

Abstract
Astrocytes can act as intermediaries between neurons and cerebral arterioles to regulate vascular tone in response to neuronal activity. Release of glutamate from presynaptic neurons increases blood flow to match metabolic demands. CO is a gasotransmitter that can be related to neural function and blood flow regulation in the brain. The present study addresses the hypothesis that glutamatergic stimulation promotes perivascular astrocyte CO production and pial arteriolar dilation in the newborn brain. Experiments used anesthetized newborn pigs with closed cranial windows, piglet astrocytes and cerebrovascular endothelial cells in primary culture, and immunocytochemical visualization of astrocytic markers. Pial arterioles and arteries of newborn pigs are ensheathed by astrocytes visualized by glial fibrillary acidic protein (GFAP) staining. Treatment (2h) of astrocytes in culture with L-2-alpha aminoadipic acid (L-AAA), followed by 14 hr in toxin free medium, dose-dependently increased cell detachment suggesting injury. Conversely, 16h of continuous exposure to L-AAA caused no decrease in endothelial cell attachment. In vivo, topical L-AAA (2mM, 5h) disrupted the cortical glia limitans histologically. Such treatment also eliminated pial arteriolar dilation to the astrocyte-dependent dilator, ADP, and to glutamate, but not to isoproterenol or CO. Glutamate stimulated CO production by the brain surface that also was abolished following L-AAA. In contrast, tetrodotoxin blocked dilation to NMDA, but not to glutamate, isoproterenol, or CO, or the glutamate-induced increase in CO. The concurrent loss of CO production and pial arteriolar dilation to glutamate following astrocyte injury suggests astrocytes may employ CO as a gasotransmitter for glutamatergic cerebrovascular dilation.
Keywords: glia limitans, cerebrovascular circulation, gasotransmitter, glia toxin
INTRODUCTION
In the cerebrovascular circulation, signals to vascular smooth muscle can come from endothelium, nerves, astrocytes, or pericytes, which interact to form a neurovascular unit (15). L-glutamic acid (glutamate) is the principal excitatory neurotransmitter in the central nervous system (38). It is a dilator of cerebral arterioles in vivo (8, 12), although direct effects of glutamate on the vascular smooth muscle are uncertain. Release of glutamate from presynaptic neurons dilates cerebral arterioles causing blood flow to increase to match metabolic demands (37).
Astrocytes are the most abundant cell type in the higher mammalian brain. They function as intermediaries between neurons and cerebral arterioles in regulation of cerebral vascular tone in response to neuronal activity, thereby adjusting cerebral blood flow to metabolism (15, 25). Whether astrocytes are involved in dilation in response to glutamate released at excitatory synapses and the nature of astrocyte-derived vasodilator(s) employed remain uncertain.
The gas, CO, is produced physiologically by catabolism of heme to CO, iron, and biliverdin (36). This reaction is catalyzed by heme oxygenase (HO) with oxidation of NADPH. Astrocytes, neurons, and cerebromicrovascular endothelium all strongly express HO-2 (49). CO is a gasotransmitter that can be related to neural function (4) and blood flow regulation in the brain (31). In vivo, topical CO dilates newborn pial arterioles (30). Glutamate and glutamatergic seizures increase CO production by the piglet brain surface (9) and dilation to glutamate is selectively inhibited following topical treatment with chromium mesoporphyrin (CrMP) that blocks CO production (30).
Therefore, the present study was undertaken to address the hypothesis that glutamatergic stimulation promotes perivascular astrocyte CO production and pial arteriolar dilation in the newborn brain.
METHODS
All procedures that involve animals were reviewed and approved by the Animal Care and Use Committee of The University of Tennessee Health Science Center.
Cranial windows in vivo
Newborn pigs (1-3 d old) (1-2.5 kg) were anesthetized with ketamine hydrochloride (33 mg/kg i.m.) and acepromazine (3.3 mg/kg i.m.) and maintained on α-chloralose (50 mg/kg i.v.) The animals were intubated and ventilated with air. Catheters were inserted into the femoral vein for anesthesia, fluid, and drug injections and into the femoral artery to record blood pressure and draw samples for blood gas and pH analysis. Blood gases, pH, and body temperature were maintained within normal ranges. The scalp was retracted and a hole, 2 cm in diameter, was made in the skull over the parietal cortex. The dura was cut without touching the brain, and all cut edges were retracted over the bone so that the periarachnoid space was not exposed to bone or damaged membranes. A stainless steel and glass cranial window was placed in the hole and cemented into place with dental acrylic. The space under the window was filled with artificial CSF (aCSF) that was equilibrated with 6% CO2 and 6% O2 that produced gases and pH within the normal range for CSF (pH = 7.33-7.40, PCO2 = 42-46 mmHg, and PO2 = 43-50 mmHg). Fluid under the window was exchanged via needle ports on the sides of the window. Pial vessels were observed with a dissecting microscope. Diameters were measured with a video micrometer coupled to a television camera mounted on the microscope and a video monitor. Data from one arteriole of approximately 60μm diameter are reported from each piglet.
Astrocyte culture
Collection of astrocyte-enriched brain cortex isolates was accomplished by gentle homogenization of the piglet cortex in DMEM (1:10) followed by sequential filtration through 300μm, 60μm, 40μm, 30μm and 20μm nylon mesh filters. Cerebral vessels and microvessels are retained on the 300 and 60μm filters and neurons are retained on the 40μm, 30μm, and 20μm filters. The 20μm filtrate is an astrocyte-enriched fraction of cerebral cortex. Following homogenation, serial filtration and centrifugation, pelleted brain filtrate was suspended in astrocyte growth-supporting media (DMEM with antibiotic/antimycotic, 10 ng/ml EGF, and 20% fetal bovine serum (FBS)). Astrocytes were grown in 75-ml flasks for 10-14 days, changing the media twice a week and replated to 12 well plates and grown in astrocyte media for 4-6 days to confluence. The astrocytes were identified by immunostaining for glial fibrillary acidic protein (GFAP) and aquaporin-4, the major water channel expressed in brain perivascular astrocyte processes (41). Such staining indicates these procedures produce pure astrocyte cultures. Cell damage was estimated by the number of detached floating cells. Floating cells were pelleted at 3,000 g for 10 min at 4°C and counted using a microscope counting chamber.
Cerebrovascular endothelial cell primary cultures (43) Following gentle homogenation of piglet brain cortex, cerebral microvessels were collected by filtration of the homogenate through 300-μm and collection on 60-μm nylon mesh screens consecutively. Microvessels were treated with collagenase-dispase (2 mg/ml for 2 h at 37°C), and dissociated cerebral microvascular endothelial cells (CMVEC) were separated from other cells and tissue on a Percoll density gradient. CMVEC were plated on Matrigel-coated plates (3 × 104 cells/well) and grown at 37°C (5% CO2-95% air) in DMEM with 20% FBS, 30 μg/ml endothelial cell growth supplement, 1 U/ml heparin, and antibiotic/antimycotic mixture for 5-6 days until confluent. Endothelial cells identified by immunostaining for von Willebrand factor accounted for at least 95% of the total cell population. As above for astrocytes, cell injury was estimated by the number of detached floating cells. Floating cells were pelleted at 3,000 g for 10 min at 4°C and counted using a microscope counting chamber.
MATERIALS
CO was purchased as compressed gas (99.5%). Water was saturated with CO to produce a 10−3M stock solution. The stock was diluted in aCSF without air contact for injection under the cranial window at a concentration of 10−7M.
Rhodamine-phalloidin for F-actin staining was from Molecular Probes, Invitrogen Corporation (Carlsbad, CA). Antibodies against GFAP and aquaporin-4 were from US Biological (Swampscott, MA) and Abcam Inc. (Cambridge, MA), respectively. Second antibodies were from Vector Laboratories (Burlingame, CA).
Other reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted.
EXPERIMENTS
Vascular responses
Isoproterenol (10−7 M), adenosine 5′-diphosphate (ADP, 10−4 M), glutamate (10−4 M), NMDA (10−4 M), and CO (10−7 M) were applied directly to pial arterioles and the maximal diameter attained over a 5-minute period is reported as the response to each dose. The 5-minute period was selected because, with these agonists, onset of dilation upon topical application is rapid with maximal diameter typically achieved within 3 minutes. The window was flushed with aCSF between treatments and the pial arteriolar diameters allowed to return to about control diameters before the next agonist was applied. Control responses were compared to the same treatments after astrocyte injury or tetrodotoxin (TTX).
Astrocyte injury
Injury to the superficial cortical glia limitans under the cranial window was produced by treatment with the selective glia toxin L-2-alpha aminoadipic acid (L-AAA) (24, 55, 56). The high cellular specificity of L-AAA apparently results from the rapid uptake of the toxin by the cystine-glutamate antiporter expressed by glia and not other brain cells (16, 47). The precise mechanism of injury remains uncertain. The method we used was modified from one developed to produce removal of the influence of glia limitans on pial arteriolar responses in the adult rat (55,56). L-AAA (2mM) was placed under the cranial window for 5h and removed by flushing with aCSF. Khurgel (24) reported before that astrocyte injury occurred 4 h following intracerebral injection of L-AAA in rats. As a control amino acid we used D-AAA that does not cause degenerative changes in astrocytes or any other cell type (24).
Blockade of neural conduction was accomplished with topical application of TTX (0.1 μM). Efficacy of TTX blockade on neuronal conduction was demonstrated by blockade of dilation to NMDA because in piglets TTX blocks dilation to NMDA without affecting dilations to isoproterenol (32). Different investigators have used a variety of concentrations to produce selective removal of neural input. The most commonly used concentration has been 1μM (17), including an earlier report by us (32). Higher concentrations have also been used and still seem to be specific (e.g. 10 μM (57). However, Liu and Lee (34) showed that 0.1μM blocked dilation of pig cerebral arterial rings to transmural nerve stimulation.
Cerebral CO production
Collections of CSF from under the cranial window were made during control and subsequent glutamate treatment before and after treatment with L-AAA. Collections were made after the CSF had been under the window for 5min. To collect the CSF, fresh aCSF was injected into one needle port on the cranial window and 400 μl of displaced CSF was collected in a glass vial through a metal spout on another port. By placing known concentrations of CO under the cranial window and then collecting that CSF for CO measurement, we have determined this collection method has an efficiency of approximately 100% (e.g. 10−6M placed under the window resulted in 1.00±0.09 × 10−6M measured in the collections, n=30). The total volume was increased to 1.4 ml, 31CO standard added, and the vial was sealed with a rubber and Teflon cap. CO in the headspace gas was measured by GC-MS and quantified by comparison to the 31CO standard as we have described before (48).
Immunocytochemistry
Immunocytochemical staining was performed on slices of the cortical surface obtained after removal of the cranial windows from control and L-AAA-exposed pigs. Paraformaldehyde-fixed brain cortex was embedded in paraffin, and sections were cut at 5 microns. After deparaffinization and rehydration through xylene and graded alcohols, brain cortex slices were heat-treated for antigen unmasking. Endogenous peroxidase was quenched with 3% peroxide for 45 min, and non-specific antigen binding was blocked with normal horse serum (30 min, room temperature). Immunostaining for GFAP was performed using monoclonal anti-porcine GFAP (US Biological, Swampscott, MA) and visualized with biotinylated goat anti-mouse IgG using the avidinbiotinylated enzyme complex technique (Vectastain ABC Kit, Vector Laboratories, Burlingam, CA). Hematoxylin was used for counterstaining. Sections were rinsed in water, alcohol-dehydrated, and covered with a coverslip.
Immunofluorescence
GFAP immunofluorescence was investigated in pial arteries and arterioles and in cultured brain cortex astrocytes. Pial arteries and arterioles were dissected from the brain surface using a dissecting microscope. Care was taken to preserve cells adhering to the vessels. Dissected cerebral vessels were placed on microscope slides and air-dried. Astrocytes in culture were replated and cultured on Matrigel-coated coverlips placed in 6-well plates to confluence. Cerebral vessels and astrocytes were fixed in 3.7% paraformaldehyde (pH 7.4) for 20 min at room temperature, and permeabilized with 0.1 % Triton X-100 in PBS for 20 min. Non-specific binding sites were blocked with 5%BSA-PBS for 1 h. GFAP was immunostained with monoclonal anti-porcine GFAP (US Biological, Swampscott, MA) and visualized with fluorescein–conjugated anti-mouse IgG (Vector Laboratories). Aquaporin-4 immunostaining of astrocytes was performed using monoclonal antibodies from Abcam Inc. For F-actin labeling and visualization, we used rhodamine phalloidin. Slides were briefly rinsed with water, air-dried, and covered with slips using Vectashield anti-fade mounting medium (Vector Laboratories). Slides were viewed using a Nikon Diaphot microscope with fluorescein and rhodamine filters, and images were deconvolved using IPLab spectrum software for image collection.
Statistical analysis
Values for each variable are presented as mean ± SEM. Comparisons among populations within each experimental group used ANOVA with repeated measures. The Tukey-Kramer Multiple Comparisons test was used to isolate differences between groups. P < 0.05 was considered significant.
RESULTS
The pial arterioles and arteries of newborn pigs are ensheathed by astrocytes as visualized with positive staining for the astrocyte specific marker GFAP (Fig 1).
Fig 1.
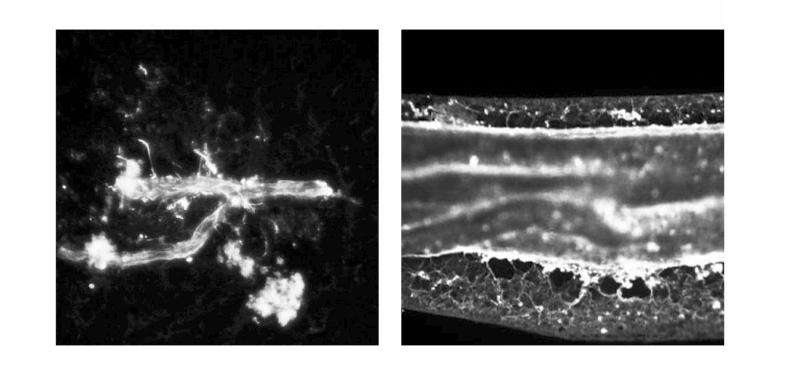
Freshly isolated newborn pial arterioles (left) and small artery (right) immunostained for GFAP. Note the complete envelopment of the vessels with GFAP positive processes.
Using piglet astrocytes (Fig 2) and cerebrovascular endothelial cells in primary culture, we investigated the specificity of the purportedly selective astrocyte toxin, L-AAA. Two hours treatment of astrocytes with L-AAA (0.2-2mM) followed by 14 hr in toxin free medium dose-dependently increased cell detachment with a nearly 3-fold elevation at 2 mM (Fig 3). Astrocytes treated with 2 mM L-AAA showed marked retraction, loss of cell-to-cell contacts and cytoskeletal changes at 1hr treatment with complete absence of normal cell structure and substantial cell loss by 4.5hr (Fig 4). Conversely, even 16h of continuous exposure to 2 mM L-AAA caused no increase in endothelial cell detachment (Fig 3) or detectible morphologic changes.
Fig 2.
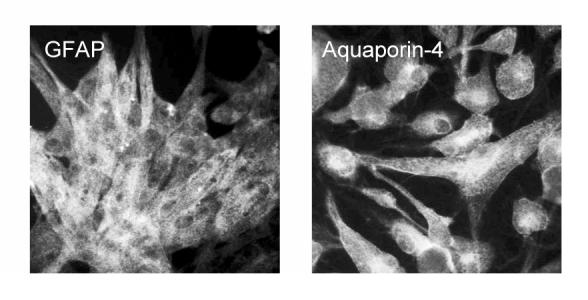
Piglet astrocytes in primary culture, immunostained for GFAP (left) and aquaporin-4 (right) (shown subconfluent for contrast).
Fig 3.
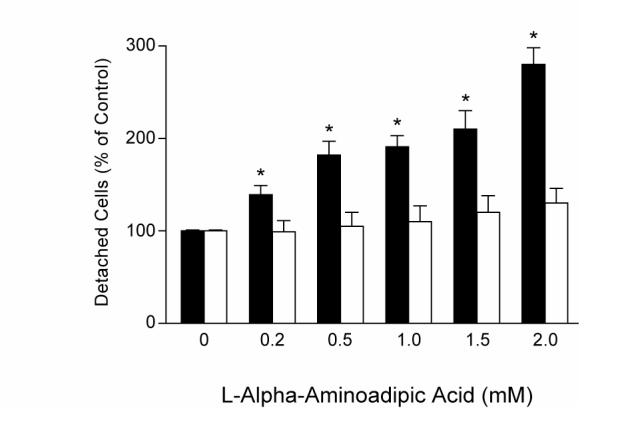
Effects of L-AAA on numbers of detached astrocytes (black bars) (2h treatment followed by 14h) and endothelial cells (white bars)(16h continuous treatment). *P<0.05 compared to no L-AAA. N=6 wells for each cell type.
Fig 4.
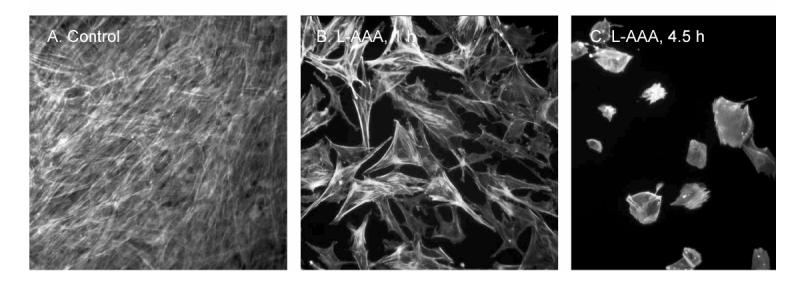
Effect of the astrocyte toxin, L-AAA (2mM), on confluent piglet astrocytes in primary culture. Immunostaining is for F-actin to show the cytoskeletal structure. Note the strong retractions of the astrocyte processes in the two panels treated with L-AAA (1 and 4.5h) as compared to the control panel on the left.
To selectively injure astrocytes in vivo, we treated the cortical surface with L-AAA (2 mM). Five-hour treatment produced histological evidence of injury to the superficial glia limitans detected as disruption of the confluent layer of GFAP positive cells that were seen in control (Fig 5). Such treatment also eliminated pial arteriolar dilation to the putative astrocyte-dependent dilator, ADP (Fig 6). L-AAA also abolished dilation of pial arterioles to glutamate (Fig 6). It appears that the injury was limited to the astrocytes because dilations to isoproterenol, that increases cAMP via vascular smooth muscle β-adrenergic receptors and CO that activates vascular smooth muscle KCa channels, were intact following L-AAA treatment (Fig 6). The endothelium also appears to be intact because dilation to CO is endothelial dependent due to the necessity for endothelial-derived permissive mediators (3). The control amino acid, D-AAA, administered exactly as was L-AAA, had no effect on pial arteriolar responses to glutamate, ADP, isoproterenol, or CO (data not shown).
Fig 5.
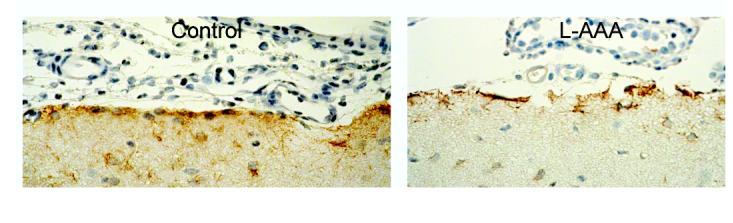
Parietal cortical surfaces of newborn pig brains immunostained for GFAP. The left panel is control and the right panel is 2 mM L-AAA for 5h showing disruption of the glia limitans.
Fig 6.
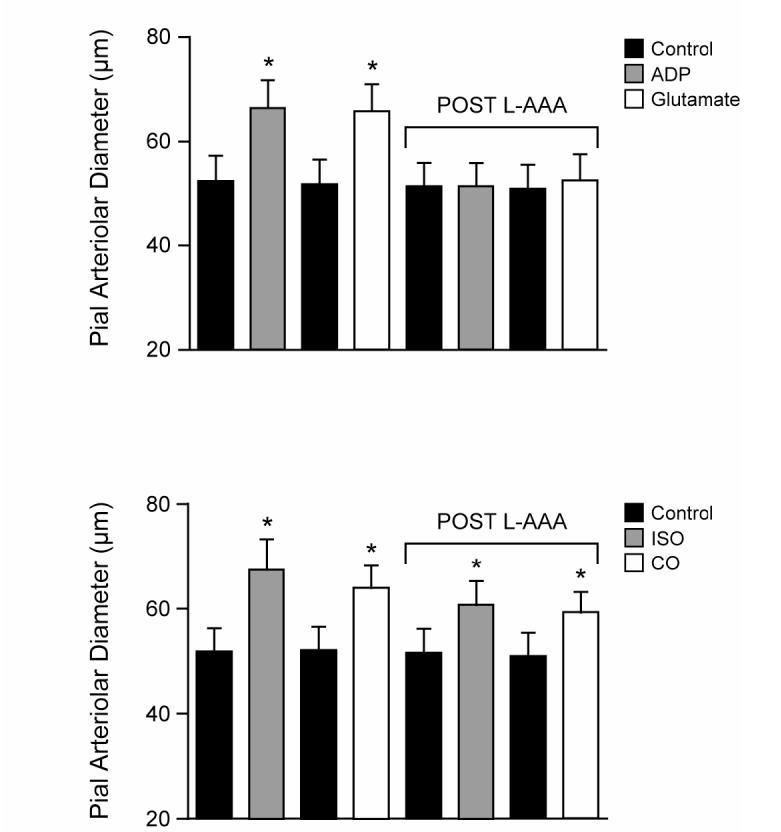
Effects of L-AAA (2mM) on pial arteriolar responses to ADP (10−4 M), glutamate (10−4 M), isoproterenol (10−7 M), and CO (10−7 M). Responses are shown before and after 5 h treatment with L-AAA. Note that dilations to isoproterenol that directly activates β-adrenergic receptors on the vascular smooth muscle and CO that activates vascular smooth muscle KCa channels remain statistically unchanged following L-AAA, while responses to the putative glia dependent dilator, ADP, and glutamate are completely abolished. N=8. *P<0.05 compared to previous bar.
CO could be a mediator of the astrocyte-induced dilations because HO inhibitors attenuate glutamatergic dilation of newborn pig arterioles (30). Treatment with glutamate stimulated CO production by the brain surface as detected in the aCSF collected from beneath the cranial window (Fig 7). Glutamate stimulation of CO production was abolished following L-AAA-induced astrocyte injury (Fig 7) but was unaffected by D-AAA (data not shown).
Fig 7.
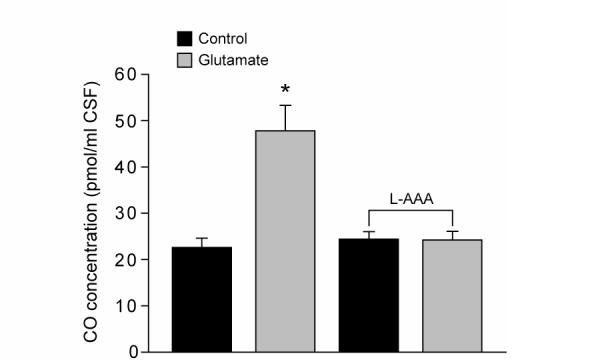
Effect of glutamate on piglet cerebral cortical surface production of CO before and after treatment with L-AAA (2mM, 5h). Glutamate increases CO production by the cortical surface and that stimulation is abolished following astrocyte toxin treatment. N=5. *P<0.05 compared to previous bar.
In contrast, tetrodotoxin (TTX, 0.1μM), that blocked dilation to NMDA, did not inhibit pial arteriolar dilation to glutamate (Fig 8), nor the increase in CO (128% and 255% increase in CO before and after TTX, respectively. N=4. P>0.05). TTX did not inhibit vasodilation to exogenously applied isoproterenol either (Fig 8).
Fig 8.

Effect of TTX (0.1μM) on piglet pial arteriolar dilations to NMDA (10−4 M), glutamate (10−4 M), and isoproterenol (10−7 M). TTX blocked dilation to NMDA, demonstrating efficacy, but did not block dilations to glutamate or isoproterenol. N=7. *P<0.05 compared to previous bar.
DISCUSSION
The new findings of the current study of newborn pigs are: 1. Pial arterioles are ensheathed in astrocyte processes; 2. L-AAA severely alters cultured astrocyte morphology and causes astrocyte detachment but has no detectable effect on cultured endothelial cells; 3. L-AAA causes histologically evident disruption of the glia limitans, in vivo; 4. L-AAA abolishes pial arteriolar dilation to glutamate as well as to the astrocyte-dependent dilator, ADP, but does not alter responses to isoproterenol or CO, endothelial-independent and -dependent dilators, respectively; 5. TTX blocks dilation of pial arterioles to NMDA but not to glutamate; and 6. L-AAA, but not TTX, blocks the ability of glutamate to stimulate CO production by the cerebral cortical surface. The concurrent loss of CO production and pial arteriolar dilation to glutamate following astrocyte injury suggests that the astrocytes may employ CO as a gasotransmitter for glutamatergic cerebrovascular dilation.
Glutamate is a dilator in the cerebral circulation in vivo (8, 11, 39). Release of glutamate from presynaptic neurons increases blood flow to match the increased metabolic demands of stimulated neurons (37). Neuronal activation in response to excitatory neurotransmitters may affect brain vessels via neurally-derived vasorelaxant factors, including NO (11, 39, 51) and CO (2). Glutamate and selective NMDA- and AMPA/kainate receptor agonists increase CO production by piglet cerebral microvessels (29, 43) and astrocytes (52). We found pressurized pial arteries respond to glutamate by endothelium-dependent vasodilation (14), although less strongly than in vivo (48). Of note, others have failed to detect glutamate-receptor-mediated responses at all in isolated cerebral arteries (50). The smaller or absent dilatory response to glutamate in vitro compared with the in vivo response, coupled with similar dilator responses to isoproterenol in vitro and in vivo (2, 14, 53), suggests that cells underrepresented in the isolated arteriole contribute to the dilation in response to glutamate in vivo. The present data suggest that astrocytes, although immunodetectable on our isolated pial arterioles, could be the underrepresented cell type when the arteriole is removed from the intact brain. It is conceivable that the reason our pressurized pial arterioles dilated in response to glutamate while those of Simandle et al (50) did not could relate to selection and handling of the vessels that resulted in the presence and absence of astrocytes, respectively.
In the cerebrovascular circulation, signals to vascular smooth muscle can come from endothelium, nerves, astrocytes, or pericytes, which interact to form a neurovascular unit (15). Astrocytes are the most abundant cell type in the higher mammalian brain. Parenchymal arterioles are ensheathed by astrocyte end-feet (15, 25) and pial arterioles are coated by astrocyte processes and end feet (present study). Astrocytes can act as intermediaries between neurons and cerebral blood vessels in regulation of cerebral vascular tone in response to neuronal activity, thereby adjusting cerebral blood flow to metabolism (15, 25). Astrocytes possess both metabotropic glutamate receptors and ionotropic glutamate receptors, but most studies have been unable to detect NMDA receptor mediated responses in astrocytes (40). Metabotropic glutamate receptors and AMPA receptors are stimulated by glutamate to produce Ca2+ oscillations in astrocytes (45). Brain slice experiments have demonstrated that glutamate can elevate astrocyte [Ca2+]c and reduce adjacent vascular smooth muscle [Ca2+]c producing vasodilation (13). However, the mechanism by which increased astrocyte [Ca2+]c produces decreased vascular smooth muscle [Ca2+]c is not known. We hypothesize that astrocyte-derived CO increases smooth muscle KCa channel activity hyperpolarizing the myocytes, which decreases myocyte [Ca2+]c, thereby providing a mechanism by which glutamate stimulation of the astrocyte can cause dilation of the arteriole.
The present results suggest pial vasodilation to glutamate is not mediated via the neuronal component of the neurovascular unit because TTX did not inhibit dilation to glutamate. TTX was effective because, as we showed before (32), TTX did block the dilatory response to NMDA. These data further suggest that the principle mechanism by which the topical glutamate produces dilation is not via activation of neuronal NMDA receptors. The inhibitory effect of TTX on NMDA-induced cerebrovascular dilation suggests activation of NMDA receptors on neurons causes release of a substance(s) that promotes dilation (11). Since Meng et al (39) reported blockade of dilation to NMDA by LNA, a potential neuronal derived dilator is NO generated by nNOS. We recently demonstrated that NO can increase CO production by cerebral microvessels (28), so neuronally derived NO could stimulate CO production in astrocytes, neurons, endothelium, and/or vascular smooth muscle to cause dilation.
Neuronal control of cerebrovascular circulation can be mediated via NO. In adult animals, NO may be involved in neurally-mediated dilation (15) and its role may be a permissive one (33). In piglets, neurally-mediated dilation (39) also appears to involve NO. In adult rats, the origin of the NO involved in glutamatergic and hypoxic cerebral dilation appears to be nNOS rather than eNOS (46).
In the present study, L-AAA abolished dilation to ADP. This is in contrast to previous research using adult rats where, although producing similar histological evidence of glia limitans disruption, L-AAA reduced dilation to ADP about 50% with the other 50% apparently being endothelial/eNOS-dependent (55,56). The reason for this difference is not known but could relate to increased envelopment of pial vessels observed in piglets as compared to adult rats where astrocyte processes are only found on the half of the vessel toward the brain (55,56, present study). Whether the difference is related to species or age is unknown. However, an age dependent component should be considered because eNOS dependent cerebrovascular control mechanisms increase with age in the pig (53).
CO is produced physiologically by HO catabolized breakdown of heme to CO, iron, and biliverdin (35). Of the three known HO isoforms, only HO-2 expression is detected on Western blots of newborn pig brain (44). Glutamate stimulation of HO-2 catalytic activity results from metabotropic glutamate receptors in neurons (6) and ionotropic glutamate receptors in cerebral microvessels and endothelial cells (43). In both cerebral microvessels and cortical neurons, calmidizolium, that inhibits calmodulin, decreases HO-2 catalytic activity and blocks glutamate stimulation of CO production (5, 29).
The predominant mechanism responsible for CO-induced cerebral vasodilation is KCa channel activation (31). CO activates KCa channels in arterial smooth muscle by binding to channel-bound ferrous heme and changing the association of the heme with the channel leading to channel activation (21). In vascular smooth muscle cells, local intracellular Ca2+ transients termed “Ca2+ sparks” activate KCa channels (22). Summation of transient KCa currents induces a membrane hyperpolarization that reduces voltage-dependent Ca2+ channel activity, and thus, intracellular Ca2+ concentration. CO elevates KCa channel Ca2+ sensitivity (54) enhancing the effective coupling of Ca2+ sparks to KCa channels (20).
Whether the apparently increased vasodilatory sensitivity to CO observed in newborn pig cerebrovascular circulation when compared to adult cerebral arteries (1, 26) is due to age, species, or both remains uncertain. However, developmental differences seem plausible considering low sensitivity to CO is found in isolated cerebral arteries from adult rats (1), rabbits (7), and dogs (26). Developmental mechanisms would not be surprising. In the newborn pig cyclooxygenase activity, that stimulates HO-2 (23) and CO, that inhibits NOS (19), are elevated, which could contribute to the reduced functional significance of NO and the increased importance of CO in the neonatal cerebrovascular circulation. Further, HO levels in cerebrum are developmentally regulated with maximal HO expression in the mature fetus, as compared to the immature fetus or adult (10).
Astrocyte toxin blocked not only pial arteriolar dilation but also stimulation of CO production in response to glutamate. Conversely, L-AAA did not affect responses to exogenous CO or isoproterenol. These data suggest glutamate could act on astrocyte glutamate receptors to stimulate CO production that can, in turn, cause dilation of pial arterioles. That responses to exogenous CO were not changed by L-AAA suggest no depression in necessary endothelial-derived permissive factors (3, 31) is caused by L-AAA. Unaltered responses to isoproterenol indicate normal smooth muscle sensitivity to β adrenergic receptor stimulation and cAMP. Specific glutamate receptors involved in elevating CO production and dilation in response to glutamate in astrocytes are not yet known. That TTX blocks vasodilation in response to NMDA, but not to glutamate, suggests a large component of glutamate-induced CO production and dilation is independent of NMDA receptors. We have found in endothelial cells that all of the ionotropic glutamate receptors, but not the metabotropic glutamate receptors, stimulate CO production (43), which is in contrast with data from rat neurons implicating metabotropic glutamate receptors (4). However, in the intact adult rat, pial arteriolar dilation to AMPA, but not to NMDA, is inhibited by treatment with the HO inhibitor, chromium mesoporphyrin (42), suggesting AMPA receptor stimulation may increase CO production that contributes to vasodilation. These authors present data suggesting adenosine may be in the pathway between AMPA receptor stimulation and CO production.
In contrast to the effects of functional removal of astrocytes, TTX did not affect responses to any of the dilator stimuli used with the exception of NMDA. Since TTX blocks dilation to NMDA, but not glutamate, it appears the glutamate receptors on astrocytes that produce vascular dilation are different from those on neurons that cause dilation to NMDA. This is not surprising because most data suggest astrocytes do not express NMDA receptors (40).
Even though we have shown that endothelial cells have ionotropic glutamate receptors that are functionally linked to CO production (43), the present study suggests the major source of the excitatory amino acid-induced CO production is astrocytes. Although there are multiple potential cerebral sources of CO, the astrocytes are uniquely positioned to receive glutamatergic stimulation from topical glutamate receptor agonists, and also excitatory nerves (18).
ACKNOWLEDGEMENTS
Research supported by NHLBI/NIH and NINDS/NIH. We thank G. Short for assistance with preparation of figures.
REFERENCES
- 1.Andresen J, Bryan RM., Jr. Mouse cerebral arterioles dilate to carbon monoxide. FASEB J. 2006;20:A292. [Google Scholar]
- 2.Baranano DE, Snyder SH. Neural roles for heme oxygenase: contrasts to nitric oxide synthase. Proc Natl Acad Sci U S A. 2001;98:10996–11002. doi: 10.1073/pnas.191351298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barkoudah E, Jaggar JH, Leffler CW. The permissive role of endothelial NO in CO-induced cerebrovascular dilation. Am J Physiol. 2004;287:H1459–H1465. doi: 10.1152/ajpheart.00369.2004. [DOI] [PubMed] [Google Scholar]
- 4.Boehning D, Moon C, Sharma S, Hurt KJ, Hester LD, Ronnett GV, Shugar D, Snyder SS. Carbon monoxide neurotransmission activated by CK2 phosphorylation of heme oxygenase-2. Neuron. 2003;40:129–137. doi: 10.1016/s0896-6273(03)00596-8. [DOI] [PubMed] [Google Scholar]
- 5.Boehning D, Sedaghat L, Sedlak TW, Snyder SS. Heme oxygenase-2 is activated by calcium-calmodulin. J Biol Chem. 2004;279:30927–30930. doi: 10.1074/jbc.C400222200. [DOI] [PubMed] [Google Scholar]
- 6.Boehning D, Snyder SH. Novel neural modulators. Ann Rev Neurosci. 2003;26:105–131. doi: 10.1146/annurev.neuro.26.041002.131047. [DOI] [PubMed] [Google Scholar]
- 7.Brian JE, Jr., Heistad DD, Faraci FM. Effects of carbon monoxide on rabbit cerebral arteries. Stroke. 1994;25:639–643. doi: 10.1161/01.str.25.3.639. [DOI] [PubMed] [Google Scholar]
- 8.Busija DW, Leffler CW. Dilator effects of amino acid neurotransmitters on piglet arterioles. Am J Physiol. 1989;257:H1200–H1203. doi: 10.1152/ajpheart.1989.257.4.H1200. [DOI] [PubMed] [Google Scholar]
- 9.Carratu P, Pourcyrous M, Fedinec A, Leffler CW, Parfenova H. Endogenous heme oxygenase prevents impairment of cerebral vascular functions caused by seizures. Am J Physiol. 2003;285:H1148–H1157. doi: 10.1152/ajpheart.00091.2003. [DOI] [PubMed] [Google Scholar]
- 10.Cook MN, Marks GS, Vreman HJ, Nakatsu K, Stevenson DK, Brien JF. Ontogeny of heme oxygenase activity in the hippocampus, frontal cerebral cortex, and cerebellum of the guinea pig. Brain Res Dev Brain Res. 1996;92:18–23. doi: 10.1016/0165-3806(95)00194-8. [DOI] [PubMed] [Google Scholar]
- 11.Faraci FM, Breese KR. Nitric oxide mediates vasodilatation in response to activation of N-methyl-D-aspartate receptors in the brain. Circ Res. 1993;72:476–480. doi: 10.1161/01.res.72.2.476. [DOI] [PubMed] [Google Scholar]
- 12.Fergus A, Lee KS. Regulation of cerebral microvessels by glutamatergic mechanisms. Brain Res. 1997;754:35–45. doi: 10.1016/s0006-8993(97)00040-1. [DOI] [PubMed] [Google Scholar]
- 13.Filosa JA, Bonev AD, Nelson MT. Calcium dynamics in cortical astrocytes and arterioles during neurovascular coupling. Circ Res. 2004;95:e73–81. doi: 10.1161/01.RES.0000148636.60732.2e. [DOI] [PubMed] [Google Scholar]
- 14.Fiumana E, Parfenova H, Jaggar JH, Leffler CW. Carbon monoxide mediates vasodilator effects of glutamate in isolated pressurized cerebral arterioles of newborn pigs. Am J Physiol. 2003;284:H1073–1079. doi: 10.1152/ajpheart.00881.2002. [DOI] [PubMed] [Google Scholar]
- 15.Girouard H, Iadecola C. Neurovascular coupling in the brain and hypertension, stroke, and Alzheimer disease. J Appl Physiol. 2006;100:328–335. doi: 10.1152/japplphysiol.00966.2005. [DOI] [PubMed] [Google Scholar]
- 16.Huck S, Grass F, Hortnagl The glutamate analog a-aminoadipic acid is taken up by astrocytes before exerting its gliotoxic effect in vitro. J Neurosci. 1984;4:2650–2657. doi: 10.1523/JNEUROSCI.04-10-02650.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iadecola C, Sherry JL, Yang G. Neural mechanisms of blood flow regulation during synaptic activity in cerebellar cortex. J Neurophys. 1996;75:940–950. doi: 10.1152/jn.1996.75.2.940. [DOI] [PubMed] [Google Scholar]
- 18.Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nature Rev Neurosci. 2004;5:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- 19.Ishikawa M, Kajimura M, Adachi T, Maruyama K, Makino N, Goda N, Yamaguchi T, Sekizuki E, Suematsu M. Carbon monoxide from heme oxygenase-2 is a tonic regulator against NO-dependent vasodilation in the adult rat cerebral microcirculation. Circ Res. 2005;97:e104–14. doi: 10.1161/01.RES.0000196681.34485.ec. [DOI] [PubMed] [Google Scholar]
- 20.Jaggar JH, Leffler CW, Cheranov SY, Tcheranova D,ES, Cheng X. Carbon monoxide dilates cerebral arterioles by enhancing the coupling of Ca2+ sparks to Ca2+-activated K+ channels. Circ Res. 2002;91:610–617. doi: 10.1161/01.res.0000036900.76780.95. [DOI] [PubMed] [Google Scholar]
- 21.Jaggar JH, Li A, Parfenova H, Liu J, Umstot ES, Dopico AM, Leffler CW. Heme is a carbon monoxide receptor for large-conductance Ca2+-activated K+ channels. Circ Res. 2005;97:805–812. doi: 10.1161/01.RES.0000186180.47148.7b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaggar JH, Nelson MT. Differential regulation of Ca2+ sparks and Ca2+ waves by UTP in rat cerebral artery smooth muscle cells. Am J Physiol. 2000;279:C1528–C1539. doi: 10.1152/ajpcell.2000.279.5.C1528. [DOI] [PubMed] [Google Scholar]
- 23.Kanu A, Gilpin D, Fedinec AL, Leffler CW. Cyclooxygenase products stimulate carbon monoxide production by piglet cerebral microvessels. Exp Biol Med. 2006;231:181–185. doi: 10.1177/153537020623100208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khurgel M, Koo AC, Ivy GO. Selective ablation of astrocytes by intracerebral injections of alpha-aminoadipate. Glia. 1996;16:351–358. doi: 10.1002/(SICI)1098-1136(199604)16:4<351::AID-GLIA7>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 25.Koehler RC, Gebremedhin D, Harder DR. Role of astrocytes in cerebrovascular regulation. J Appl Physiol. 2006;100:307–317. doi: 10.1152/japplphysiol.00938.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Komuro T, Borsody MK, Onon S, Marton LS, Weir BK, Zhang ZD, Paik E, MacDonald RL. The vasorelaxation of cerebral arteries by carbon monoxide. Exp Biol Med. 2001;226:860–865. doi: 10.1177/153537020122600909. [DOI] [PubMed] [Google Scholar]
- 27.Leffler CW, Balabanova L, Fedinec AL, Waters CM, Parfenova H. Mechanism of glutamate stimulation of CO production in cerebral microvessels. Am J Physiol. 2003;285:H74–H80. doi: 10.1152/ajpheart.01081.2002. [DOI] [PubMed] [Google Scholar]
- 28.Leffler CW, Balabanova L, Fedinec AL, Parfenova H. Nitric oxide increases carbon monoxide production by piglet cerebral microvessels. Am J Physiol. 2005;289:H1442–H1447. doi: 10.1152/ajpheart.00464.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leffler CW, Balabanova L, Sullivan CD, Wang X, Fedinec AL, Parfenova H. Regulation of CO production in cerebral microvessels of newborn pigs. Am J Physiol. 2003;285:H292–H297. doi: 10.1152/ajpheart.01059.2002. [DOI] [PubMed] [Google Scholar]
- 30.Leffler CW, Nasjletti A, Yu C, Johnson RA, Fedinec AL, Walker N. Carbon monoxide and cerebral microvascular tone in newborn pigs. Am J Physiol Heart Circ Physiol. 1999;276:H1641–H1646. doi: 10.1152/ajpheart.1999.276.5.H1641. [DOI] [PubMed] [Google Scholar]
- 31.Leffler CW, Parfenova H, Jaggar JH, Wang R. Carbon monoxide and hydrogen sulfide: gaseous messengers in cerebrovascular circulation. J Appl Physiol. 2006;100:1065–1076. doi: 10.1152/japplphysiol.00793.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leffler CW, Smith JS, Edrington JL, Zuckerman SL, Parfenova H. Mechanisms of hypoxia-induced cerebrovascular dilation in the newborn pig. Am J Physiol. 1997;272:H1323–H1332. doi: 10.1152/ajpheart.1997.272.3.H1323. [DOI] [PubMed] [Google Scholar]
- 33.Lindauer U, Megow D, Matsuda H, Dirnagl U. Nitric oxide: a modulator, but not a mediator, of neurovascular coupling in rat somatosensory cortex. Am J Physiol. 1999;277:H799–H811. doi: 10.1152/ajpheart.1999.277.2.H799. [DOI] [PubMed] [Google Scholar]
- 34.Liu J, Lee TJF. Mechanism of prejunctional muscarinic receptor-mediated inhibition of neurogenic vasodilation in cerebral arteries. Am J Physiol. 1999;276:H194–H204. doi: 10.1152/ajpheart.1999.276.1.H194. [DOI] [PubMed] [Google Scholar]
- 35.Maines MD. Carbon monoxide: an emerging regulator of cGMP in the brain. Mol Cell Neurosciences. 1993;4:389–397. doi: 10.1006/mcne.1993.1049. [DOI] [PubMed] [Google Scholar]
- 36.Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. 1997;37:517–554. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- 37.Meldrum BS. Concept of activity-induced cell death in epilepsy: historical and contemporary perspectives. Prog Brain Res. 2002;135:3–11. doi: 10.1016/S0079-6123(02)35003-9. [DOI] [PubMed] [Google Scholar]
- 38.Meldrum BS. Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J Nutr. 2000;130:1007S–1015S. doi: 10.1093/jn/130.4.1007S. [DOI] [PubMed] [Google Scholar]
- 39.Meng W, Tobin JR, Busija DW. Glutamate-induced cerebral vasodilation is mediated by nitric oxide through N-methyl-D-aspartate receptors. Stroke. 1995;26:857–862. doi: 10.1161/01.str.26.5.857. [DOI] [PubMed] [Google Scholar]
- 40.Nedergaard M, Takano T, Hansen AJ. Beyond the role of glutamate as a neurotransmitter. Nat Rev Neurosci. 2002;3:748–55. doi: 10.1038/nrn916. [DOI] [PubMed] [Google Scholar]
- 41.Nicchia GP, Nico B, Camassa LM, Mola MG, Loh N, Dermietzel R, Spray DC, Svelto M, Frigeri A. The role of aquaporin-4 in the blood-brain barrier development and integrity: studies in animal and cell culture models. Neuroscience. 2004;129:935–45. doi: 10.1016/j.neuroscience.2004.07.055. [DOI] [PubMed] [Google Scholar]
- 42.Ohata H, Cao S, Koehler RC. Contribution of adenosine A2A and A2B receptors and heme oxygenase to AMPA-induced dilation of pial arterioles in rats. Am J Physiol. 2006 doi: 10.1152/ajpregu.00757.2005. In press. Articles in PresS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parfenova H, Fedinec A, Leffler CW. Ionotropic glutamate receptors in cerebral microvascular endothelium are functionally linked to heme oxygenase. J Cereb Blood Flow Metab. 2003;23:190–197. doi: 10.1097/01.WCB.000004823561824.C4. [DOI] [PubMed] [Google Scholar]
- 44.Parfenova H, Neff RA, III, Alonso JS, Shlopov BV, Jamal CN, Sarkasova SA, Leffler CW. Cerebrovascular endothelial heme oxygenase: expression, intracellular compartmentalization, and activation by glutamate. Am J Physiol. 2001;281:C1954–C1963. doi: 10.1152/ajpcell.2001.281.6.C1954. [DOI] [PubMed] [Google Scholar]
- 45.Pasti L, Volterra A, Pozzan T, Carmignoto G. Intracellular calcium oscillations in astrocytes: a highly plastic, bidirectional form of communication between neurons and astrocytes in situ. J Neurosci. 1997;17:7817–30. doi: 10.1523/JNEUROSCI.17-20-07817.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pelligrino DA, Wang Q, Koenig HM, Albrecht RF. Role of nitric oxide, adenosine, N-methyl-D-aspartate receptors, and neuronal activation in hypoxia-induced pial arteriolar dilation in rats. Brain Res. 1995;704:61–70. doi: 10.1016/0006-8993(95)01105-6. [DOI] [PubMed] [Google Scholar]
- 47.Pow DV. Visualizing the activity of the cysteine-glutamate antiporter in glial cells using antibodies to aminoadipic acid, a selectively transported substrate. Glia. 2001;34:27–38. doi: 10.1002/glia.1037. [DOI] [PubMed] [Google Scholar]
- 48.Robinson JS, Fedinec AL, Leffler CW. Role of CO in glutamate receptor-induced dilation of newborn pig pial arterioles. Am J Physiol. 2002;282:H2371–H2376. doi: 10.1152/ajpheart.00911.2001. [DOI] [PubMed] [Google Scholar]
- 49.Scapagnini G, D'Agata V, Calabrese V, Pascale A, Colombrita C, Alkon D, Cavallaro S. Gene expression profiles of heme oxygenase isoforms in the rat brain. Brain Res. 2002;954:51–59. doi: 10.1016/s0006-8993(02)03338-3. [DOI] [PubMed] [Google Scholar]
- 50.Simandle SA, Kerr BA, Lacza Z, Eckman DM, Busija DW, Bari F. Piglet pial arteries respond to N-methyl-d-aspartate in vivo but not in vitro. Microvasc Res. 2005;70:76–83. doi: 10.1016/j.mvr.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 51.Snyder SH, Ferris CD. Novel neurotransmitters and their neuropsychiatric relevance. Am J Psychiatry. 2000;157:1738–1751. doi: 10.1176/appi.ajp.157.11.1738. [DOI] [PubMed] [Google Scholar]
- 52.Tcheranova D, Basuroy S, Parfenova H, Leffler CW. Regulation of CO production by glutamate in cultured astrocytes from cerebral cortex. FASEB J. 2006;20:A292. [Google Scholar]
- 53.Willis AP, Leffler CW. NO and prostanoids: age dependence of hypercapnia- and histamine-induced dilations of pig pial arterioles. Am J Physiol. 1999;277:H299–H307. doi: 10.1152/ajpheart.1999.277.1.H299. [DOI] [PubMed] [Google Scholar]
- 54.Xi Q, Tcheranova D, Parfenova H, Horowitz B, Leffler CW, Jaggar JH. Carbon monoxide activates KCa channels in newborn arteriole smooth muscle cells by increasing apparent Ca2+ sensitivity of alpha-subunits. Am J Physiol. 2004;286:H610–H618. doi: 10.1152/ajpheart.00782.2003. [DOI] [PubMed] [Google Scholar]
- 55.Xu H, Koenig HM, Ye S, Feinstein DL, Pelligrino DA. Influence of the glia limitans on pial arteriolar relaxation in the rat. Am J Physiol. 2004;287:H331–H339. doi: 10.1152/ajpheart.00831.2003. [DOI] [PubMed] [Google Scholar]
- 56.Xu HL, Ye S, Baughman VL, Feinstein DL, Pelligrino DA. The role of glia limitans in ADP-induced pial arteriolar relaxation in intact and ovariectomized female rats. Am J Physiol. 2005;288:H382–H388. doi: 10.1152/ajpheart.00727.2004. [DOI] [PubMed] [Google Scholar]
- 57.Yang G, Iadecola C. Activation of cerebellar climbing fibers increases cerebral blood flow: role of glutamate receptors, nitric oxide, and cGMP. Stroke. 1998;29:499–508. doi: 10.1161/01.str.29.2.499. [DOI] [PubMed] [Google Scholar]