

Abstract
Open reading frame 45 (ORF45) of Kaposi's sarcoma-associated herpesvirus (KSHV) encodes an immediate-early protein. This protein is also present in virions as a tegument protein. ORF45 protein interacts with interferon regulatory factor 7 (IRF-7) and inhibits virus-induced type I interferon production by blocking activation of IRF-7. To define further the function of ORF45 and the mechanism underlying its action, we constructed an ORF45-null recombinant virus genome (BAC-stop45) by using a bacterial artificial chromosome (BAC) system. Stable 293T cells carrying the BAC36 (wild type) and BAC-stop45 genomes were generated. When monolayers of 293T BAC36 and 293T BAC-stop45 cells were induced with 12-O-tetradecanoylphorbol-13-acetate and sodium butyrate, no significant difference was found between them in overall viral gene expression and lytic DNA replication, but induced 293T BAC-stop45 cells released 10-fold fewer virions to the medium than did 293T BAC36 cells. When ORF45-null virus was used to infect cells, lower infectivity was observed than for wild-type BAC36. These results suggest that KSHV ORF45 plays roles in both early and late stages of viral infection, probably in viral ingress and egress.
Kaposi's sarcoma-associated herpesvirus (KSHV), also known as human herpesvirus 8, is a human DNA tumor virus (7, 27). It is associated with the endothelial neoplasm Kaposi's sarcoma (KS), as well as the B-cell lymphoproliferative disorders primary effusion lymphoma and multicentric Castleman disease (9, 10). Like all herpesviruses, KSHV displays two alternative life cycles, latent and lytic. During latent infection, the viral genome is maintained as an episome, and only a few viral genes are expressed. Under appropriate conditions, latent genomes can be activated to express a full panel of viral genes, which leads to release of progeny virus particles. In KS lesions, most spindle cells of endothelial origin are latently infected with KSHV, but in a small percentage of the cells, viruses spontaneously undergo lytic replication. Several observations suggest that the lytic life cycle of KSHV is crucial for KS development. For example, the antiviral drugs that specifically block herpesviral lytic replication dramatically reduce the incidence of KS development in high-risk individuals (24). Lytic infection of KSHV helps formation of KS lesions by facilitating virus spread to the target sites and expressing paracrine factors (encoded by viral lytic genes) to support the growth of KS tumor cells (2, 5, 10). Recent data also showed that KSHV episomes in latently infected cells are unstable and can be rapidly lost as infected cells proliferate. KSHV lytic replication and constant infection of fresh cells are therefore essential to maintaining the population of infected cells and critical for viral pathogenesis (14, 35).
Because the lytic cycle of KSHV plays critical roles in viral pathogenesis, we sought to identify viral factors that control viral infection and lytic replication. For this purpose, we identified and characterized immediate-early genes of KSHV, including open reading frame 50 (ORF50) (replication and transcription activator [RTA]), K8 (KbZIP), ORF45, K4.2, and others (31, 39). Recently, we also identified virion proteins of KSHV by a systematic proteomic approach (38). We believe that immediate-early proteins and some virion proteins, especially tegument proteins, might have regulatory roles in viral infection and lytic replication because they exert their functions at a very early stage in viral infection and reactivation. One viral protein, ORF45, was found to be expressed as an immediate-early protein and also to be present in KSHV virions as a tegument protein.
ORF45 is unique to gammaherpesviruses and has no homologue in the alpha- or betaherpesviruses. The homology of this protein among members of the Gammaherpesvirinae subfamily is very limited and restricted mostly to the very ends of the N and C termini. The length of the protein also differs dramatically in different members of the subfamily. KSHV ORF45 is 407 amino acids (aa) in length, whereas its counterparts in Epstein-Barr virus (EBV), rhesus rhadinovirus (RRV), and murine herpesvirus 68 (MHV-68) are only 217 aa, 353 aa, and 206 aa long, respectively. ORF45 homologues have been identified as virion protein components in all gammaherpesviruses characterized so far, including KSHV, EBV, RRV, and MHV-68 (4, 18, 29, 38, 41). Studies with MHV-68 have shown that disruption of ORF45 expression by RNA interference or targeted mutagenesis in the viral genome inhibits MHV-68 viral replication (16, 17). ORF45-deficient MHV-68 also has a defect in primary infection and is not able to express early lytic genes (16). KSHV ORF45 has been shown to be able to complement the defect of ORF45-null MHV-68 (16), so ORF45 analogues in different gammaherpesviruses may share a common function even though the ORF45 function in the viral life cycle has not been identified.
Previously, we demonstrated that KSHV ORF45 interacts with interferon regulatory factor 7 (IRF-7), a master regulator of type I interferon (IFN) expression (15, 40). As a result, ORF45 efficiently suppresses virus-mediated IFN gene expression and may have a role in viral immune evasion (40). However, the ORF45 counterparts of EBV and RRV were found neither to interact with IRF-7 nor to inhibit IRF-7-mediated transcription of IFN promoters (our unpublished data), suggesting that interaction with IRF-7 and inhibition of its activation in response to viral infection are probably functions unique to KSHV ORF45.
To study the role of KSHV ORF45 in the viral life cycle, we generated an ORF45-null recombinant virus with bacterial artificial chromosome (BAC) and recombineering technology (21, 37). Our results show that disruption of ORF45 causes a decrease in yield of progeny viruses and reduced infectivity. These results demonstrate that ORF45 has important roles at both early and late stages of the viral lytic life cycle.
MATERIALS AND METHODS
Cells, Escherichia coli strains, and chemicals.
BCBL-1 cells, a primary effusion lymphoma cell line latently infected with KSHV, were cultured in RPMI 1640 medium with 10% heat-inactivated fetal bovine serum (FBS) and antibiotics. Human embryonic kidney (HEK) 293T cells were obtained from ATCC. Human foreskin fibroblasts (HFF) (HFF2441) were kindly provided by Meenhard Herlyn at the Wistar Institute. Both 293T cells and HFF were cultured in Dulbecco's modified Eagle's medium supplemented with 10% FBS, 2 mM l-glutamine, and antibiotics. E. coli strain EL350 was obtained from Neal Copeland and Nancy Jenkins at NCI-Frederick (21). BAC36, which carries the entire KSHV genome, has been described previously (37). 12-O-Tetradecanoylphorbol-13-acetate (TPA), sodium butyrate, and Polybrene were purchased from Sigma (St. Louis, MO). Hygromycin was purchased from Roche (Indianapolis, IN). Turbo DNase I was obtained from Ambion (Austin, TX).
Genetic manipulation of KSHV BAC genome.
Mutagenesis of BAC36 was performed by using a recombineering system (http://recombineering.ncifcrf.gov). BAC36 was first introduced into EL350 by electroporation. The EL350 strain contains a defective λ prophage that harbors the recombination genes exo, beta, and gam under tight control of a temperature-sensitive cl857 repressor. Recombination functions can be supplied transiently by transfer of the culture to 42°C for 15 min (21). To generate an ORF45 deletion mutant, we replaced the ORF45 coding sequence of BAC36 with a kanamycin (Kan)/SacB cassette by homologous recombination. The Kan/SacB cassette was amplified from plasmid pBS-Kan/SacB by PCR with primers ORF45-Kan/SacB-5′ (5′-CCTAGCGGTCAACCCCGTACAAGGCCATGGCGATGTTTGTGAGGACCTCGGCATGCGACGTCCACATATAC-3′)and ORF45-Kan/SacB-3′ (5′-ATGAGACTTGACACCTATAATGGTCTGTATTGACACCATTCTTTTATTTACTACCGCACAGA TGCGTAAGG-3′). Each primer contains 21 nucleotides (nt) homologous to the antibiotic resistance cassette Kan/SacB at its 3′ end and 50 nt homologous to the first 50 nt next to the start or stop codons of ORF45 at the 5′ end. These two primers were used to amplify the Kan/SacB cassette, which contains the kanamycin resistance gene and SacB, by PCR. PCR was carried out at 94°C for 30 s, 60°C for 30 s, and 72°C for 2 min for 30 cycles with Roche Expand high-fidelity Taq polymerase. The PCR product was treated with DpnI to remove the plasmid template. The digested product was gel purified with a QIAEX II gel extraction kit (QIAGEN). The purified PCR fragment was electroporated into BAC36-containing EL350 cells that had been induced at 42°C for 15 min. The parameters for electroporation were set at 1.75 kV, 276 Ω, and 50 μF in a 0.1-mm cuvette (BTX). The recombinant clones were selected at 32°C on LB plates containing 12.5 μg of chloramphenicol and 50 μg of kanamycin per ml and then characterized by PCR and Southern blot analyses. The sensitivity to sucrose (Suc) was tested by plating the same amount of overnight culture on plates containing LB plus Kan and plates containing LB plus 7% sucrose. The resultant BAC was designated BAC-del45.
To make a revertant mutant, we replaced the Kan/SacB cassette in BAC-del45 with a wild-type ORF45 sequence by a homologous recombination strategy similar to that described above. The wild-type ORF45 sequence was amplified by PCR, with pEBN9 plasmid as a template and oligonucleotides ORF45wt5′ (5′-TTTCCGCCCCTAGCGGTCAACCCCGTACAAGGCCATGGCGATGTTTGTG AGGACCTCGTCTAGCACACACGATGAAGA-3′) and ORF45wt3′ (5′-CTGATGTGTTTGGGAATAAAGCATGAGACTTGACACCTATAATGGTCTGTA TTGACACCATTCTTTTATTTA-3′) as primers. pEBN9 is a pBluescript-derived plasmid in which the KSHV genomic sequence between nucleotides 66444 and 69094 was cloned at the EcoRI and BamHI sites. In addition, a mutant containing a premature stop codon was constructed. The Kan/SacB cassette in BAC-del45 was replaced with an ORF45 sequence-of-stop-codon mutant, which was generated by PCR with primers ORF45stop5′ (5′-TTTCCGCCCCTAGCGGTCAACCCCGTACAAGGCCATGGCGATGTTTGTGAGGACCtagTCTAGCACAC ACGATGAAGA-3′) and ORF45wt3′. The lowercase letters “tag” represent a single point mutation which is introduced at the eighth codon and replaces the TCG codon with a stop codon.
About 200 ng of gel-purified PCR fragments was electroporated into induced EL350 cells carrying BAC-del45. The transformants were selected at 32°C on LB plates containing 12.5 μg/ml of chloramphenicol and 7% sucrose. Because of frequent undesired recombination, many of the Kans/Sucr colonies did not have the expected recombination. To identify the clones that did, we performed in situ colony hybridization with the wild-type ORF45 coding sequence as a probe. The positive clones were expanded, and the BAC DNAs were extracted and analyzed by PCR and Southern blot analyses. All BAC clones were also sequenced with primer ORF45up (5′-CCAACGACTATTTGACTCGCC-3′). The BAC DNAs with the proper recombination were prepared from overnight cultures with a large-construct kit (QIAGEN).
Reconstitution of recombinant viruses.
The freshly prepared BAC DNAs were introduced into 293T cells by means of a QIAGEN Effectene transfection kit. In brief, 1 μg of BAC DNA was used to transfect 293T cells of 40 to 60% confluence in a 60-mm dish. Two days after transfection, the cells were examined by fluorescence microscopy and then subcultured in T150 flasks with fresh medium containing 200 μg/ml hygromycin. When the colonies were visible (usually 10 to 14 days after transfection), the cell colonies were dislodged and seeded into a new T150 flask. When the monolayer reached 80 to 90% confluence, cells from each flask were split into three new T150 flasks. Three days later, cells were split again and induced with 20 ng/ml TPA and 0.3 mM sodium butyrate. Usually, 20 or more T150 flasks (150 cm2) of cells were induced for 4 to 5 days, and released virion particles were purified from the supernatant. The induced culture media were collected and filtered through 0.45-μm filters. The virions were then pelleted by ultracentrifugation on a 25% sucrose cushion at 100,000 × g for 1 h with a Beckman SW28 rotor. The pellets were dissolved in 1% of the original volume of 1× phosphate-buffered saline (PBS) or Dulbecco's modified Eagle's medium and stored at −80°C.
Preparation of genomic and virion DNAs.
Total DNAs were prepared from viral stocks or cells with a DNeasy tissue kit (QIAGEN). Monolayers of infected cells in 60-mm dishes were trypsinized, washed, and resuspended in 200 μl of 1× PBS. Total DNA was prepared according to the manufacturer's instructions. For the preparation of DNA from intact virions, 200 μl of virus stocks was pretreated with 10 μl of Turbo DNase I (Ambion) for 1 h at 37°C. The reaction was stopped by addition of EDTA followed by heat inactivation at 70°C. Then, 20 μl of proteinase K solution and 200 μl of buffer ALfrom the DNeasy kit (QIAGEN) were added. The mixture was kept at 70°C for 15 min and then extracted with phenol-chloroform. The DNA was ethanol precipitated with glycogen, and the DNA pellet was dissolved in 40 μl of Tris-EDTA buffer.
Quantification of viral genomic DNA and virion proteins.
Copy numbers of KSHV genomic DNA in viral stocks and in the infected cells were estimated by real-time DNA PCR. Briefly, DNA was prepared with a DNeasy tissue kit (QIAGEN), and DNA samples were subjected to real-time DNA PCR with a Roche LightCycler and a LightCycler FastStart DNA MasterPlus SYBR green kit. Viral DNA copy numbers were calculated from external standards of known concentrations of BAC36 DNA. The primers ORF73-LCN(5′-CGCGAATACCGCTATGTACTCA-3′) and ORF73-LCC (5′-GGAACGCGCCTCATACGA-3′) were previously described by Krishnan et al. (20).
Virion proteins in the preparations were analyzed by Western blotting. In brief, virus stocks were diluted to 200 μl of PBS, and then equal volumes of 20% polyethylene glycol (PEG) 6000 were added (final concentration of PEG was 10%). The mixtures were vortexed thoroughly and spun at 13,000 × g for 10 min. The resulting pellets of viral particles were dissolved in sodium dodecyl sulfate (SDS) loading buffer and analyzed by Western blotting with antibodies against ORF45, ORF21 (thymidine kinase [TK]), and ORF65 (small capsomer-interacting protein [SCIP])(38).
Western blotting.
Cells were washed with 1× PBS and lysed with cell lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 1 mM sodium orthovanadate [Na3VO4], 20 mM sodium pyrophosphate, 100 mM sodium fluoride, 10% glycerol, 1 mM EDTA, 5 μg/ml of aprotinin, 5 μg/ml of leupeptin, 5 mM benzamidine, and 1 mM phenylmethylsulfonyl fluoride). The cell lysates were homogenized and centrifuged at 13,000 rpm for 5 min at 4°C. Fifty micrograms of whole-cell extracts was resolved by SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. The membranes were blocked in 5% dried milk in 1× PBS plus 0.2% Tween 20 and then incubated with diluted primary antibodies for 2 h at room temperature or overnight at 4°C. Anti-rabbit or anti-mouse immunoglobulin G antibodies conjugated to horseradish peroxidase (Amersham) were used as the secondary antibodies. An enhanced chemiluminescence system (Amersham) was used for detection of antibody-antigen complexes.
Gardella gel assay.
Approximately 5 × 105 293T BAC36 cells were collected 3 days after induction for lytic KSHV replication. The cells were pelleted and resuspended in 100 μl of sample buffer A, containing 15% Ficoll, 40 μg/ml RNase A, and 0.01% bromophenol blue in 1× Tris-borate-EDTA, and loaded into 3-mm by 10-mm slots of a 160-mm-long by 200-mm-wide by 3-mm-thick vertical 0.8% agarose gel. A total of 100 μl of lysis buffer, containing 5% Ficoll, 1% SDS, 1 mg/ml pronase, and 0.05% xylene cyanol, was carefully overlaid on the cell suspension, and the gel was electrophoresed at 0.8 V/cm for 3 h and then at 5 V/cm for 12 h at 4°C with 1× Tris-borate-EDTA running buffer. After electrophoresis, the gel was treated with 0.25 M HCl for 15 min and then with 0.5 M NaOH-1.5 M NaCl and finally neutralized with 0.5 M Tris-HCl (pH 7.5)-1.5 M NaCl. The DNA was transferred with 10× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) onto a Nytran membrane and hybridized with a [32P]-labeled KSHV-specific probe.
Infection.
293T cells plated in 24-well plates were incubated with concentrated virus plus Polybrene (4 μg/ml) and spun at 2,500 rpm for 1 h at room temperature. The inocula were then removed and replaced with fresh media with 5% FBS. The following day, the media were replaced with fresh media containing 1% FBS. Green fluorescent protein (GFP) expression was used to monitor infection 2 days after infection. The cells were then split into 100-mm dishes and cultured in media with 200 μg/ml hygromycin. Colonies were scored 10 or 14 days after infection.
Real-time reverse transcription-PCR analysis.
HFF in six-well plates were infected with viruses at 50 genomes per cell. Six hours later, cells were lysed with TRIzol reagent (Invitrogen), and total RNAs were isolated according to the protocol provided by the manufacturer. Residual DNA contamination was eliminated with Turbo DNase I (Ambion). cDNA was generated from total RNA with a SuperScript first-strand synthesis system (Invitrogen), primed with random hexamers. The cDNA samples were used for real-time PCR with a FastStart DNA MasterPlus SYBR green kit and gene-specific primers. The primers were ORF73-LCN (5′-CGCGAATACCGCTATGTACTCA-3′), ORF73-LCC (5′-GGAACGCGCCTCATACGA-3′), ORF45+ (5′-GGGATGGGTTAGTCAGGATG-3′), ORF45− (5′-CCTCGTCGTCTGAAGGTGA-3′), K5+ (5′-TGAACTGTTTCTGCTGATGTCTG-3′), K5− (5′-AGCGTCCAGGTGCACAAC-3′), ORF21+ (5′-CTGGTGCGTCTTTGATAGGC-3′), ORF21− (5′-AGGTGCATGAGAGGGAACAC-3′), ORF50+ (5′-CGCAATGCGTTACGTTGTTG-3′), ORF50− (5′-GCCCGGACTGTTGAATCG-3′), GAPDH+ (5′-AGCCACATCGCTCAGACAC-3′), and GAPDH− (5′-GCCCAATACGACCAAATCC-3′). The amounts of mRNA were determined by comparison with the standard templates of cloned cDNAs of known copy number. The expression levels were then normalized with GAPDH (glyceraldehyde-3-phosphate dehydrogenase).
RESULTS
Generation of ORF45-null recombinant KSHV.
To investigate the roles of ORF45 in the viral lytic life cycle, we decided to construct an ORF45-null KSHV recombinant virus. Cloning of the KSHV genome as a BAC has facilitated genetic manipulation of the KSHV genome in E. coli. A BAC-cloned KSHV (BAC36) was constructed and reported previously (37). BAC36 carries the full KSHV genome, and infectious KSHV can be reconstituted from it by transfection of BAC36 DNAs into 293 cells (37). BAC36 contains GFP and hygromycin resistance gene markers, allowing easy detection of eukaryotic cells containing the KSHV genomes. To generate an ORF45-null mutant virus, we employed a two-step replacement procedure (Fig. 1). In the first step, the ORF45 coding sequence in BAC36 was replaced with a Kan/SacB cassette by means of a recently developed technology known as recombineering (recombination-mediated genetic engineering). Briefly, a bacterial double selection cassette, Kan/SacB, flanked by sequences homologous to the first 50 nt after the initiation codon of ORF45 and the last 50 nt next to the stop codon of ORF45 at two ends, was synthesized by PCR and transformed into E. coli EL350 cells carrying BAC36. Induction of recombination activity in the EL350 cells at 42°C resulted in the replacement of ORF45 by the Kan/SacB cassette. Transformants were selected by kanamycin resistance (Kan+). DNAs were isolated from kanamycin-resistant colonies, digested with restriction enzymes, and analyzed on 0.8% agarose gels. Digestion of the wild-type BAC36 DNA with KpnI generated two fragments, 3.7 kb and 2.2 kb, at the ORF45 locus (Fig. 2A and B). Replacement of the ORF45 coding sequence with the Kan/SacB cassette shifted the two fragment sizes to 4.3 kb and 3.4 kb. Digestion of BAC36 DNA with BamHI yielded a 3.3-kb double band at the ORF45 locus. A unique band of 5.1 kb was detected in the ORF45 deletion clones, as expected. To confirm further that the altered digestion pattern of the BAC mutants was the result of the expected recombination, we carried out Southern blot hybridizations. The restriction-digested DNAs were transferred onto nylon membranes and then probed with 32P-labeled Kan/SacB DNA and ORF45 coding sequence, respectively. When probed with ORF45 coding sequence, two KpnI fragments of 2.2 kb and 3.7 kb and a BamHI fragment of 3.3 kb were detected only in wild-type BAC36 and not in the BAC mutant. When the same blot was probed with Kan/SacB cassette DNA, the signals were seen only in the deletion mutant BAC. The KpnI-restricted mutant BAC showed distinct signals of 3.4 kb and 4.3 kb, and the BamHI-restricted mutant had a signal of 5.1 kb (Fig. 2B). The result confirmed that the ORF45 coding sequence was successfully replaced with a Kan/SacB cassette. The mutant was designated BAC-del45.
FIG. 1.

Strategy for constructing ORF45-null and revertant recombinant viruses. ORF45-null virus was constructed in a two-step procedure. In the first step, a 3.0-kb Kan/SacB cassette flanked by ORF45 sequence (50 bp at each end) was PCR amplified and electroporated into heat-induced EL350 cells harboring BAC36. Recombinants were selected with chloramphenicol and kanamycin. An ORF45 deletion virus (BAC-del45) was generated (left panel). In the second step, PCR fragments of wild-type ORF45 (middle panel) and an ORF45 sequence with a premature stop codon at the eighth amino acid (right panel) were electroporated into heat-induced EL350 cells harboring BAC-del45. The transformants were plated on LB plates containing chloramphenicol and 7% sucrose. Colonies were then subjected to in situ colony hybridization with the ORF45 coding sequence as a probe. An ORF45 and a revertant virus were generated under this strategy.
FIG. 2.

Construction and analyses of ORF45 mutant BACs. (A) Schematic diagrams of the structures of ORF45 and its neighboring ORFs in the wild-type or mutant BACs. The nucleotide sequences refer to GenBank accession number U75698. (B) Agarose gel electrophoresis and Southern analysis of wild-type (BAC36) and ORF45 deletion mutant (BAC-del45) DNAs. BAC36 (lanes 1 and 3) and BAC-del45 (lanes 2 and 4) DNAs were digested with KpnI (lanes 1 and 2) or BamHI (lanes 3 and 4). The digested DNAs were electrophoresed, blotted, and hybridized with a [32P]dCTP-labeled ORF45 fragment (middle panel) or a Kan/SacB fragment as indicated. (C) Agarose gel electrophoresis of restricted DNAs of BAC36 (lane 1), BAC-del45 (lane 2), BAC-res45 (lane 3), and BAC-stop45 (lane 4). The BAC DNAs were digested with KpnI and NotI. The restricted DNAs were resolved on an 0.8% agarose gel and stained with ethidium bromide. TR, terminal repeat. (D) Sequences of the wild-type and mutant BAC DNAs at the ORF45 locus. The BAC DNAs were purified with a QIAGEN large-construct kit and subjected to automatic DNA sequencing. The sequence chromatograms and deduced amino acids of ORF45 are shown. The sequences of the Kan/SacB cassette and the single point mutation from TCG to stop codon TAG are boxed.
In the second step, we generated a rescued revertant virus and a stop codon mutant. A PCR fragment of the wild-type ORF45 sequence was introduced into BAC-del45-bearing EL350 cells. A desired recombination should replace the Kan/SacB cassette in BAC-del45 with a wild-type ORF45 sequence. The Kan/SacB cassette has a double selection property, permitting both positive and negative selection. SacB, originally from Bacillus subtilis, encodes an enzyme that converts sucrose to levan, which is toxic to bacteria and causes cell death (12). Bacteria that express SacB are killed in medium containing sucrose but grow normally in medium without it. The revertant mutants can thus be screened for both sucrose-resistant and kanamycin-sensitive phenotypes. However, our initial experiment showed that the majority of Suc+/Kan− colonies had lost the cassette and a large portion of the viral genome, presumably because of the repeat sequences in the viral genome (data not shown). To solve this problem, we performed in situ colony hybridization using the ORF45 coding sequence as a probe to identify the clones with a proper recombination. Positive clones were verified by restriction enzyme digestion and Southern blot analysis. The clone that displayed the expected pattern was designated BAC-res45. Similarly, a mutant that carries a premature stop codon was also constructed by the same strategy and designated BAC-stop45. All of these BAC DNAs, including BAC36 (wild type), BAC-del45, BAC-res45, and BAC-stop45, were digested with KpnI and NotI. The restriction digestion patterns of these BAC constructs are shown in Fig. 2C. In addition, purified DNAs of these BAC constructs were sequenced. The sequencing data confirmed that BAC-res45 has exactly the same sequence around the recombination junction as does wild-type BAC36, that BAC-del45 lacks the ORF45 coding region after the eighth codon, and that BAC-stop45 carries a TCG-to-TAG single codon mutation at the eighth codon (Fig. 2D).
Reconstitution of BAC-cloned KSHV and the derived mutants in 293T cells.
To reconstitute recombinant viruses, we transfected 293T cells with BAC36 and the mutant BAC DNAs. The presence of hygromycin resistance and GFP markers in the BAC36 genomes enabled us to monitor the transfection efficiencies by GFP detection and to enrich transfected cells by hygromycin selection. Because KSHV typically establishes latent infection by default, the cells harboring the BAC genomes can be enriched and maintained in medium containing hygromycin. When almost 100% of cells became positive for GFP expression after selection (Fig. 3), the cells were treated with TPA and butyrate to induce viral lytic replication. Whole-cell lysates were prepared from induced and uninduced cells and analyzed for expression of viral genes of different kinetic categories by Western blot analysis using antibodies against various KSHV gene products. ORF45 was not detected in 293T BAC-stop45 cells (Fig. 4) or 293T BAC-del45 cells (data not shown), whereas it could be detected easily in 293T BAC36 cells (Fig. 4) and 293T BAC-res45 cells (data not shown). Second, the Western analyses detected no significant difference between wild-type KSHV (BAC36 and BAC-res45) and ORF45-null virus (BAC-stop45) in expression of latency-associated nuclear antigen (LANA), ORF50 (RTA), K8, or ORF59 (polymerase processivity factor [PPF]). This suggests that the absence of ORF45 expression in a mutant KSHV appears to have no effect on latent (LANA), immediate-early (RTA), or delayed-early (K8 and PPF) gene expression. Furthermore, the study also showed that a late gene product, ORF64, which encodes a large tegument protein, was expressed in BAC-stop45 at a level similar to that in BAC36, suggesting that the lack of ORF45 had no effect on late gene expression (Fig. 4).
FIG. 3.

Transfection of 293T cells with BAC36 and BAC-stop45 DNAs. Cells were transfected with BAC36 (A and C) and BAC-stop45 (B and D) DNAs with an Effectene transfection kit. GFP expression levels were monitored by fluorescent microscopy 2 days after transfection (A and B). Then, the transfected cells were split and selected with hygromycin. The hygromycin-resistant cells were pooled and passed three to four times. The cells were examined for GFP expression (C and D).
FIG. 4.
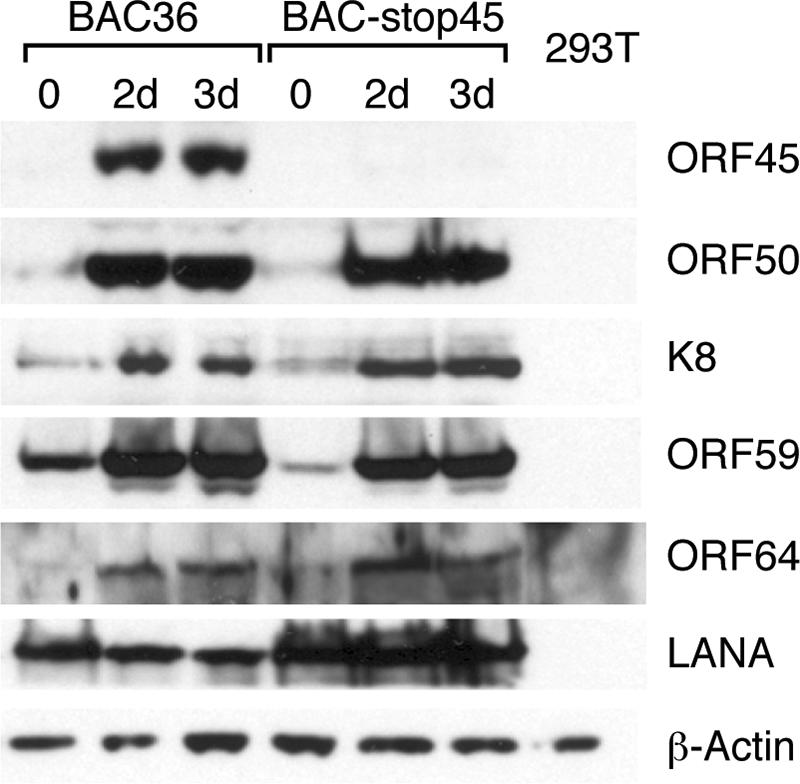
Comparison of levels of viral gene expression of BAC36 and BAC-stop45 in 293T cells. The pooled hygromycin-resistant 293T BAC36 and 293T BAC-stop45 cells were induced with TPA and sodium butyrate for 0 days, 2 days (2d), and 3 days (3d). The induced cells as well as KSHV-free parental 293T cells were lysed, and the whole-cell lysates were immunoblotted with the indicated antibodies against viral proteins of different kinetic categories. The same blot was also reprobed with anti-β-actin antibody to ensure equal loading of each sample.
Viral lytic DNA replication is not affected by ORF45 deficiency.
Next, we tested for an effect on viral lytic DNA replication in ORF45-null KSHV mutants. We carried out a Gardella gel assay to assess the impact of ORF45 deficiency on viral lytic DNA replication. This assay uses a native vertical agarose gel electrophoresis that can distinguish between linear viral DNA, presumably the result of viral DNA replication, and circular KSHV DNA, representing the episomal version of the viral DNA (11). After TPA and butyrate induction, dramatic increases of the replicating (linear) form of viral DNA were seen with both wild-type BAC36 and rescued BAC-res45. A similar level of replication was detected in BAC-stop45 (Fig. 5). In contrast, the level of viral lytic replication (linear viral DNA) in BAC-del45 was found to be significantly lower than those of the wild type and BAC-stop45. In addition, the levels of intracellular viral DNA in induced cells harboring BAC36, BAC-res45, and BAC-stop45 were measured by a real-time PCR analysis, and no significant differences were detected (data not shown). These data suggest that viral lytic DNA replication is not significantly affected by the absence of ORF45; the lack of lytic replication in BAC-del45 might be due to a large deletion of the ORF45 coding sequence, which affects the expression of neighboring genes. BAC-stop45 therefore represents a better ORF45-null mutant.
FIG. 5.

Gardella gel analysis of viral lytic DNA replication. The pooled hygromycin-resistant cells were induced with TPA for 3 days. Approximately 5 × 105 of TPA-induced (+) and uninduced (−) BAC-containing 293T cells were loaded on a 0.8% vertical agarose gel. Approximately 5 × 104 TPA-induced and uninduced BCBL-1 cells were included as positive controls. Samples were overlaid with lysis buffer and electrophoresed initially at 0.8 V/cm for 3 h and then at 4.5 V/cm for 12 h at 4°C. Gels were subsequently transferred onto nylon membranes and probed with a [32P]-labeled DNA fragment of the LANA coding sequence. The positions corresponding to the circular DNA form and the linear replicating form of KSHV are indicated.
ORF45-null mutant produces significantly fewer progeny viruses.
The pooled hygromycin-resistant and GFP-positive cells were induced with TPA and butyrate for 4 days. Viral particles were collected from the culture media, passed through 0.45-μm filters, and concentrated by ultracentrifugation over a 25% sucrose cushion. Because a typical plaque assay is not available for KSHV, we estimated the amounts of virus by quantifying encapsidated viral DNA and virion proteins in preparations.
We first treated the concentrated viruses with Turbo DNase I (Ambion) at 37°C for 1 h to remove any contaminating DNA outside viral particles. The viral DNAs were extracted and analyzed with real-time PCR. From a standard curve with known amounts of BAC36 DNA, the copy numbers of viral genomes in induced media were estimated. For both wild-type BAC36 and rescued BAC-res45, the viral copy number was about 3 × 106/ml. The BAC-stop45 mutant produced about 1/10 as many viral particles as BAC36 (Fig. 6A). The viral DNAs were also analyzed by Southern blotting. The viral DNAs were digested with NotI, resolved on 0.8% agarose gels, and subjected to Southern analysis with a probe of the KSHV terminal repeat sequence (the NotI fragments of 0.8 kb) (Fig. 6B). The result was consistent with that of the real-time PCR analysis. The signals from BAC36 and BAC-res45 were about the same, whereas that of BAC-stop45 was 10-fold less than that of the wild-type viruses.
FIG. 6.

Extracellular progeny viruses produced by BAC36 and BAC-stop45. The pooled hygromycin-resistant cells were induced with TPA and butyrate for 4 days, and viruses in the supernatants were concentrated 100-fold. Virus stocks (200 μl) were treated with Turbo DNase I for 1 h at 37°C, and viral DNAs were extracted. (A) Viral DNAs were analyzed by a real-time PCR assay using primers to LANA. A serial dilution of a known amount of BAC36 DNA was used to construct a standard curve. Copy numbers were normalized and are expressed as copy number per milliliter of supernatant. (B) Viral DNAs were digested with NotI, resolved on a 0.8% agarose gel, and subjected to Southern analysis with a probe of the KSHV terminal repeat sequence. (C) Virion proteins in the preparations were analyzed by Western blotting. Samples of 15 μl of 100-fold-concentrated viruses of BAC36 (lane 1) and BAC-stop45 (lane 2) were subjected to Western blot analysis with antibodies against KSHV virion proteins as indicated. Equal numbers of virions (1 × 107) of BAC36 (lane 3) and BAC-stop45 (lane 4), based on the viral genomic copy number determined by real-time PCR, were diluted to equal volumes with 1× PBS, and viral particles were precipitated with PEG. The pellets were dissolved in SDS loading buffer and analyzed by Western blotting.
To assess the integrity of the BAC-stop45 recombinant viral particle and rule out the possibility that the BAC-stop45 preparation does not contain intact virions but only nucleocapsids from lysed cells, we compared the ratios of viral DNA to tegument proteins in the virion preparations from wild-type BAC36 and BAC-stop45 recombinant viruses. As illustrated in Fig. 6C, when the virion particles from equal volumes of induced culture media were analyzed by Western blotting for capsid protein ORF65 (SCIP) and tegument protein ORF21 (TK), significantly less virion protein was found in the BAC-stop45 preparation (Fig. 6C, left panel). However, when virion preparations corresponding to the same number of viral genomes (1 × 107), determined by real-time PCR, were analyzed, similar levels of capsid protein ORF65 and tegument protein ORF21 were detected in wild-type BAC36 and BAC-stop45 preparations by Western analysis (Fig. 6C, right panel). Thus, the copy numbers of encapsidated viral DNA proportionally represent the numbers of virion particles in preparations, and such estimates are used in our studies below.
Taken together, our data showed that ORF45 deficiency does not affect viral DNA replication but that the ORF45-null mutant released 10-fold fewer viral particles to the media, suggesting a possible role of ORF45 in virion assembly or viral egress.
Infectivity of ORF45-null recombinant virus.
In our study of the infectivity of the ORF45-null virus, the concentrated BAC-stop45 viral particles as well as wild-type viruses (BAC36 and BAC-res45) were used to infect 293T cells at 50 genomes per cell. The wild-type viruses were highly infectious, as revealed by the appearance of green cells upon inoculation of fresh 293T cells (Fig. 7). BAC-stop45 produced significantly fewer green cells (on average, 40-fold fewer) than BAC36 and BAC-res45. The infected cells were subcultured in 100-mm dishes with fresh media containing 200 μg/ml hygromycin. After 2 weeks of selection, colonies representing an infection unit were scored. Cells infected with BAC36 produced hundreds of colonies, whereas cells infected with BAC-stop45 produced only three colonies (Fig. 7B). These results suggest that ORF45-null virus is less infectious than wild-type viruses.
FIG. 7.

Infectivity of reconstituted BAC36 and BAC-stop45 viruses. (A) 293T cells plated in 24-well plates were incubated with concentrated virus (50 genome copies per cell) plus Polybrene (4 μg/ml) and spun at 2,500 rpm for 1 h at room temperature. The inocula were then removed and replaced with fresh media plus 5% FBS. The following day, the media were replaced with fresh media containing 1% FBS. GFP expression was examined under a fluorescent microscope 2 days after infection. (B) The cells were then split and cultured with media with 200 μg/ml hygromycin. Colonies were stained with 0.1% crystal violet after 10 days.
Next, we investigated viral gene expression of the ORF45-null virus during primary infection. HFF were infected with BAC-stop45 as well as with wild-type viruses at 50 viral DNA copies per cell. Six hours after infection, total RNAs were isolated, converted to cDNAs, and analyzed by real-time reverse transcription-PCR for viral transcripts. In BAC36 virus, transcriptions of ORF50, K5, ORF21, and LANA were easily detected (Fig. 8). This result is consistent with a previous report by Chandran's group that concurrent expression of latent and lytic genes occurs during KSHV primary infection (20). In contrast, in the cells infected with BAC-stop45, the expression levels of ORF50, K5, ORF21, and LANA were found to be significantly lower (Fig. 8). These results suggest that deficiency of ORF45 causes a defect in the early stage of KSHV de novo infection.
FIG. 8.

Viral gene transcription of BAC36 and BAC-stop45 viruses after a de novo infection. HFF were infected with BAC36 and BAC-stop45 viruses (50 genome copies per cell). Six hours after infection, total RNAs were extracted, treated with DNase I, and reverse transcribed to cDNAs. Real-time PCRs were performed with specific primers to each viral gene as indicated. The level of expression was normalized with GAPDH.
DISCUSSION
In this report, we have generated an ORF45-null KSHV with a BAC system. Using recombineering technology, we first generated a deletion mutant by replacing the entire coding sequence of ORF45 with a Kan/SacB selection cassette. The deletion mutant had a defect in viral lytic DNA replication. Because the neighboring ORF44 (helicase, HEL) and ORF46 (uracil DNA glucosidase, UDG) are only 93 bp and 63 bp away from ORF45, respectively, the deletion of the entire ORF45 coding region may affect the expression of the neighboring genes. The defect of BAC-del45 might therefore be caused by loss of function of the neighboring genes. To avoid this problem, we designed and generated a stop codon mutant (BAC-stop45), in which a translation stop codon was introduced into the coding region of ORF45 near the N terminus. In BAC-stop45, the overall viral gene expression appears not to be altered, except for ORF45, during reactivation induced by TPA and butyrate. The viral lytic replication during reactivation also seems normal in comparison to that of wild-type virus (BAC36), but even though the wild type and the mutant are similar in lytic DNA replication efficiencies and intracellular viral DNA levels, BAC-stop45 produced 10-fold fewer extracellular virions than did BAC36. This result suggests a function of ORF45 in the late stage of viral lytic replication, probably in viral maturation and egress. When equal amounts of wild-type and mutant viruses were used to infect 293T cells, BAC-stop45 displayed lower infectivity, as demonstrated by production of 40-fold fewer green cells upon infection than were produced by wild-type BAC36. In the cells infected with mutant BAC-stop45, viral gene expression, either latent or lytic, was ablated. These results suggest that ORF45 also has a function in the early stage of de novo infection.
ORF45 is a relatively abundant tegument protein in KSHV virions (38, 41). The functions of tegument proteins in gammaherpesviruses have not been well studied, and current knowledge of tegument proteins comes mainly from studies of alpha- and betaherpesviruses. In general, tegument proteins have three essential functions in the viral life cycle. First, they are involved in modulation of the host cellular environment during the immediate-early phase of infection. For example, herpes simplex virus type 1 (HSV-1) VP16 (UL48) is transported to the nucleus and acts as a transcriptional activator for viral immediate-early genes (3). The HSV-1 virion host shutoff protein (UL41) is known to degrade host mRNA and shut down the host translation program (32). Cytomegalovirus abundant tegument protein pp65 is able to suppress a subset of interferon-stimulated genes during infection (1, 6). Second, some tegument proteins play roles in transport of capsids to the nucleus after the virus enters a host cell. Incoming capsids and associated tegument proteins are believed to be transported to the nucleus along microtubules (33). It has been demonstrated that targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins (23). Recent in vitro studies showed that the inner tegument promotes HSV capsid motility along microtubules (22, 36). Third, tegument proteins participate in virion assembly and egress. The assembly and egress of herpesviruses are very complex, following envelopment-deenvelopment-reenvelopment processes (25, 26). The newly synthesized viral DNA is incorporated into preformed capsids, and the nucleocapsids leave the nucleus by first budding through the inner nuclear membrane, formatting primary enveloped virions in the perinuclear space. The primary envelope then fuses with the outer leaflet of the nuclear membrane, thereby releasing nucleocapsids into the cytoplasm. Final envelopment, including the acquisition of many tegument and envelope glycoproteins, occurs by budding into Golgi body-derived vesicles. Mature virions are released after fusion of the vesicle membrane with the plasma membrane of the cell. Tegument proteins are known to be involved in various steps of viral assembly and egress progress. Disruption of the major tegument protein of HSV-1 UL36, which is conserved among all herpesviruses, resulted in accumulation of unenveloped DNA-filled capsids in the cytoplasm (8). Null mutation of HSV-1 UL48 (VP16) interferes with viral egress downstream of the primary envelopment, presumably by impairing virus assembly in the cytoplasm (28).
Because ORF45 has no homologue in alpha- and betaherpesviruses, the mechanisms of ORF45 function in these processes cannot be inferred from those of any known tegument proteins of other herpesviruses. The data presented here and in other studies indicate that KSHV ORF45 might contribute to all three functions mentioned above.
(i) The first function is the regulatory role of ORF45 in modulation of host cell environment. Some viral tegument proteins are released into infected cells during infection, allowing the virus to initiate quick responses to hostile host antiviral defenses. ORF45 has been found to interact with cellular IRF-7 and to suppress its activation in response to viral infection (40). IRF-7 is known as the master regulator for type I IFN-dependent immune responses (15, 40). By suppressing IRF-7, ORF45 inhibits virus-induced type I IFN production and helps the virus to evade the host antiviral responses and ensure a successful infection. Using BAC-stop45, we recently demonstrated that ORF45 plays a critical role in the virus's evasion of host antiviral responses (42).
(ii) Second is the possible function of ORF45 in virion assembly and egress. Null mutation of KSHV ORF45 does not affect the level of intracellular viral DNA but causes a 10-fold decrease in the levels of extracellular virion DNA, suggesting that ORF45 plays an important role in viral maturation or egress. Similar phenotypes have been described for alphaherpesviruses. US3 is unique and seen only in the alphaherpesviruses. The US3 tegument proteins of pseudorabies virus and HSV-1 are present in both the primary virions and the mature extracellular virus particles (13, 30). Deletion of the US3 gene reduces infectivity by almost 10-fold. In the absence of US3, primary virions accumulate in the perinuclear space (19, 30, 34).
(iii) Third is the possible function of ORF45 in transport of capsid and ingress. ORF45 is required in the early stage of primary infection, as BAC-stop45 mutant virus neither expresses any of its genes (including the GFP marker) after infecting 293T cells nor establishes latency. ORF45 may play a role in delivery of the viral genome to the nucleus, where transcription takes place. Because ORF45 is tightly associated with the capsid and probably located in the inner layer of tegument (41), ORF45 may well be found to be either involved in KSHV capsid transport or required for releasing viral genomic DNA from capsid into the nucleoplasm.
A study of MHV-68 by Jia et al. demonstrated that disruption of ORF45 blocked viral immediate-early and early gene expression (16). Furthermore, KSHV ORF45 was found to be capable of partial rescue of an MHV-68 ORF45-deficient mutant, suggesting that certain functions of ORF45 are conserved among gammaherpesviruses. Although suppression of IRF-7 seems unique to KSHV, the functions involving ingress and egress are possibly conserved among gammaherpesviruses. Because of the low yield of KSHV in our system, detailed structural and morphological study with electron microscopy is still challenging. We estimated that each 293T cell produces on average only two to five viral particles. If the functions of ORF45 in ingress or egress are conserved among gammaherpesvirus, electron microscopy studies with RRV or MHV-68 ORF45-null mutants would yield useful information.
In summary, with the 293T KSHV BAC system, we demonstrated that deficiency of ORF45 did not affect overall viral gene expression or lytic DNA replication during reactivation but that 10-fold fewer extracellular progeny virions were produced by an ORF45-null mutant virus than by wild-type BAC36. The mutant progeny virus also displayed lower infectivity. These results suggest that ORF45 has important functions at both early and late stages of infection. The additive effects of defects at both the early and late stages cause infectivity at least 2 to 3 orders of magnitude lower in the ORF45-null mutant virus, supporting the hypothesis that ORF45 plays critical roles in the KSHV lytic life cycle.
Acknowledgments
We thank Nancy Jenkins and Neal Copeland at NIH for providing the recombineering system, Meenhard Herlyn at the Wistar Institute for HFF2441 cells, Erle Robertson at the University of Pennsylvania for pTR, which contains the KSHV terminal repeat sequence, and Anne Thistle at Florida State University for excellent editorial assistance. We also thank all members of the Yuan lab for critical reading of the manuscript and helpful discussion.
This work was supported by National Institutes of Health grants R01CA86839 to Y.Y. and R01DE016680 to F.Z.
Footnotes
Published ahead of print on 11 October 2006.
REFERENCES
- 1.Abate, D. A., S. Watanabe, and E. S. Mocarski. 2004. Major human cytomegalovirus structural protein pp65 (ppUL83) prevents interferon response factor 3 activation in the interferon response. J. Virol. 78:10995-11006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bais, C., A. Van Geelen, P. Eroles, A. Mutlu, C. Chiozzini, S. Dias, R. L. Silverstein, S. Rafii, and E. A. Mesri. 2003. Kaposi's sarcoma associated herpesvirus G protein-coupled receptor immortalizes human endothelial cells by activation of the VEGF receptor-2/KDR. Cancer Cell 3:131-143. [DOI] [PubMed] [Google Scholar]
- 3.Batterson, W., and B. Roizman. 1983. Characterization of the herpes simplex virion-associated factor responsible for the induction of alpha genes. J. Virol. 46:371-377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bortz, E., J. P. Whitelegge, Q. Jia, Z. H. Zhou, J. P. Stewart, T. T. Wu, and R. Sun. 2003. Identification of proteins associated with murine gammaherpesvirus 68 virions. J. Virol. 77:13425-13432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boshoff, C., Y. Endo, P. D. Collins, Y. Takeuchi, J. D. Reeves, V. L. Schweickart, M. A. Siani, T. Sasaki, T. J. Williams, P. W. Gray, P. S. Moore, Y. Chang, and R. A. Weiss. 1997. Angiogenic and HIV-inhibitory functions of KSHV-encoded chemokines. Science 278:290-294. [DOI] [PubMed] [Google Scholar]
- 6.Browne, E. P., and T. Shenk. 2003. Human cytomegalovirus UL83-coded pp65 virion protein inhibits antiviral gene expression in infected cells. Proc. Natl. Acad. Sci. USA 100:11439-11444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang, Y., E. Cesarman, M. S. Pessin, F. Lee, J. Culpepper, D. M. Knowles, and P. S. Moore. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865-1869. [DOI] [PubMed] [Google Scholar]
- 8.Desai, P. J. 2000. A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J. Virol. 74:11608-11618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dourmishev, L. A., A. L. Dourmishev, D. Palmeri, R. A. Schwartz, and D. M. Lukac. 2003. Molecular genetics of Kaposi's sarcoma-associated herpesvirus (human herpesvirus-8) epidemiology and pathogenesis. Microbiol. Mol. Biol. Rev. 67:175-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ganem, D. 2006. KSHV infection and the pathogenesis of Kaposi's sarcoma. Annu. Rev. Pathol. Mech. Dis. 1:273-296. [DOI] [PubMed] [Google Scholar]
- 11.Gardella, T., P. Medveczky, T. Sairenji, and C. Mulder. 1984. Detection of circular and linear herpesvirus DNA molecules in mammalian cells by gel electrophoresis. J. Virol. 50:248-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gay, P., D. Le Coq, M. Steinmetz, E. Ferrari, and J. A. Hoch. 1983. Cloning structural gene sacB, which codes for exoenzyme levansucrase of Bacillus subtilis: expression of the gene in Escherichia coli. J. Bacteriol. 153:1424-1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Granzow, H., B. G. Klupp, and T. C. Mettenleiter. 2004. The pseudorabies virus US3 protein is a component of primary and of mature virions. J. Virol. 78:1314-1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grundhoff, A., and D. Ganem. 2004. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis. J. Clin. Investig. 113:124-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Honda, K., H. Yanai, H. Negishi, M. Asagiri, M. Sato, T. Mizutani, N. Shimada, Y. Ohba, A. Takaoka, N. Yoshida, and T. Taniguchi. 2005. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434:772-777. [DOI] [PubMed] [Google Scholar]
- 16.Jia, Q., V. Chernishof, E. Bortz, I. McHardy, T. T. Wu, H. I. Liao, and R. Sun. 2005. Murine gammaherpesvirus 68 open reading frame 45 plays an essential role during the immediate-early phase of viral replication. J. Virol. 79:5129-5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jia, Q., and R. Sun. 2003. Inhibition of gammaherpesvirus replication by RNA interference. J. Virol. 77:3301-3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johannsen, E., M. Luftig, M. R. Chase, S. Weicksel, E. Cahir-McFarland, D. Illanes, D. Sarracino, and E. Kieff. 2004. Proteins of purified Epstein-Barr virus. Proc. Natl. Acad. Sci. USA 101:16286-16291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klupp, B. G., H. Granzow, and T. C. Mettenleiter. 2001. Effect of the pseudorabies virus US3 protein on nuclear membrane localization of the UL34 protein and virus egress from the nucleus. J. Gen. Virol. 82:2363-2371. [DOI] [PubMed] [Google Scholar]
- 20.Krishnan, H. H., P. P. Naranatt, M. S. Smith, L. Zeng, C. Bloomer, and B. Chandran. 2004. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi's sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 78:3601-3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee, E. C., D. Yu, J. Martinez de Velasco, L. Tessarollo, D. A. Swing, D. L. Court, N. A. Jenkins, and N. G. Copeland. 2001. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics 73:56-65. [DOI] [PubMed] [Google Scholar]
- 22.Lee, G. E., J. W. Murray, A. W. Wolkoff, and D. W. Wilson. 2006. Reconstitution of herpes simplex virus microtubule-dependent trafficking in vitro. J. Virol. 80:4264-4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luxton, G. W., S. Haverlock, K. E. Coller, S. E. Antinone, A. Pincetic, and G. A. Smith. 2005. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc. Natl. Acad. Sci. USA 102:5832-5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martin, D. F., B. D. Kuppermann, R. A. Wolitz, A. G. Palestine, H. Li, and C. A. Robinson. 1999. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. N. Engl. J. Med. 340:1063-1070. [DOI] [PubMed] [Google Scholar]
- 25.Mettenleiter, T. C. 2002. Herpesvirus assembly and egress. J. Virol. 76:1537-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mettenleiter, T. C. 2004. Budding events in herpesvirus morphogenesis. Virus Res. 106:167-180. [DOI] [PubMed] [Google Scholar]
- 27.Moore, P. S., and Y. Chang. 2001. Kaposi's sarcoma-associated herpesvirus, p. 2803-2833. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, and S. E. Straus (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, Pa. [Google Scholar]
- 28.Mossman, K. L., R. Sherburne, C. Lavery, J. Duncan, and J. R. Smiley. 2000. Evidence that herpes simplex virus VP16 is required for viral egress downstream of the initial envelopment event. J. Virol. 74:6287-6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O'Connor, C. M., and D. H. Kedes. 2006. Mass spectrometric analyses of purified rhesus monkey rhadinovirus reveal 33 virion-associated proteins. J. Virol. 80:1574-1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reynolds, A. E., E. G. Wills, R. J. Roller, B. J. Ryckman, and J. D. Baines. 2002. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J. Virol. 76:8939-8952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saveliev, A., F. Zhu, and Y. Yuan. 2002. Transcription mapping and expression patterns of genes in the major immediate-early region of Kaposi's sarcoma-associated herpesvirus. Virology 299:301-314. [DOI] [PubMed] [Google Scholar]
- 32.Smiley, J. R. 2004. Herpes simplex virus virion host shutoff protein: immune evasion mediated by a viral RNase? J. Virol. 78:1063-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sodeik, B., M. W. Ebersold, and A. Helenius. 1997. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 136:1007-1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wagenaar, F., J. M. Pol, B. Peeters, A. L. Gielkens, N. de Wind, and T. G. Kimman. 1995. The US3-encoded protein kinase from pseudorabies virus affects egress of virions from the nucleus. J. Gen. Virol. 76:1851-1859. [DOI] [PubMed] [Google Scholar]
- 35.Wang, C. Y., and B. Sugden. 2004. New viruses shake old paradigms. J. Clin. Investig. 113:21-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wolfstein, A., C. H. Nagel, K. Radtke, K. Dohner, V. J. Allan, and B. Sodeik. 2006. The inner tegument promotes herpes simplex virus capsid motility along microtubules in vitro. Traffic 7:227-237. [DOI] [PubMed] [Google Scholar]
- 37.Zhou, F. C., Y. J. Zhang, J. H. Deng, X. P. Wang, H. Y. Pan, E. Hettler, and S. J. Gao. 2002. Efficient infection by a recombinant Kaposi's sarcoma-associated herpesvirus cloned in a bacterial artificial chromosome: application for genetic analysis. J. Virol. 76:6185-6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu, F. X., J. M. Chong, L. Wu, and Y. Yuan. 2005. Virion proteins of Kaposi's sarcoma-associated herpesvirus. J. Virol. 79:800-811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu, F. X., T. Cusano, and Y. Yuan. 1999. Identification of the immediate-early transcripts of Kaposi's sarcoma-associated herpesvirus. J. Virol. 73:5556-5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu, F. X., S. M. King, E. J. Smith, D. E. Levy, and Y. Yuan. 2002. A Kaposi's sarcoma-associated herpesviral protein inhibits virus-mediated induction of type I interferon by blocking IRF-7 phosphorylation and nuclear accumulation. Proc. Natl. Acad. Sci. USA 99:5573-5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu, F. X., and Y. Yuan. 2003. The ORF45 protein of Kaposi's sarcoma-associated herpesvirus is associated with purified virions. J. Virol. 77:4221-4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu, F. X., and Y. Yuan. Unpublished data.