

Abstract
ClpA, a bacterial member of the Clp/Hsp100 chaperone family, is an ATP-dependent molecular chaperone and the regulatory component of the ATP-dependent ClpAP protease. To study the mechanism of binding and unfolding of proteins by ClpA and translocation to ClpP, we used as a model substrate a fusion protein that joined the ClpA recognition signal from RepA to green fluorescent protein (GFP). ClpAP degrades the fusion protein in vivo and in vitro. The substrate binds specifically to ClpA in a reaction requiring ATP binding but not hydrolysis. Binding alone is not sufficient to destabilize the native structure of the GFP portion of the fusion protein. Upon ATP hydrolysis the GFP fusion protein is unfolded, and the unfolded intermediate can be sequestered by ClpA if a nonhydrolyzable analog is added to displace ATP. ATP is required for release. We found that although ClpA is unable to recognize native proteins lacking recognition signals, including GFP and rhodanese, it interacts with those same proteins when they are unfolded. Unfolded GFP is held in a nonnative conformation while associated with ClpA and its release requires ATP hydrolysis. Degradation of unfolded untagged proteins by ClpAP requires ATP even though the initial ATP-dependent unfolding reaction is bypassed. These results suggest that there are two ATP-requiring steps: an initial protein unfolding step followed by translocation of the unfolded protein to ClpP or in some cases release from the complex.
Keywords: molecular chaperones, green fluorescent protein, protein unfolding
Clp proteins form a large family of homologous ATPases that participate in many cellular functions, including DNA replication, tolerance to heat stress, control of gene expression, protein degradation, and inheritance of prion-like factors (1, 2). ATP-dependent protein unfolding and remodeling have been implicated in each of these processes, and biochemical studies revealed that the Clp ATPases are classical molecular chaperones. For example, Escherichia coli ClpA remodels inactive dimers of the plasmid P1 initiator protein, RepA, into monomers that bind DNA specifically and prevents irreversible heat inactivation of proteins (3). Similarly, ClpX of E. coli disassembles protein complexes (4) and aggregates (5) and dissociates dimers of the initiator protein of plasmid RK2 (6). Hsp104, a yeast Clp ATPase, and ClpB, its E. coli homologue, participate in disaggregating heat-denatured proteins in vivo and in vitro (7–11). Some of the Clp ATPases assemble in complexes with proteolytic components where their ATP-dependent unfolding activity regulates proteolysis (12, 13). For example, ClpA enables ClpP to degrade specific proteins. ClpX also activates ClpP to degrade other specific proteins. Taken together, the results show that substrate recognition resides in the ATPase component of the protease.
Clp ATPases, including ClpA, ClpX, ClpB, Hsp104, and HslU, self-assemble into oligomeric rings in the presence of ATP or nonhydrolyzable ATP analogs (14–18). When complexed to a proteolytic component, such as ClpP or HslV in E. coli, the ATPase rings are at either or both ends of the proteolytic core, forming structures resembling the eukaryotic 26S proteasomes (14). The crystal structures of ClpP and HslV reveal that, similar to the proteolytic core of the 26S proteasome, the proteolytic sites are in an internal chamber of stacked rings of identical subunits (19–22). Access to the proteolytic chamber appears to be through narrow pores at either end of the chamber. With this arrangement the active sites of the protease are sequestered from the cytoplasm and entry is regulated by the ATPase. The interaction of a substrate, RepA, with ClpAP has recently been visualized by electron microscopy. RepA is seen bound to the outside ends of ClpA, away from ClpP, in complexes made with chemically inactivated ClpP and in the presence of adenosine 5′-O-(3-thiotriphosphate) (ATP[γS]) (T. Ishikawa, F. Beuron, M. Kessel, S.W., M.R.M., and A. C. Steven, unpublished data). ATP causes conformational changes in ClpA and RepA is seen in the central ClpP chamber.
From the biochemical and structural data, it was proposed that the ATPase components flanking the proteolytic core unfold the substrate and then translocate the unfolded polypeptide through the small pore into the proteolytic chamber (19, 25, 26). We have shown that in fact ClpA translocates substrates from their binding sites on ClpA to ClpP (24). Horwich and colleagues (27) have recently shown that ClpA catalyzes protein unfolding. They used a variant of green fluorescent protein (GFP), GFP11, in which the 11 amino acid SsrA recognition tag is fused to the carboxyl terminus of GFP. In vivo, SsrA tags are added to polypeptides that have become stalled on ribosomes (28, 29). The released tagged proteins are then degraded by ClpAP or ClpXP (30, 31). In vitro, a large decrease in fluorescence was observed when ClpA and ATP were incubated with GFP11 in reaction mixtures containing GroEL “trap” (32), a mutant which binds unfolded proteins but does not release them. Deuterium–hydrogen exchange experiments confirmed that ClpA completely unfolded GFP11.
In the current study, we explored the interaction of ClpA with native and unfolded proteins with and without ClpA recognition signals. We examined intermediates in the reaction and the role of ATP in protein unfolding by ClpA and release from ClpA or translocation to ClpP.
Methods
Materials.
ATP, adenylyl imidodiphosphate (AMP-PNP), and ATP[γS] were obtained from Boehringer Mannheim. Restriction endonucleases, T4 DNA ligase, and T4 polynucleotide kinase were obtained from New England Biolabs. Rhodanese was from Sigma. Buffer A contained 150 mM Tris⋅HCl at pH 7.5, 20 mM MgCl2, 50 mM KCl, 2.5 mM DTT, 5% (vol/vol) glycerol, 0.05 mM EDTA, and 200 μg/ml BSA. Buffer B contained 20 mM Tris⋅HCl at pH 7.5, 20 mM MgCl2, 100 mM KCl, 5 mM DTT, 10% glycerol, 0.1 mM EDTA, and 200 μg/ml BSA.
Plasmid and Strains.
Plasmid pBAD-GFP was constructed by PCR amplification of the GFPuv gene from pGFPuv (CLONTECH) by using 5′ KpnI oligonucleotide JH.054 (5′-TAATGGTACCCAGTAAAGGAGAAGAACTTTTCACTGG-3′) and 3′ XbaI oligonucleotide JH.055 (5′-TAATTCTAGATTATTTGTAGAGCTCATCCATGCC-3′). The PCR product was digested with KpnI and XbaI and ligated into KpnI- and XbaI-digested pBAD24 (33). The pBAD-RepGFP plasmid was constructed by PCR amplification of DNA coding for RepA amino acids 1–70 by using mJH2 (34) and oligonucleotides JH.063 (5′-TAATGAATTCACCATGAATCAATCATTTATCTCCG-3′) and JH.064 (5′-TAATGGTACCGCACCCTCGGCTTTAGCTATCTCCAG-3′), followed by ligation into pBAD-GFP digested with EcoRI and KpnI. The sequences of the gene fusions were verified by DNA sequencing.
The following araΔ derivatives of MC4100 were kindly constructed by Susan Gottesman (National Institutes of Health): SG22215, clpP∷cat; SG22216, clpX∷kan; and SG22217, clpA∷kan.
Proteins.
ClpA (35), ClpP (35), and RepA (36) were isolated as described.
GFP and the fusion protein RepGFP were isolated from SG22215 harboring pBAD-GFP or pBAD-RepGFP. Cells were grown overnight with arabinose, collected, disrupted with a French pressure cell, and centrifuged for 1 hr at 83,000 × g. For GFP the soluble fraction was extracted with ethanol and chromatographed on a phenyl-Sepharose column essentially as described (37). RepGFP was extracted from the insoluble fraction by 50 mM Tris⋅HCl, pH 7.5/150 mM NaCl/5 mM EDTA containing 6 M urea and dialyzed against 50 mM Tris⋅HCl, pH 7.5/150 mM NaCl/5 mM EDTA. Ammonium sulfate was added to 1 M and the material was then centrifuged. The supernatant was applied to a phenyl-Sepharose column and RepGFP was eluted with a linear gradient of 1 to 0 M ammonium sulfate in 20 mM Tris⋅HCl, pH 7.5/1 mM EDTA.
GFP, RepGFP, RepA, and rhodanese were labeled in vitro as described (24). Protein concentrations are expressed as molar amounts of ClpA hexamers, RepA dimers, ClpP tetradecamers, and GFP, RepGFP, and rhodanese monomers.
GFP, RepGFP, and rhodanese were acid-denatured by incubating 40 pmol of protein in 50 μl of 25 mM HCl, pH 1.5, for 5 min at 25°C. The proteins were guanidine⋅HCl-denatured by incubating 1.5 mg/ml protein in 6 M guanidine⋅HCl containing 25 mM Tris⋅HCl at pH 8.0, 10 mM DTT, and 1 mM EDTA for 2 hr at 25°C. The unfolded proteins were diluted directly into reaction mixtures and used immediately.
Protein Degradation Assays.
Reaction mixtures (100 μl) contained 10 mM ATP, 20 mM creatine phosphate, 6 μg of creatine kinase, 160 pmol of ClpA, 200 pmol of ClpP, and 40 pmol of [3H]GFP (1,200 cpm/pmol), [3H]RepGFP (150 cpm/pmol), [3H]rhodanese (690 cpm/pmol), or unlabeled RepGFP in buffer B. The mixtures were incubated at 25°C for 20 min. Trichloroacetic acid (TCA) was added to 20% (wt/vol) and acid-soluble radioactivity was measured. Alternatively, TCA pellets were analyzed by SDS/PAGE and RepGFP was quantitated by densitometry.
To measure degradation of acid-unfolded proteins, 50 μl of acid-denatured 3H-labeled protein (40 pmol) was diluted into reaction mixtures (100 μl final volume) containing buffer A, 20 mM creatine phosphate, 6 μg of creatine kinase, 160 pmol of ClpA, 200 pmol of ClpP, and 1 mM ATP[γS]. After 5 min at 25°C, 10 mM ATP was added. After an additional 20 min, degradation was determined by measuring acid-soluble radioactivity. Degradation assays using guanidine⋅HCl-denatured proteins were carried out as described for acid-denatured proteins with the exception that buffer B replaced buffer A and 1 μl of guanidine⋅HCl-denatured 3H-labeled protein (40 pmol) was diluted into 100-μl reaction mixtures.
Protein Unfolding Assay.
RepGFP (40 pmol) and 200 pmol of ClpA were incubated in 100-μl reaction mixtures with buffer A containing 10 mM ATP, 20 mM creatine phosphate, and 3 μg of creatine kinase. Fluorescence was measured at 25°C with excitation at 395 nm and emission at 515 nm by using a Perkin–Elmer LS50B luminescence spectrophotometer equipped with a well plate reader.
Results
ClpAP Degrades a Native GFP Fusion Protein with an N-Terminal ClpA Recognition Signal.
To study the binding and unfolding of a native protein by ClpA, we constructed a derivative of GFP containing a ClpA recognition signal. GFP was chosen because it fluoresces in its native state but not in nonnative conformations (38). We joined the amino-terminal 70 amino acids of RepA to GFP to specifically target GFP for recognition by ClpA, because previous work had shown that ClpA binds RepA with high affinity (3) and recognizes a signal residing in the amino-terminal portion of RepA (J.H. and S.W., unpublished observation). We anticipated that the fusion protein, referred to as RepGFP, would be a substrate for unfolding by ClpA and degradation by ClpAP. In a control experiment, E. coli wild-type colonies expressing GFP fluoresced when viewed under UV light (Fig. 1A). There was no apparent difference in the fluorescent intensity of the wild-type colonies when compared with clpAΔ, clpPΔ, or clpXΔ colonies. In contrast, when RepGFP was expressed in the same strains, the clpAΔ and clpPΔ colonies appeared much more fluorescent than the wild-type or clpXΔ colonies (Fig. 1B). These results suggest that the amino-terminal portion of RepA specifically targets GFP for degradation by ClpAP in vivo.
Figure 1.

Degradation of RepGFP in vivo. E. coli wild-type, clpAΔ, clpPΔ, and clpXΔ strains carrying plasmids pBAD-GFP (A) or pBAD-RepGFP (B) were grown on LB agar containing 100 μg/ml ampicillin and 0.02% arabinose at 25°C for 72 hr and photographed using UV transillumination (312 nm).
We purified RepGFP and GFP and tested them as substrates for degradation by ClpAP. Degradation of RepGFP but not GFP was observed, as measured by a decrease in fluorescence with time (data not shown). Analysis of the reaction mixtures by SDS/PAGE showed directly that the RepGFP protein disappeared with time when incubated with ClpAP and ATP, whereas GFP did not (Fig. 2). The reaction required both ClpAP and ATP. Thus, although ClpAP does not recognize native GFP, the addition of the amino-terminal 70 amino acids of RepA to GFP targets the fusion protein for degradation by ClpAP.
Figure 2.

Degradation of RepGFP by ClpAP in vitro. Degradation of RepGFP and [3H]GFP was determined as described in the text by measuring acid solubilization of [3H]GFP (hatched bars) and by analyzing the products of the reaction with RepGFP (yellow bars) by SDS/PAGE and quantitating RepGFP by densitometry.
ClpA Binds RepGFP Without Perturbing the Native Conformation of GFP.
Previous work showed that binding of RepA to ClpA involves a time- and temperature-dependent switch from a weaker, salt-sensitive, complex to a tighter, salt-insensitive, one (39), suggesting that some conformational change in RepA may accompany the tight binding reaction. To know whether RepGFP is structurally perturbed or unfolded upon binding to ClpA, we incubated [3H]RepGFP with ClpA and ATP[γS] under conditions that generate stable ClpA–RepA complexes (39) and then analyzed the mixtures by gel filtration. About 60% of the RepGFP eluted in the excluded volume of the column with ClpA, indicating that the RepGFP fusion protein, like RepA, has a high affinity for ClpA (Fig. 3). The ratio of fluorescence to radioactivity of the isolated ClpA-RepGFP complex was unchanged relative to that of free RepGFP. In a control experiment in the absence of ClpA, RepGFP eluted as expected in the partially included volume (data not shown). These results show that the GFP portion of the fusion protein is not converted to a nonnative form simply upon binding ClpA. Similar results were obtained with a weaker-binding fusion protein containing the first 15 amino acids of RepA joined to GFP. Thus a 15 amino acid tag is sufficient to direct the binding of GFP to ClpA without destabilizing the GFP portion of the fusion protein.
Figure 3.

Isolation of RepGFP-ClpA complexes by gel filtration. [3H]RepGFP (160 pmol) was incubated with 200 pmol of ClpA for 15 min at 24°C in 100-μl reaction mixtures containing buffer B and 1 mM ATP[γS]. The mixtures were then applied to a 0.7 × 15 cm column of Sephacryl S200 HR at 24°C equilibrated with 20 mM Tris⋅HCl, pH 7.5/10% (vol/vol) glycerol/100 mM KCl/5 mM DTT/0.1 mM EDTA/5 mM MgCl2/0.5 mM ATP[γS]. Fractions (150 μl) were collected into microtiter plates. Relative fluorescence (blue circles) and radioactivity (open black squares) were measured in aliquots of the fractions.
ClpA Unfolds RepGFP in the Presence of ATP.
We next wanted to know whether ClpA unfolds native RepGFP in the presence of ATP. When RepGFP was incubated with ClpA and ATP, we found that the fluorescence intensity of RepGFP decreased about 50% (Fig. 4A). After about 40 min, RepGFP regained 80–90% of its original fluorescence, indicating that the protein was released and refolded into its native conformation (Fig. 4A). The time required to recover the initial fluorescence was increased by adding more phosphocreatine and was decreased by omitting the ATP regenerating system (data not shown). Addition of ADP at any time during the unfolding reaction caused a rapid regain of fluorescence, suggesting release and refolding (Fig. 4A). When an excess of another ClpA substrate, α-casein (Fig. 4A) or native RepA (data not shown), was added during the unfolding reaction, fluorescence rapidly returned. Thus the decrease in fluorescence of RepGFP in the presence of ClpA and ATP is reversible and the apparent “trapping” of the unfolded protein by ClpA reflects repeated ATP-driven cycles of binding, unfolding, release, and refolding of RepGFP.
Figure 4.

Unfolding and trapping of RepGFP by ClpA. (A) RepGFP was incubated with ClpA in the presence of 2 mM ATP and an ATP-regenerating system (red triangles) in unfolding assays as described in the text. ADP (10 mM) was added to a reaction mixture that had been incubated for 9 min with ATP (black open squares). α-Casein (40 μM) was added to another reaction mixture that had been incubated with ATP for 9 min (green circles). Fluorescence was measured at the times indicated. (B) RepGFP was incubated with ClpA in the presence of 2 mM ATP and an ATP-regenerating system (red triangles) as in A. After 9 min of incubation with ATP, either 10 mM ATP[γS] (blue squares) or 10 mM ATP[γS] and 40 μM α-casein (open black circles) were added to the reaction mixtures. Fluorescence was measured with time at 25°C and the initial fluorescence was set equal to 1.
ClpA Sequesters Unfolded RepGFP in the Presence of ATP[γS] and Release of Unfolded RepGFP Requires ATP.
We wanted to know whether the RepGFP unfolded by ClpA could be trapped on ClpA in its nonnative form. To address this, ClpA and RepGFP were incubated with ATP for a short time to unfold RepGFP and then a 10-fold molar excess of ATP[γS] to ATP was added, knowing from previous experiments that ATP rapidly exchanges with ATP[γS] bound to ClpA (39). We found that the addition of ATP[γS] prevented the recovery of fluorescence observed with the time-dependent depletion of ATP (Fig. 4B). Similar results were obtained when adenylyl imidodiphosphate (AMP-PNP) was used in place of ATP[γS]. Recovery of fluorescence could be prevented for more than 15 hr. However, fluorescence could be recovered by incubating the mixtures with low concentrations of denaturants.
It was possible that either an unfolded protein intermediate in the reaction was trapped on ClpA by the exchange of ATP with ATP[γS] or the unfolded RepGFP was released and then rebound to ClpA upon nucleotide exchange. To distinguish between these possibilities, we added α-casein at the same time as ATP[γS]. If the unfolded protein were free at the time of ATP[γS] addition, α-casein should compete with the released protein and prevent its rebinding as it did when added in the presence of ATP (Fig. 4A). The results showed that there was only a very slow increase in fluorescence, suggesting that the unfolded RepGFP was associated with ClpA, trapped in the act of unfolding (Fig. 4B). These results suggest that both unfolding and release of the bound unfolded protein require ATP.
ClpA Binds Unfolded GFP That Lacks a ClpA Recognition Tag.
The ability to trap unfolding intermediates on ClpA led us to investigate whether the requirement for a specific recognition motif could be bypassed by presenting ClpA with an unfolded untagged protein. We acid-denatured GFP, which in its native conformation is not recognized by ClpA (ref. 27 and Fig. 2), and then diluted it into reaction mixtures with and without ClpA and ATP[γS]. Without ClpA, unfolded GFP rapidly regained full fluorescence (t1/2 ≅ 1.5 min) (Fig. 5A). The addition of ClpA in the absence of nucleotide did not affect the rate of refolding (Fig. 5A). In contrast, only about 40% of the initial fluorescence of GFP was recovered after dilution into reaction mixtures with ClpA and ATP[γS], indicating that ClpA captured much of the unfolded GFP before it refolded (Fig. 5A). When unfolded GFP was diluted into reaction mixtures with ClpA and ATP, the kinetics of recovery of fluorescence were only slightly slower than in the absence of ClpA.
Figure 5.

Sequestration of unfolded GFP by ClpA. (A) GFP was acid-denatured as described in the text, and 40 pmol (50 μl) was added to reaction mixtures (100 μl final volume) containing buffer A with 20 mM creatine phosphate and 6 μg of creatine kinase in the absence of ClpA (green circles), in the presence of 160 pmol of ClpA and no nucleotide (black squares), in the presence of 160 pmol of ClpA and 10 mM ATP (red triangles), and in the presence of 160 pmol of ClpA and 1 mM ATP[γS] (open blue circles). Fluorescence was measured with time at 24°C. Fluorescence intensity of native GFP of the same concentration was taken as 1. (B) Reaction mixtures were as in A. Acid-denatured GFP was incubated for 5 min with 160 pmol of ClpA and 1 mM ATP[γS] and then 10 mM ADP (open black triangles), or 10 mM ATP (red squares) was added and incubations were continued. In a control, acid-denatured GFP was added alone to a reaction mixture (green circles).
We then tested whether the GFP sequestered on ClpA in ATP[γS] could be released by the addition of ATP. The addition of excess ATP after a 5-min incubation with ATP[γS] caused fluorescence to quickly return, indicating that ClpA released GFP upon ATP hydrolysis, and GFP regained its native conformation (Fig. 5B). Exchange of ATP[γS] with ADP did not result in an increase in fluorescence, suggesting that ATP was required for release (Fig. 5B). Results obtained with GFP denatured by treatment with guanidine⋅HCl were identical to those with acid-denatured GFP (results not shown).
To confirm that the unfolded GFP was associated with ClpA in the presence of ATP[γS], ClpA was incubated with unfolded [3H]GFP and ATP[γS] and the products were analyzed by gel filtration (Fig. 6A). About half of the labeled GFP eluted in the excluded volume with ClpA and the rest eluted as expected for free GFP. No fluorescence was detected in the fractions containing ClpA-GFP complexes, although the free GFP in the included volume was fluorescent. When the fractions containing ClpA-GFP complexes were incubated with ATP, fluorescence was recovered, indicating release and refolding of GFP. In a control experiment, native [3H]GFP was incubated with ClpA and ATP[γS] and subjected to gel filtration. Both radioactivity and fluorescence eluted at the position of free GFP (Fig. 6B). These results show that an unfolded protein, which lacks an exposed specific ClpA recognition signal in its folded conformation, can be bound by ClpA and held in a nonnative form. Release of the nonnative protein requires ATP.
Figure 6.

Gel filtration of unfolded GFP-ClpA complex. A 160-pmol sample of acid-denatured [3H]GFP (A) or native [3H]GFP (B) was incubated with 200 pmol of ClpA in 100-μl reaction mixtures containing buffer A and 1 mM ATP[γS] for 5 min at 24°C. The mixtures were then applied to a 0.7 × 15 cm column of Sephacryl S200 HR as described in the legend of Fig. 3. Initial fluorescence (blue circles) was measured in the fractions. In A 10 mM ATP was then added to each fraction, and after 25 min at 25°C, fluorescence was measured again (red triangles). Radioactivity (open black squares) was measured in aliquots of the fractions.
To test whether or not a ClpA recognition tag influences binding of unfolded proteins by ClpA, we acid-denatured RepGFP and then diluted the denatured protein into reaction mixtures with ClpA and ATP[γS] as we had done above in Fig. 5A, using denatured GFP. We found that RepGFP, like GFP, regained about 60% of the fluorescence intensity regained in the absence of ClpA, suggesting that 40% of the protein was bound by ClpA in a nonfluorescing nonnative form (Fig. 7). When ATP was used instead of ATP[γS], again about 60% of the initial fluorescence was attained (Fig. 7). After about 15 min, the fluorescence increased to the initial level. This is in contrast to the rapid refolding of denatured untagged GFP seen in the presence of ClpA and ATP (Fig. 5A). To determine whether the unfolded RepGFP was released slowly from ClpA compared with unfolded GFP or was released rapidly, but then refolded and became a native tagged substrate for the unfolding reaction, α-casein was added 5 min after the addition of ClpA and ATP. If unfolded RepGFP were slow to release, there should be little effect of the competitor. We found that fluorescence returned rapidly, showing that the apparent “trapping” of unfolded RepGFP by ClpA in the presence of ATP is due to steady-state unfolding. In a control experiment, there was no effect of the addition of ovalbumin, a protein that is not recognized by ClpA (24). Thus, although ClpA binds both denatured GFP and RepGFP, only RepGFP is rebound after ATP-mediated release (compare Figs. 5A and 7). These results suggest that GFP released from ClpA, although nonfluorescent and thus at least partially unfolded, is no longer in a conformation recognized by ClpA.
Figure 7.

Sequestration of unfolded RepGFP by ClpA. Acid-denatured RepGFP was diluted into reaction mixtures containing ClpA and 10 mM ATP (red triangles), ClpA and 1 mM ATP[γS] (open blue circles), or ClpA alone (green circles) as described in the legend of Fig. 5. To other mixtures, either 40 μM α-casein (black squares) or 23 μM ovalbumin (open black squares) was added after RepGFP, ClpA, and ATP had been incubated for 5 min. Fluorescence was measured with time.
Native Tagged Substrates Compete with Unfolded Proteins for Binding to ClpA.
To determine whether unfolded GFP binds to the same site on ClpA as specific native proteins, we carried out competition experiments. Native RepA was mixed with ClpA, at several different molar ratios, in the presence of ATP[γS]. After 2 min, unfolded GFP was added and fluorescence was measured. The amount of fluorescence recovered increased with the amount of RepA added, showing competition (Fig. 8). Ovalbumin had no effect on the recovery of fluorescence (Fig. 8). In another experiment, ClpA and ATP[γS] were incubated in the absence (Fig. 9A) or presence of unfolded GFP (Fig. 9B) and then [3H]RepA was added. Gel filtration analysis of the mixtures showed that preincubation with unfolded GFP significantly decreased the amount of RepA complexed to ClpA. These experiments indicate that the same or overlapping sites on ClpA bind unfolded proteins and native tagged proteins.
Figure 8.
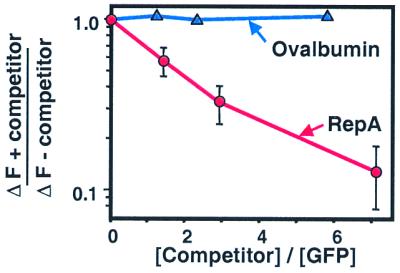
Competition between native and unfolded proteins for ClpA binding. ClpA (40 pmol) was incubated in 50-μl reaction mixtures containing buffer A with 2 mM ATP[γS] and 0, 60, 115, or 285 pmol of RepA (red circles) or 0, 50, 90, or 230 pmol of ovalbumin (blue triangles) for 2 min at 24°C. Then 40 pmol of acid-denatured GFP (in 50 μl) was added to each and fluorescence was measured after 10 min. Results are means (±SEM) of three independent experiments for RepA and a single representative experiment for ovalbumin.
Figure 9.

Inhibition of RepA-ClpA complex formation by unfolded GFP. (A) ClpA (100 pmol) was incubated (in 100 μl) of buffer A containing 2 mM ATP[γS] for 5 min at 24°C. Then 100 pmol of [3H]RepA (1,170 cpm/pmol) was added and the reaction mixture was incubated for 10 min at 25°C. The mixture was applied to a 0.7 × 15 cm column of Sephacryl S200 HR as described in the legend of Fig. 3. Radioactivity was measured in aliquots of the fractions (red squares). (B) ClpA (100 pmol) was incubated as in A with the exception that 300 pmol of acid-denatured GFP was added to the initial reaction mixture. After incubation with [3H]RepA as in A, the sample was analyzed by gel filtration as above. Radioactivity was measured (red squares). Initial fluorescence of the fractions was measured (open blue triangles). Then, 5 mM ATP and an ATP-regenerating system were added and fluorescence was measured after 15 min at 24°C (open black circles; A. U., arbitrary units).
Unfolded Proteins Lacking Recognition Tags Are Degraded by ClpAP.
Having found that ClpA binds unfolded substrates, thereby bypassing the unfolding step, we wanted to know whether ClpAP is able to degrade unfolded proteins without recognition tags. To test this possibility, unfolded [3H]GFP was diluted into reaction mixtures with ClpAP and ATP[γS] to trap the substrate and then a 10-fold molar excess of ATP to ATP[γS] was added. After a short incubation, degradation was measured. We found that ClpAP degraded unfolded GFP but not native GFP (Fig. 10). ATP stimulated degradation about 10-fold, even though the ATP-mediated unfolding reaction apparently had been bypassed. ClpA was essential, indicating that the unfolded protein could not simply diffuse into ClpP and be degraded. When the first incubation with ATP[γS], designed to trap the substrate, was omitted, a similar amount of degradation was seen (Fig. 10). GFP denatured in guanidine was similarly trapped and degraded by ClpAP.
Figure 10.

Degradation of unfolded GFP and rhodanese by ClpAP. Acid-denatured [3H]GFP (blue bars), native [3H]GFP (blue hatched bars), acid-denatured [3H]rhodanese (yellow bars), and native [3H]rhodanese (hatched yellow bars) were incubated as described in the text with ClpAP and 1 mM ATP[γS] for 5 min to bind the unfolded protein. Then 10 mM ATP was added and after 20 min, degradation was measured. Where indicated, ATP, ClpP, ClpA, or ATP[γS] was omitted. Results are means (±SEM) of three independent experiments.
To test whether ClpAP could recognize and degrade other unfolded proteins, we performed a similar experiment with unfolded [3H]rhodanese. We found that unfolded rhodanese was degraded by ClpAP but native rhodanese was not. The requirements for degradation were the same as for unfolded GFP (Fig. 10). There was no difference in the degradation of rhodanese unfolded with guanidine⋅HCl compared with rhodanese unfolded by acid treatment (data not shown).
These experiments demonstrate that ClpAP degrades untagged substrates if the initial unfolding step is bypassed. The observation that degradation is stimulated 10-fold by ATP suggests that there is a second ATP-requiring step in the degradation pathway in addition to ATP-dependent unfolding. One possible energy-requiring reaction may be the translocation of unfolded proteins from ClpA to ClpP.
Discussion
The current results add several features to the model of protein unfolding by ClpA and degradation by ClpAP (Fig. 11). Briefly, native proteins with exposed ClpA recognition signals are bound by ClpA in a reaction requiring ATP binding but not hydrolysis and in which the role of ATP can be mimicked by ATP[γS]. We have shown that the act of binding to ClpA does not necessarily destabilize the native structure of a protein fused to a ClpA recognition signal. Upon ATP hydrolysis, ClpA mediates unfolding of a bound native substrate. If ATP hydrolysis is interrupted by addition of a nonhydrolyzable ATP analog, ClpA sequesters the unfolded protein intermediate and its release requires additional ATP. It remains to been seen where on ClpA this reaction is taking place, but it is tempting to speculate that it is in the cavity of ClpA by analogy to GroEL (40). Release or translocation to ClpP requires ATP, suggesting another ATP-dependent step in the pathway in addition to ATP-dependent unfolding.
Figure 11.

Working model showing steps in the pathway of degradation by ClpAP. See Discussion.
We have shown that in a reaction bypassing the initial specific binding and ATP-dependent unfolding reactions, unfolded proteins lacking specific recognition signals are bound by ClpA and maintained in a nonnative conformation. This suggests that there are other recognition sites, perhaps simply hydrophobic regions, that become exposed after the initial binding reaction that contribute to the stability of the complex between ClpA and the substrate. Interestingly, ClpX is unable to bind unfolded proteins that lack specific recognition signals (41). However, unfolded tagged proteins are bound by ClpX with higher affinity than native tagged proteins (41). Those observations in combination with the ones presented here suggest that there are additional substrate binding sites on substrate proteins that become exposed during the unfolding reaction. Sauer, Baker, and their colleagues proposed that SSD (sensor and substrate discrimination) domains discriminate between specific and nonspecific substrates, binding only the specific substrates (28, 42, 43). The finding that the initial binding of specific proteins competes with binding of unfolded nonspecific proteins implies that binding to specific sites may occlude binding at unfolded protein sites. Alternatively, the initial substrate binding may not be to the SSD domain. Further study is necessary to answer this question.
The in vitro finding that ClpA binds unfolded proteins and ClpAP degrades unfolded proteins raises the question as to whether this reaction may occur in vivo. It has previously been shown that ClpA mutants are slightly defective in degrading abnormal canavanyl proteins and that ClpA levels increase during heat stress, although not in a σ32-dependent manner (44). Is has also been shown that abnormal proteins synthesized during starvation are stabilized in ClpA mutants (23). Based on the high-affinity interaction observed between unfolded proteins and ClpA in vitro, ClpA may play at least a limited role in vivo by interacting with unfolded proteins that escape surveillance by the predominant chaperones.
Acknowledgments
We dedicate this paper to the memory of Paul Sigler. We thank Susan Gottesman for constructing E. coli strains for us.
Abbreviations
- ATP[γS]
adenosine 5′-O-(3-thiotriphosphate)
- GFP
green fluorescent protein
References
- 1.Schirmer E C, Glover J R, Singer M A, Lindquist S. Trends Biochem Sci. 1996;21(8):289–296. [PubMed] [Google Scholar]
- 2.Gottesman S, Wickner S, Maurizi M. Genes Dev. 1997;11:815–823. doi: 10.1101/gad.11.7.815. [DOI] [PubMed] [Google Scholar]
- 3.Wickner S, Gottesman S, Skowyra D, Hoskins J, McKenney K, Maurizi M R. Proc Natl Acad Sci USA. 1994;91:12218–12222. doi: 10.1073/pnas.91.25.12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levchenko I, Luo L, Baker T A. Genes Dev. 1995;9:2399–2408. doi: 10.1101/gad.9.19.2399. [DOI] [PubMed] [Google Scholar]
- 5.Wawrzynow A, Wojtkowiak D, Marszalek J, Banecki B, Jonsen M, Graves B, Georgopoulos C, Zylicz M. EMBO J. 1995;14:1867–1877. doi: 10.1002/j.1460-2075.1995.tb07179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Konieczny I, Helinski D R. Proc Natl Acad Sci USA. 1997;94:14378–14382. doi: 10.1073/pnas.94.26.14378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parsell D A, Kowal A S, Singer M A, Lindquist S. Nature (London) 1994;372:475–478. doi: 10.1038/372475a0. [DOI] [PubMed] [Google Scholar]
- 8.Glover J R, Lindquist S. Cell. 1998;94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- 9.Zolkiewski M. J Biol Chem. 1999;274:28083–28086. doi: 10.1074/jbc.274.40.28083. [DOI] [PubMed] [Google Scholar]
- 10.Mogk A, Tomoyasu T, Goloubinoff P, Rudiger S, Roder D, Langen H, Bukau B. EMBO J. 1999;18:38017–38026. doi: 10.1093/emboj/18.24.6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Motohashi K, Watanabe Y, Yohda M, Yoshida M. Proc Natl Acad Sci USA. 1999;96:7184–7189. doi: 10.1073/pnas.96.13.7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmidt A, Lupas A N, Finley D. Curr Opin Chem Biol. 1999;3:584–591. doi: 10.1016/s1367-5931(99)00013-7. [DOI] [PubMed] [Google Scholar]
- 13.Wickner S, Maurizi M, Gottesman S. Science. 1999;286:1888–1893. doi: 10.1126/science.286.5446.1888. [DOI] [PubMed] [Google Scholar]
- 14.Kessel M, Maurizi M R, Kim B, Kocsis E, Trus B L, Singh S K, Steven A C. J Mol Biol. 1995;250:587–594. doi: 10.1006/jmbi.1995.0400. [DOI] [PubMed] [Google Scholar]
- 15.Parsell D A, Sanchez Y, Stitzel J D, Lindquist S. Nature (London) 1991;353:270–273. doi: 10.1038/353270a0. [DOI] [PubMed] [Google Scholar]
- 16.Grimaud R, Kessel M, Beuron F, Steven A C, Maurizi M R. J Biol Chem. 1998;273:12476–12481. doi: 10.1074/jbc.273.20.12476. [DOI] [PubMed] [Google Scholar]
- 17.Rohrwild M, Pfeifer G, Santarius U, Muller S A, Huang H C, Engel A, Baumeister W, Goldberg A L. Nat Struct Biol. 1997;4:133–139. doi: 10.1038/nsb0297-133. [DOI] [PubMed] [Google Scholar]
- 18.Kessel M, Wu W, Gottesman S, Kocsis E, Steven A C, Maurizi M R. FEBS Lett. 1996;398:274–278. doi: 10.1016/s0014-5793(96)01261-6. [DOI] [PubMed] [Google Scholar]
- 19.Wang J, Hartling J A, Flanagan J M. Cell. 1997;91:447–456. doi: 10.1016/s0092-8674(00)80431-6. [DOI] [PubMed] [Google Scholar]
- 20.Lowe J, Stock D, Jap B, Zwickl P, Baumeister W, Huber R. Science. 1995;268:533–539. doi: 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]
- 21.Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik H D, Huber R. Nature (London) 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 22.Bochtler M, Ditzel L, Groll M, Huber R. Proc Natl Acad Sci USA. 1997;94:6070–6074. doi: 10.1073/pnas.94.12.6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Damerau K, St. John A C. J Bacteriol. 1993;175:53–63. doi: 10.1128/jb.175.1.53-63.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoskins J R, Pak M, Maurizi M R, Wickner S. Proc Natl Acad Sci USA. 1998;95:12135–12140. doi: 10.1073/pnas.95.21.12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gottesman S, Maurizi M R, Wickner S. Cell. 1997;91:435–438. doi: 10.1016/s0092-8674(00)80428-6. [DOI] [PubMed] [Google Scholar]
- 26.Larsen C N, Finley D. Cell. 1997;91:431–434. doi: 10.1016/s0092-8674(00)80427-4. [DOI] [PubMed] [Google Scholar]
- 27.Weber-Ban E U, Reid B G, Miranker A D, Horwich A L. Nature (London) 1999;401:90–93. doi: 10.1038/43481. [DOI] [PubMed] [Google Scholar]
- 28.Keiler K C, Waller P R, Sauer R T. Science. 1996;271:990–993. doi: 10.1126/science.271.5251.990. [DOI] [PubMed] [Google Scholar]
- 29.Herman C, Thevenet D, Bouloc P, Walker G C, D'Ari R. Genes Dev. 1998;12:1348–1355. doi: 10.1101/gad.12.9.1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gottesman S, Roche E, Zhou Y, Sauer R T. Genes Dev. 1998;12:1338–1347. doi: 10.1101/gad.12.9.1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roche E D, Sauer R T. EMBO J. 1999;18:4579–4589. doi: 10.1093/emboj/18.16.4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fenton W A, Kashl Y, Furtak K, Horwich A L. Nature (London) 1994;371:614–619. doi: 10.1038/371614a0. [DOI] [PubMed] [Google Scholar]
- 33.Guzman L M, Belin D, Carson M J, Beckwith J. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wickner S, Hoskins J, Chattoraj D, McKenney K. J Biol Chem. 1990;265:11622–11627. [PubMed] [Google Scholar]
- 35.Maurizi M R, Thompson M W, Singh S K, Kim S H. Methods Enzymol. 1994;344:314–331. doi: 10.1016/0076-6879(94)44025-5. [DOI] [PubMed] [Google Scholar]
- 36.Wickner S H. Proc Natl Acad Sci USA. 1990;87:2690–2694. doi: 10.1073/pnas.87.7.2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yakhnin A V, Vinokurov L M, Surin A K, Alakhov Y B. Protein Expr Purif. 1998;14:382–386. doi: 10.1006/prep.1998.0981. [DOI] [PubMed] [Google Scholar]
- 38.Ward W W, Bokman S H. Biochemistry. 1982;21:4535–4540. doi: 10.1021/bi00262a003. [DOI] [PubMed] [Google Scholar]
- 39.Pak M, Wickner S. Proc Natl Acad Sci USA. 1997;94:4901–4906. doi: 10.1073/pnas.94.10.4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bukau B, Horwich A L. Cell. 1998;92:351–366. doi: 10.1016/s0092-8674(00)80928-9. [DOI] [PubMed] [Google Scholar]
- 41.Singh S K, Grimaud R, Hoskins J R, Wickner S, Maurizi M R. Proc Natl Acad Sci USA. 2000;97:8898–8903. doi: 10.1073/pnas.97.16.8898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Levchenko I, Smith C K, Walsh N P, Sauer R T, Baker T A. Cell. 1997;91:939–947. doi: 10.1016/s0092-8674(00)80485-7. [DOI] [PubMed] [Google Scholar]
- 43.Smith C K, Baker T A, Sauer R T. Proc Natl Acad USA. 1999;96:6678–6682. doi: 10.1073/pnas.96.12.6678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Katayama Y, Gottesman S, Pumphrey J, Rudikoff S, Clark W P, Maurizi M R. J Biol Chem. 1988;263:15226–15236. [PubMed] [Google Scholar]