for HVEMt and nectin-1t were essentially the same. We already knew that this was the case with WT gDt (26, 61). HVEM and nectin-1 have no known structural relatedness, so that such similar binding is remarkable and suggests that gD binds to the two receptors in a similar manner. This contrasts with measles virus haemaggluttinin, which binds to two different receptors via overlapping sites but with significantly different kinetics for each (46). It is possible that the gD-binding site on nectin-1 has some conformational identity to the site on HVEM, but this will be revealed only when the structure of the gD-nectin-1 complex becomes available.
The FRI mutant gD(∇34t) was unable to bind to HVEMt but did bind to nectin-1t. The structure of the gD-HVEM complex (3) revealed that when gD is bound to HVEM, the first 37 residues of gD form a hairpin loop, bending at residue 21. There is no hairpin in the unbound form of gD, arguing that HVEM binding causes the hairpin to form or stabilizes it. Figure 8 shows a detailed depiction of the gD-HVEM interface. FRI (gD residues 27 to 43 [Fig. 1 and 8]) includes part of the hairpin loop in the HVEM-bound form of gD. Although the linker-insertion in gD(∇34t) is downstream of the last HVEM contact residue (Pro-32), it may disrupt the interface between gD and HVEM. The linker-insertion may distort gD so as to prevent formation of the intramolecular β-sheet formed between β-strands in HVEM (residues 35 to 37 [pink in Fig. 8]) and at the start of FRI (residues 27 to 29). Alternatively, the hairpin loop might be displaced, preventing insertion of HVEM Tyr-23 into its binding crevice formed by residues 11, 14, and 25 of gD (3, 7). A third possibility is that the linker prevents conformational changes associated with hairpin loop formation, thus blocking HVEM binding.
FIG. 8.

Detail of the gD/HVEM interface. Residues 4 to 105 (boxed numbers) of HVEM and residues 1 to 43 of gD are shown. The bulk of gD has been removed to allow this view; the viewer is looking through the N-terminal hairpin loop of gD onto the apposed surface of HVEM. The approximate viewing angle is indicated by the pink arrow in Fig. 1. HVEM is shown in blue, gD FRI is in red, and the rest of gD is grey. HVEM residue 23 (shown in space-filling format in green) is visible through the binding pocket formed by gD residues 11, 14, and 25 (each shown in yellow). A β-strand of HVEM (residues 35 to 37 [shown in pink]) forms an intermolecular antiparallel β-sheet with gD residues 27 to 29, which are at the start of FRI.
Although gD(∇34t) bound to nectin-1t, this interaction did not fit the 1:1 model used for analysis of biosensor data. This is consistent with previous ELISA studies in which the gD(∇34t) binding curve was close but not identical to the WT curve (25). The differential effect on binding to the two receptors emphasizes that the binding sites on gD for the two receptors are distinct (25, 38).
The differences in KD seen with the FR mutants were due to alterations in the on-rate, that is, the rate at which the complex forms. Variations of on-rate over a wide range are not unprecedented in virus-receptor interactions; the on-rate for human immunodeficiency virus env-coreceptor interactions varies over 2 orders of magnitude (10). Notably, the gD of a viable HSV-1 mutant, Rid 1 (9), as well as that of another alphaherpesvirus, pseudorabies virus, binds to nectin-1 with 10-fold-higher affinity than that of WT HSV gD. In each case, the affinity increase is largely due to an increased on-rate (8, 26). The kinetics for the interaction of gD(∇34t) with nectin-1 remain unknown, but both on- and off-rates could differ significantly from WT rates while combining to give the WT affinity suggested by Scatchard analysis.
We do not know whether the formation and dissociation of the gD-receptor complex represent different events in virus entry. If the gD-receptor complex dissociates during entry, alterations in the off-rate might be highly significant; however, none of the FR mutants had off-rates significantly different from WT rates. It is possible that high-affinity attachment of virions to proteoglycans (44) anchors them to the cell surface so that increased dissociation of the gD complex would have little effect. If so, linker insertions causing alterations in the off-rate might not have been identified as having impaired entry function in the initial screen.
What is the relationship between affinity and function?
Both assays of gD function, i.e., complementation and cell fusion, gave similar results, affirming the utility of cell fusion as a model of glycoprotein function. However, neither assay showed a clear correlation between gD function and receptor-binding affinity, suggesting a degree of flexibility in the involvement of gD in virus entry. Interactions between gD and receptor would seem more likely to occur with higher gD-receptor affinity. Why, then, is there no obvious correlation between gD receptor-binding affinity and virus entry?
If the interaction between virion gD and cell surface receptor occurs as a simple equilibrium, excess receptor might mask defects in gD function in complementation and fusion. Changes in receptor-binding affinity would affect entry only when the receptor levels on the target cell are limiting (10), as seen with influenza virus HA (29). The receptor-defined cell lines used here overexpress receptors, but we found, using transient-expression assays, that reduced levels of receptor did not alter the results obtained with the four gD mutants in fusion (data not shown).
A correlation might also be masked by high levels of gD. There are about 34,000 molecules of gD per WT HSV-1 virion (19). This effectively increases the valency of the gD-receptor interaction and its avidity, perhaps allowing defects in the affinity of individual receptor-ligand interactions to be overcome. The viral glycoproteins are expressed at high levels in the fusion assay, and we have not tested the effect of reduced expression levels or of changes in their relative amounts. We do not know how many gD-receptor interactions are required per virion to initiate entry, but the HSV mutant FUS6kan, which contains 500-fold less gD than does WT virus, is viable only on cells that express large amounts of receptor (21).
We assume that our biosensor data are valid estimates of the affinities of virion-associated forms of gD for cell surface receptor; however, this may not be the case. Virion association per se did not affect our results, since the pattern of activity was essentially the same in cell fusion as in complementation (compare Fig. 4A and B with Fig. 5B and C). However, the transmembrane and cytoplasmic domains may affect receptor binding, although the latter domain is not essential for infectivity (12, 37).
The gD(∇243) mutant has an altered rate of entry.
It seems likely that two of the FR mutants are impaired in different stages of gD function. The fact that gD(∇34)-complemented virus formed plaques on nectin-1 cells shows that this form of gD can mediate virus entry and that, consistent with the biosensor analysis, its defect on HVEM cells is solely one of binding. By contrast, gD(∇243t) had WT affinity for both receptors, yet virus complemented with this form of gD was impaired for entry into cells bearing either receptor. This is consistent with the suggestion, based on studies of nectin-1 mutations, that gD binding is not sufficient for entry (15). We have proposed that gD(∇243) might be defective in a postbinding event (61). Indirect support for a such a defect of gD(∇243) was obtained from experiments testing the rates of virus entry.
We found that virions complemented with gD(∇126) and gD(Δ290-299) had slightly reduced rates, but the most significant effect was seen with gD(∇243). The reduction in the rate of entry of virions complemented with this form of gD is consistent with a defect in a hypothetical postbinding function and would represent the first evidence of this for gD. Two previous observations are relevant in this regard. First, some gD-specific monoclonal antibodies (for example, DL6) do not block the interaction of gD with HVEM or nectin-1 but neutralize virus infectivity on cells expressing these receptors (11, 22, 25, 38). Second, gD(∇243t) blocks HSV entry with the same efficiency as WT gDt does (39). Assuming that blocking with soluble gD relies on simple competition for receptor, this supports a postbinding defect of gD(∇243) in complementation and fusion. Further analysis of gD(∇243) will best be carried out using a virus with this mutation in its genome.
How do we account for the entry phenotype of gD(∇34)?
One issue that remains unexplained is the inability of gD(∇34) to complement the infectivity of KOSgDβ on VD60 cells. This was surprising because this form of gD was able to mediate the entry of this virus into cells expressing human nectin-1 and to mediate the fusion of cells expressing Vero nectin-1. Since gD(∇34t) is unable to bind to HVEMt or use this receptor for entry, one explanation for its inactivity on VD60 cells is that HVEM is the preferred HSV receptor on these cells. We think this unlikely because other HSV mutants that are unable to bind to or use HVEM, such as Rid 1 and HSV1 F-dl2 (which lacks residues 7 to 21 of gD), can enter Vero cells (9, 12). However, Rid 1 could potentially use nectin-2 to enter Vero cells (56). It is not clear whether HSV1 F-dl2 can use this receptor.
A second explanation for the impairment of gD(∇34)-complemented virus on VD60 cells is that HSV uses different pathways to enter Vero-derived and B78H1-derived cells, for example, pH independent entry, such as is thought to be usual for HSV entry (62), versus pH-dependent entry (37a). If this is the case, gD(∇34) might provide a useful differentiating reagent. Another possibility, not necessarily exclusive of the previous one, is that there is some aspect of the presentation of nectin-1 on VD60 cells that prevents its use by gD(∇34)-complemented virus. For example, an interaction between nectin-1 and nectin-3 (48) or between nectin-1 and an unknown cell surface protein might occlude the gD-binding site so that nectin-1 is still able to bind WT gD but is unable to bind gD(∇34). This would be consistent with the linker in gD(∇34) being adjacent to the nectin-1-binding domain on gD.
In summary, the data presented in this paper suggest that receptor-binding affinity, at least within the broad range measured here, does not have a direct bearing on virus entry. It follows that as long as binding occurs to a functional receptor, regardless of its affinity, entry will progress. This flexibility in virus entry may be advantageous for virus evolution.
Acknowledgments
This work was supported by Public Health Service grant AI-18289 from the National Institute of Allergy and Infectious Diseases (to G.H.C.) and grants NS-36731 and NS-30606 (to R.J.E.) from the National Institute of Neurological Disorders and Stroke. Sheri L. Hanna was supported by Public Health Service Training grant T32-GM-007229.
We thank Patricia Spear, Peter Pertel, and David Johnson for reagents; Bruce Shenker and Lisa Pankoski for help with the luminometer; and Sarah Connolly, Claude Krummenacher, and Chuck Whitbeck for helpful comments on the manuscript.
REFERENCES
- 1.Cai, W., S. Person, S. C. Warner, J. Zhou, and N. A. DeLuca. 1987. Linker-insertion nonsense and restriction-site deletion mutations of the gB glycoprotein gene of herpes simplex virus type 1. J. Virol. 61:714-721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campadelli-Fiume, G., F. Cocchi, L. Menotti, and M. Lopez. 2000. The novel receptors that mediate the entry of herpes simplex viruses and animal alphaherpesviruses into cells. Rev. Med. Virol. 10:305-319. [DOI] [PubMed] [Google Scholar]
- 3.Carfi, A., S. H. Willis, J. C. Whitbeck, C. Krummenacher, G. H. Cohen, R. J. Eisenberg, and D. C. Wiley. 2001. Herpes simplex virus glycoprotein D bound to the human receptor HveA. Mol. Cell 8:169-179. [DOI] [PubMed] [Google Scholar]
- 4.Chiang, H.-Y., G. H. Cohen, and R. J. Eisenberg. 1994. Identification of functional regions of herpes simplex virus glycoprotein gD by using linker-insertion mutagenesis. J. Virol. 68:2529-2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cocchi, F., L. Menotti, P. Mirandola, M. Lopez, and G. Campadelli-Fiume. 1998. The ectodomain of a novel member of the immunoglobulin subfamily related to the poliovirus receptor has the attribute of a bona fide receptor for herpes simplex virus types 1 and 2 in human cells. J. Virol. 72:9992-10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen, G. H., W. C. Wilcox, D. L. Sodora, D. Long, J. Z. Levin, and R. J. Eisenberg. 1988. Expression of herpes simplex virus type 1 glycoprotein D deletion mutants in mammalian cells. J. Virol. 62:1932-1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Connolly, S. A., D. J. Landsburg, A. Carfi, D. C. Wiley, R. J. Eisenberg, and G. H. Cohen. 2002. Structure-based analysis of the herpes simplex virus glycoprotein D binding site present on herpesvirus entry mediator HveA(HVEM). J. Virol. 76:10894-10904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Connolly, S. A., J. C. Whitbeck, A. H. Rux, C. Krummenacher, S. van Drunen Little-van den Hurk, G. H. Cohen, and R. J. Eisenberg. 2001. Glycoprotein D homologues in herpes simplex virus type 1, pseudorabies virus, and bovine herpes virus type 1 bind directly to human HveC (nectin-1) with different affinities. Virology 280:7-18. [DOI] [PubMed] [Google Scholar]
- 9.Dean, H. J., S. S. Terhune, M. Shieh, N. Susmarski, and P. G. Spear. 1994. Single amino acid substitutions in gD of herpes simplex virus 1 confer resistance to gD-mediated interference and cause cell-type-dependent alterations in infectivity. Virology 199:67-80. [DOI] [PubMed] [Google Scholar]
- 10.Doms, R. W. 2000. Beyond receptor expression: the influence of receptor conformation, density, and affinity in HIV-1 infection. Virology 276:229-237. [DOI] [PubMed] [Google Scholar]
- 11.Eisenberg, R. J., D. Long, M. Ponce de Leon, J. T. Matthews, P. G. Spear, M. G. Gibson, L. A. Lasky, P. Berman, E. Golub, and G. H. Cohen. 1985. Localization of epitopes of herpes simplex virus type 1 glycoprotein D. J. Virol. 53:634-644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feenstra, V., M. Hodaie, and D. C. Johnson. 1990. Deletions in herpes simplex virus glycoprotein D define nonessential and essential domains. J. Virol. 64:2096-2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Forrester, A., H. Farrell, G. Wilkinson, J. Kaye, N. Davis-Poynter, and T. Minson. 1992. Construction and properties of a mutant of herpes simplex virus type 1 with glycoprotein H coding sequences deleted. J. Virol. 66:341-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foster, T. P., V. N. Chouljenko, and K. G. Kousoulas. 1999. Functional characterization of the HveA homolog specified by African green monkey kidney cells with a herpes simplex virus expressing the green fluorescence protein. Virology 258:365-374. [DOI] [PubMed] [Google Scholar]
- 15.Geraghty, R. J., A. Fridberg, C. Krummenacher, G. H. Cohen, R. J. Eisenberg, and P. G. Spear. 2001. Use of chimeric nectin-1(HveC)-related receptors to demonstrate that ability to bind alphaherpesvirus gD is not necessarily sufficient for viral entry. Virology 285:366-375. [DOI] [PubMed] [Google Scholar]
- 16.Geraghty, R. J., C. R. Jogger, and P. G. Spear. 2000. Cellular expression of alphaherpesvirus gD interferes with entry of homologous and heterologous alphaherpesviruses by blocking access to a shared gD receptor. Virology 268:147-158. [DOI] [PubMed] [Google Scholar]
- 17.Geraghty, R. J., C. Krummenacher, R. J. Eisenberg, G. H. Cohen, and P. G. Spear. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor related protein 1 and poliovirus receptor. Science 280:1618-1620. [DOI] [PubMed] [Google Scholar]
- 18.Guex, N., and M. C. Peitsch. 1997. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714-2723. [DOI] [PubMed] [Google Scholar]
- 19.Handler, C. G., R. J. Eisenberg, and G. H. Cohen. 1996. Oligomeric structure of glycoproteins in herpes simplex virus type 1. J. Virol. 70:6067-6075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harman, A., H. Browne, and T. Minson. 2002. The transmembrane domain and cytoplasmic tail of herpes simplex virus type 1 glycoprotein H play a role in membrane fusion. J. Virol. 76:10708-10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huber, M. T., T. W. Wisner, N. R. Hegde, K. A. Goldsmith, D. A. Rauch, R. J. Roller, C. Krummenacher, R. J. Eisenberg, G. H. Cohen, and D. C. Johnson. 2001. Herpes simplex virus with highly reduced gD levels can efficiently enter and spread between human keratinocytes. J. Virol. 75:10309-10318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Isola, V. J., R. J. Eisenberg, G. R. Siebert, C. J. Heilman, W. C. Wilcox, and G. H. Cohen. 1989. Fine mapping of antigenic site II of herpes simplex virus glycoprotein D. J. Virol. 63:2325-2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krummenacher, C., I. Baribaud, M. Ponce de Leon, J. C. Whitbeck, H. Lou, G. H. Cohen, and R. J. Eisenberg. 2000. Localization of a binding site for herpes simplex virus glycoprotein D on the herpesvirus entry mediator C by using anti-receptor monoclonal antibodies. J. Virol. 74:10863-10872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krummenacher, C., I. Baribaud, J. F. Sanzo, G. H. Cohen, and R. J. Eisenberg. 2002. Effects of herpes simplex virus on structure and function of nectin- 1/HveC. J. Virol. 76:2424-2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krummenacher, C., A. V. Nicola, J. C. Whitbeck, H. Lou, W. Hou, J. D. Lambris, R. J. Geraghty, P. G. Spear, G. H. Cohen, and R. J. Eisenberg. 1998. Herpes simplex virus glycoprotein D can bind to poliovirus receptor-related protein 1 or herpesvirus entry mediator, two structurally unrelated mediators of virus entry. J. Virol. 72:7064-7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krummenacher, C., A. H. Rux, J. C. Whitbeck, M. Ponce de Leon, H. Lou, I. Baribaud, W. Hou, C. Zou, R. J. Geraghty, P. G. Spear, R. J. Eisenberg, and G. H. Cohen. 1999. The first immunoglobulin-like domain of HveC is sufficient to bind herpes simplex virus gD with full affinity while the third domain is involved in oligomerization of HveC. J. Virol. 73:8127-8137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ligas, M. W., and D. C. Johnson. 1988. A herpes simplex virus mutant in which glycoprotein D sequences are replaced by β-galactosidase sequences binds to but is unable to penetrate into cells. J. Virol. 62:1486-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lopez, M., F. Eberlé, M.-G. Mattei, J. Gabert, F. Birg, F. Bardin, C. Maroc, and P. Dubreuil. 1995. Complementary DNA characterization and chromosomal localization of a human gene related to the poliovirus receptor-encoding gene. Gene 155:261-265. [DOI] [PubMed] [Google Scholar]
- 29.Martin, J., S. A. Wharton, Y. P. Lin, D. K. Takemoto, J. J. Skehel, D. C. Wiley, and D. A. Steinhauer. 1998. Studies of the binding properties of influenza hemagglutinin receptor-site mutants. Virology 241:101-111. [DOI] [PubMed] [Google Scholar]
- 30.Martinez, W. M., and P. G. Spear. 2002. Amino acid substitutions in the V domain of nectin-1 (HveC) that impair entry activity for herpes simplex virus types 1 and 2 but not for pseudorabies virus or bovine herpesvirus 1. J. Virol. 76:7255-7262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mauri, D. N., R. Ebner, K. D. Kochel, R. I. Montgomery, T. C. Cheung, G.-L. Yu, M. Murphy, R. J. Eisenberg, G. H. Cohen, P. G. Spear, and C. F. Ware. 1998. LIGHT, a new member of the TNF superfamily, and lymphotoxin (LT) a are ligands for herpesvirus entry mediator (HVEM). Immunity 8:21-30. [DOI] [PubMed] [Google Scholar]
- 32.Menotti, L., F. Cocchi, and G. Campadelli-Fiume. 2002. Critical residues in the CC' ridge of the human nectin1 receptor V domain enable herpes simplex virus entry into the cell and act synergistically with the downstream region. Virology 301:6-12. [DOI] [PubMed] [Google Scholar]
- 33.Miller, C. G., C. Krummenacher, R. J. Eisenberg, G. H. Cohen, and N. W. Fraser. 2001. Development of a syngenic murine B16 cell line-derived melanoma susceptible to destruction by neuroattenuated HSV-1. Mol. Ther. 3:160-168. [DOI] [PubMed] [Google Scholar]
- 34.Milne, R. S. B., S. A. Connolly, C. Krummenacher, R. J. Eisenberg, and G. H. Cohen. 2001. Porcine HveC, a member of the highly conserved HveC/nectin 1 family, is a functional alphaherpesvirus receptor. Virology 281:315-328. [DOI] [PubMed] [Google Scholar]
- 35.Minson, A. C., T. C. Hodgman, P. Digard, D. C. Hancock, S. E. Bell, and E. A. Buckmaster. 1986. An analysis of the biological properties of monoclonal antibodies against glycoprotein D of herpes simplex virus and identification of amino acid substitutions that confer resistance to neutralization. J. Gen. Virol. 67:1001-1013. [DOI] [PubMed] [Google Scholar]
- 36.Montgomery, R. I., M. S. Warner, B. J. Lum, and P. G. Spear. 1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87:427-436. [DOI] [PubMed] [Google Scholar]
- 37.Muggeridge, M. I., W. C. Wilcox, G. H. Cohen, and R. J. Eisenberg. 1990. Identification of a site on herpes simplex virus type 1 gD that is essential for infectivity. J. Virol. 64:3617-3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37a.Nicola, A.V., McEvoy, A. M., Straus, S. E. 2003. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J. Virol. 77:5324-5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nicola, A. V., M. Ponce de Leon, R. Xu, W. Hou, J. C. Whitbeck, C. Krummenacher, R. I. Montgomery, P. G. Spear, R. J. Eisenberg, and G. H. Cohen. 1998. Monoclonal antibodies to distinct sites on the herpes simplex virus (HSV) glycoprotein D block HSV binding to HVEM. J. Virol. 72:3595-3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nicola, A. V., S. H. Willis, N. N. Naidoo, R. J. Eisenberg, and G. H. Cohen. 1996. Structure-function analysis of soluble forms of herpes simplex virus glycoprotein D. J. Virol. 70:3815-3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Niwa, H., K. Yamamura, and J. Miyazaki. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193-199. [DOI] [PubMed] [Google Scholar]
- 41.Okuma, K., M. Nakamura, S. Nakano, Y. Niho, and Y. Matsuura. 1999. Host range of human T-cell leukemia virus type I analyzed by a cell fusion-dependent reporter gene activation assay. Virology 254:235-244. [DOI] [PubMed] [Google Scholar]
- 42.Pertel, P. E., A. Fridberg, M. L. Parish, and P. G. Spear. 2001. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 279:313-324. [DOI] [PubMed] [Google Scholar]
- 43.Roop, C., L. Hutchinson, and D. C. Johnson. 1993. A mutant herpes simplex virus type 1 unable to express glycoprotein L cannot enter cells, and its particles lack glycoprotein H. J. Virol. 67:2285-2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rux, A. H., H. Lou, J. D. Lambris, H. M. Friedman, R. J. Eisenberg, and G. H. Cohen. 2002. Kinetic analysis of glycoprotein C of herpes simplex virus types 1 and 2 binding to heparin, heparan sulfate, and complement component C3b. Virology 294:324-332. [DOI] [PubMed] [Google Scholar]
- 45.Rux, A. H., S. H. Willis, A. V. Nicola, W. Hou, C. Peng, H. Lou, G. H. Cohen, and R. J. Eisenberg. 1998. Functional region IV of glycoprotein D from herpes simplex virus modulates glycoprotein binding to the herpes virus entry mediator. J. Virol. 72:7091-7098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Santiago, C., E. Bjorling, T. Stehle, and J. M. Casasnovas. 2002. Distinct kinetics for binding of the CD46 and SLAM receptors to overlapping sites in the measles virus hemagglutinin protein. J. Biol. Chem. 277:32294-3301. [DOI] [PubMed] [Google Scholar]
- 47.Sarrias, M. R., J. C. Whitbeck, I. Rooney, C. F. Ware, R. J. Eisenberg, G. H. Cohen, and J. D. Lambris. 2000. The three HveA receptor ligands, gD, LT-alpha and LIGHT bind to distinct sites on HveA. Mol. Immunol. 37:665-673. [DOI] [PubMed] [Google Scholar]
- 48.Satoh-Horikawa, K., H. Nakanishi, K. Takahashi, M. Miyahara, M. Nishimura, K. Tachibana, A. Mizoguchi, and Y. Takai. 2000. Nectin-3, a new member of immunoglobulin-like cell adhesion molecules that shows homophilic and heterophilic cell-cell adhesion activities. J. Biol. Chem. 275:10291-10299. [DOI] [PubMed] [Google Scholar]
- 49.Sisk, W. P., J. D. Bradley, R. J. Leipold, A. M. Stoltzfus, M. Ponce de Leon, M. Hilf, C. Peng, G. H. Cohen, and R. J. Eisenberg. 1994. High-level expression and purification of secreted forms of herpes simplex virus type 1 glycoprotein gD synthesized by baculovirus-infected insect cells. J. Virol. 68:766-775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spear, P. G. 1993. Membrane fusion induced by herpes simplex virus, p. 201-232. In J. Bentz (ed.), Viral fusion mechanisms. CRC Press, Inc., Boca Raton, Fla.
- 51.Spear, P. G., R. J. Eisenberg, and G. H. Cohen. 2000. Three classes of cell surface receptors for alphaherpesvirus entry. Virology 275:1-8. [DOI] [PubMed] [Google Scholar]
- 52.Takahashi, K., H. Nakanishi, M. Miyahara, K. Mandai, K. Satoh, A. Satoh, H. Nishioka, J. Aoki, A. Nomoto, A. Mizoguchi, and Y. Takai. 1999. Nectin/PRR: an immunoglobulin-like cell adhesion molecule recruited to cadherin-based adherens junctions through interaction with afadin, a PDZ domain-containing protein. J. Cell Biol. 145:539-549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takai, Y., and H. Nakanishi. 2003. Nectin and afadin: novel organizers of intercellular junctions. J. Cell Sci. 116:17-27. [DOI] [PubMed] [Google Scholar]
- 54.Terry-Allison, T., R. I. Montgomery, J. C. Whitbeck, R. Xu, G. H. Cohen, R. J. Eisenberg, and P. G. Spear. 1998. HveA (herpesvirus entry mediator A), a coreceptor for herpes simplex virus entry, also participates in virus-induced cell fusion. J. Virol. 72:5802-5810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Turner, A., B. Bruun, T. Minson, and H. Browne. 1998. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J. Virol. 72:873-875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Warner, M. S., W. Martinez, R. J. Geraghty, R. I. Montgomery, J. C. Whitbeck, R. Xu, R. J. Eisenberg, G. H. Cohen, and P. G. Spear. 1998. A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by herpes simplex virus type 2, mutants of herpes simplex virus type 1 and pseudorabies virus. Virology 246:179-189. [DOI] [PubMed] [Google Scholar]
- 57.Whitbeck, J. C., S. A. Connolly, S. H. Willis, W. Hou, C. Krummenacher, M. Ponce de Leon, H. Lou, I. Baribaud, R. J. Eisenberg, and G. H. Cohen. 2001. Localization of the gD-binding region of the human herpes simplex virus receptor, HveA. J. Virol. 75:171- [DOI] [PMC free article] [PubMed]
- 58.Whitbeck, J. C., M. I. Muggeridge, A. Rux, W. Hou, C. Krummenacher, H. Lou, A. van Geelen, R. J. Eisenberg, and G. H. Cohen. 1999. The major neutralizing antigenic site on herpes simplex virus glycoprotein D overlaps a receptor-binding domain. J. Virol. 73:9879-9890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Whitbeck, J. C., C. Peng, H. Lou, R. Xu, S. H. Willis, M. Ponce de Leon, T. Peng, A. V. Nicola, R. I. Montgomery, M. S. Warner, A. M. Soulika, L. A. Spruce, W. T. Moore, J. D. Lambris, P. G. Spear, G. H. Cohen, and R. J. Eisenberg. 1997. Glycoprotein D of herpes simplex virus (HSV) binds directly to HVEM, a member of the TNFR superfamily and a mediator of HSV entry. J. Virol. 71:6083-6093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Willis, S. H., C. Peng, M. Ponce de Leon, A. V. Nicola, A. H. Rux, G. H. Cohen, and R. J. Eisenberg. 1998. Expression and purification of secreted forms of HSV glycoproteins from baculovirus-infected insect cells. Methods Mol. Med. 10:131-156. [DOI] [PubMed] [Google Scholar]
- 61.Willis, S. H., A. H. Rux, C. Peng, J. C. Whitbeck, A. V. Nicola, H. Lou, W. Hou, L. Salvador, G. H. Cohen, and R. J. Eisenberg. 1998. Examination of the kinetics of herpes simplex virus glycoprotein D binding to the herpesvirus entry mediator, using surface plasmon resonance. J. Virol. 72:5937-5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wittels, M., and P. G. Spear. 1990. Penetration of cells by herpes simplex virus does not require a low pH-dependent endocytic pathway. Virus Res. 18:271-290. [DOI] [PubMed] [Google Scholar]
- 63.Yoon, M., and P. G. Spear. 2002. Disruption of adherens junctions liberates nectin-1 to serve as receptor for herpes simplex virus and pseudorabies virus entry. J. Virol. 76:7203-7208. [DOI] [PMC free article] [PubMed] [Google Scholar]