

Abstract
Microtubule associated protein tau, which is expressed in six alternatively spliced molecular isoforms in human brain, is abnormally hyperphosphorylated in Alzheimer disease and related tauopathies. Here we show (i) that GSK-3α and neither GSK-3β nor cdk5 can phosphorylate tau at Ser262 and phosphorylation at Ser235 by cdk5 primes phosphorylation at Thr231 by GSK-3α/β; (ii) that tau isoforms with two N-terminal inserts (τ4L, τ3L) are phosphorylated by cdk5 plus GSK-3 at Thr231 markedly more than isoforms lacking these inserts (τ4, τ3); and (iii) that Thr231 is phosphorylated ~50% more in free tau than in microtubule-bound tau, and the phosphorylation at this site results in the dissociation of tau from microtubules. These findings suggest that the phosphorylation of tau at Thr231 and Ser262 by cdk5 plus GSK-3, which inhibits its normal biological activity, is regulated both by its amino terminal inserts and its physical state.
Keywords: Alzheimer disease, glycogen synthase kinase-3, cyclin-dependent protein kinase-5, tau hyperphosphorylation, neurofibrillary degeneration, tau kinases
Abbreviations used: AD, Alzheimer disease; PHF, paired helical filaments; NFT, neurofibrillary tangles; A-kinase, cyclic AMP-dependent protein kinase; CaM Kinase II, calcium/calmodulin-dependent protein kinase; C-kinase, calcium/phospholipids-dependent protein kinase; CK-1, casein kinase-1; cdk5, cyclin-dependent protein kinase 5; GSK-3, glycogen synthase kinase-3; MAP kinase, mitogen-activated protein kinase; PDPK, proline-dependent protein kinase; MT, microtubule; PMSF, phenyl methyl sulfonyl fluoride
1. Introduction
Alzheimer disease (AD) and related tauopathies are characterized by neurofibrillary degeneration of abnormally hyperphosphorylated tau [1]. The AD abnormally hyperphosphorylated tau does not bind to tubulin but inhibits in vitro microtubule assembly and disrupts preformed microtubules (MT) by sequestering normal tau, and high molecular weight microtubule associated proteins (MAPs), MAP1 and MAP2 [2–4]. These inhibitory effects of the AD hyperphosphorylated are, however, abolished by in vitro dephosphorylation by protein phosphatases (PP), especially PP-2A [5, 6]. Despite its importance, the molecular mechanism of the abnormal hyperphosphorylation of tau is not fully understood.
To date, over 25 abnormal sites have been identified at which tau in PHF is phosphorylated [7]. The phosphorylation of tau at only some of these sites might play a significant physiological role. Tau can be phosphorylated at one or more of these abnormal sites by several protein kinases (see [8]) Among these protein kinases, GSK-3 and cdk5 are believed to be major enzymes involved in Alzheimer abnormal hyperphosphorylation of tau. Cdk5 and GSK-3 co-localize with Alzheimer neurofibrillary degeneration [9–12] and both kinases have been shown to produce neurofibrillary degeneration of hyperphosphorylated tau in transgenic mice ([13, 14].
Phosphorylation of tau at some of the sites inhibits its binding to microtubules (MT) and its ability to promote microtubule assembly [2, 15]. Thr231 and Ser262 are the two major sites phosphorylation of which by cdk5 followed by GSK-3 inhibits binding to MT [16–18]. An important factor that might influence the phosphorylation of tau at specific sites that have received little attention to date is that tau is present in the brain in different substrate forms. In adult human brain, six isoforms of human tau are present. These isoforms, which are generated from a single gene through alternate mRNA splicing [19], vary in length between 352 and 441 amino acids and differ in whether they contain three (τ3, τ3S, τ3L) or four (τ4, τ4S, τ4L) microtubule binding domains of 31 or 32 amino acids each and none (τ3, τ4), one (τ3S, τ4S) or two (τ3L, τ4L) N-terminal inserts of 29 amino acids each. Furthermore, tau in brain is present in at least two major pools, (i) as free tau and (ii) as tau bound to MT.
In the present study we compared (i) the phosphorylation of tau isoforms with or without the N-terminal inserts by cdk5 and GSK-3, singly or in combination and their binding to MT, and (ii) the phosphorylation of free and microtubule-bound taus by these two protein kinases. We found (i) that GSK-3α but neither GSK-3β nor cdk5 could phosphorylate tau at Ser262; (ii) that both cdk5 and GSK-3α could phosphorylate Ser-235; (iii) that phosphorylation of tau by cdk5 markedly enhanced the phosphorylation of tau at Ser235 and resulted in a robust phosphorylation of tau at Thr231 by GSK-3, especially the GSK-3β; and (iv) that after phosphorylation by cdk5 and GSK-3, human tau isoforms bound to MT differentially. The phosphorylation at Thr231 and Ser262 by the above kinases and the consequent inhibition of binding to MT were more in tau isoforms with the two amino terminal inserts than the isoforms lacking the inserts. The phosphorylation of Thr231 and Ser262 was decreased in MT-bound tau compared to free tau, whereas the phosphorylation of Ser46, Ser396, Ser404 and Ser422 was unchanged. These findings suggest that the phosphorylation of tau at sites which affect its biological activity is regulated significantly at the substrate level.
2. Materials and Methods
Materials
Tau monoclonal antibodies (mAb) employed were as follows: M4 to tau phosphorylated at Thr231 and Ser235 [20] was a gift from Dr. Y. Ihara (University of Tokyo, Japan), 12E8 to tau phosphorylated at Ser262 and/or Ser356 [21] was a gift from Dr. Dale Schenk of Elan Pharmaceuticals (San Francisco, CA), and PHF-1 to tau phosphorylated at Ser396 and/or Ser404 [22] was a gift from Dr. P. Davies, Albert Einstein College of Medicine (Bronx, NY). Polyclonal tau antibodies 92e to total tau and 102c to tau dephosphorylated at Ser46 were raised in rabbits, as previously reported [23, 24]. Polyclonal rabbit R145 to tau phosphorylated at Ser422 was described previously [25]. Rabbit antibodies pT231, pS236, and pS262 to tau phosphorylated at Thr231, Ser235 and Ser262, respectively, were obtained from Biosource Intl. (Camarillo, CA). Rabbit antibodies 127d to GSK-3β and 133d to GSK-3α and GSK-3β were raised in our lab [10]. Polyclonal antibody to GSK-3α was from Upstate Biotechnology, Inc. (Lake Placid, NY), and monoclonal antibody to GSK-3β was from Transduction Labs (San Diego, CA). Recombinant GSK-3β was purchased from Calbiochem (San Diego, CA). 125I-labeled anti-mouse and anti-rabbit IgG antibodies were purchased from Amersham (Arlington Heights, IL). [γ-32P]ATP was purchased from ICN Biomedicals (Costa Mesa, CA).
Methods
The human tau clones 23, 24, 39 and 40 (kindly provided by Dr. M. Goedert) that encode for the isoforms τ3, τ4, τ3L and τ4L, respectively, [19] were subcloned in E.coli and purified from cell extracts as described previously [26]. pGEX-2T plasmids containing cdk5 and p25, kind gifts from Dr. Jerry H. Wang (The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong) were expressed and purified from E.coli as GST fusion proteins [27]. The purification of GSK-3 and cdk5 was as described [28]. The GSK-3 preparation contained both α and β isoforms in 3:2 ratio [26]. Cdk5 and GSK-3 were also purified by immunoprecipitation with appropriate antibodies from rat brain extract. The 16,000 x g brain extract, 50 μg protein was incubated with 2 μg of specific antibodies in 50 mM Tris, pH 7.4, 150 mM NaCl, 10 mM NaF, 1 mM Va3VO4, 2 mM EGTA, 1 mM phenylmethyl sulfonyl fluoride (PMSF) and 5 μg/ml each of leupeptin, pepstatin and aprotinin. After incubation overnight at 4°C, 30 μl of protein G immobilized on agarose beads (Pierce, Rockford, IL) was added and the reaction mixture was further incubated for 3 hours with constant mixing. The incubated mixture was then centrifuged and the beads were washed 3 times with immunoprecipitation buffer, resuspended in 10 μl of the phosphorylation reaction buffer, and used as a kinase.
MAPs-free tubulin was isolated from rat brain extract through two temperature-dependent cycles of microtubule polymerization-depolymerization, followed by chromatography on cellulose [2]. Taxol-stabilized MT were prepared by incubating MAPs-free tubulin (4 mg/ml) with taxol (20 μM) in the presence of MES (0.1 M<, pH 6.8), EGTA (1 mM) and PMSF (1 mM) for 30 min at 37°C. The MT were collected by centrifugation over a cushion of 0.125 M sucrose at 50,000 x g for 30 min at 32°C.
Tau isoforms were phosphorylated at 30°C in a reaction mixture normally containing 4.0 μM tau, 6 mM MgCl2, 10 mM 2-mercaptoethanol, 0.25 mM [γ-32P]ATP, and 40 mm HEPES (pH 7.5). Reactions were initiated at 30°C by the addition of kinases. For determination of 32P incorporation into protein, aliquots of the reaction mixtures were removed at different times, spotted on strips of Whatman filter paper (31ET) and free [γ-32P]ATP separated from 32P-labeled protein by ascending chromatography [26]. The 32P-labeled tau on the filters was quantitated by Cerenkov counting in a scintillation counter. When prephosphorylation of tau was required, [γ-32P]ATP was replaced with unlabeled ATP. After incubation for 2 hours at 30°C, the reaction was stopped by heating at 95°C for 5 min and denatured kinases removed by centrifugation (10,000 x g for 10 min). The extent of tau phosphorylation was determined in parallel experiments using [γ-32P]ATP. The heat-stable phosphorylated tau was then used as a substrate for the second kinase. In controls, the incubation was carried out without the first kinase but otherwise treated identically as the samples for combination phosphorylation.
To analyze the binding of tau to MT, 0.47 μM tau (non-phosphorylated or phosphorylated) was incubated with 7.4 μM taxol-stabilized MT for 30 min at 37°C in buffer A (0.1 M HEPES, pH 6.8, 2 mM MgCl2, 1 mM EDTA, 1 mM PMSF, 20 μM taxol and 1 mM GTP). Tau bound to MT was separated from unbound tau by centrifugation at 50,000 x g for 30 min at 32°C on a cushion of 10% glycerol. The pellet was washed twice with buffer A and dried in a Speed Vac. The combined supernatants were precipitated by acetone, centrifuged and the precipitate was again dried by a vacuum concentrator. All samples were electrophoresed in 10% SDS-polyacrylamide gels, transferred to nitrocellulose and immunoblots probed with the rabbit polyclonal phosphorylation-independent tau antibody, 92e (1/5,000). The immunoreactive bands were quantitated. In each set, the supernatant (free tau) and the pellet (bound tau) were calculated as percent of total tau. To assay the release of MT-bound tau, the phosphorylation reaction was stopped by cooling to 4°C and immediately centrifuged (50,000 x g) to separate tau which remained bound to microtubules (pellets) from free released tau (supernatant).
Immunoblotting of tau by the different antibodies was carried out as described previously [1]. The following dilutions of the primary antibodies were used: MT (1/1,000), 12E8 (1/500), PHF-1 (1/500), 92e (1/5,000), 102c (1/1,000) and anti-P-Ser422 (1/3,000). Blots were then further probed with anti-mouse or anti-rabbit IgG secondary antibodies which were labeled with 125I. The immunoreactive bands were visualized and quantitated by Fuji 1500 Phosphorimager Imaging System.
3. Results
Phosphorylation of free and MT-bound tau by a combination of cdk5 and GSK-3
To examine the regulation of the phosphorylation of tau by its state of conformation as a substrate, we investigated the phosphorylation of free and MT-bound taus by cdk5 and GSK-3. For this purpose, we chose two tau isoforms (see Fig. 1), τ3 and τ3L as free proteins and as bound to MT stabilized with taxol. Prephosphorylation of MT-bound tau by cdk5 increased the rate of subsequent phosphorylation by GSK-3 by ~25% in the case of τ3L but not τ3 (Fig. 2). On the other hand, prephosphorylation of free τ3 and τ3L by cdk5 increased subsequent phosphorylation by GSK-3 by ~30% and ~50%, respectively (Fig. 2C). These results suggest that prephosphorylation by cdk5 stimulates the subsequent phosphorylation by GSK-3 (i) more in tau with the N-terminal inserts than the same protein without the inserts, and (ii) more in free tau than the same as MT-bound protein.
Fig. 1.
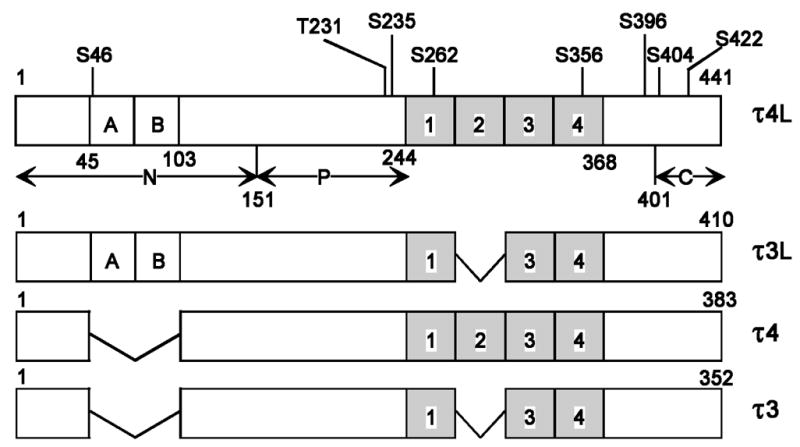
Schematic representation of the structure of the four human tau isoforms employed in the present study. Two of the isoforms (τ3, τ3L) contain three repeat microtubule-binding domains, whereas the other two isoforms (τ4, τ4L) contain four repeats (hatched areas 1, 2, 3, 4). In addition, τ3L and τ4L contain two N-terminal inserts, each 29 amino acids long (A and B). τ3 and τ4 lack such N-terminal inserts. Bar diagram of τ4L shows locations of eight Ser/Thr sites phosphorylated by cdk5 plus GSK-3. N = N-terminal domain, P = proline-rich region, and C = C-terminal tail. τ4L, τ3L, τ4 and τ3 are 441, 410, 383, and 352 amino acid long, respectively.
Fig. 2.
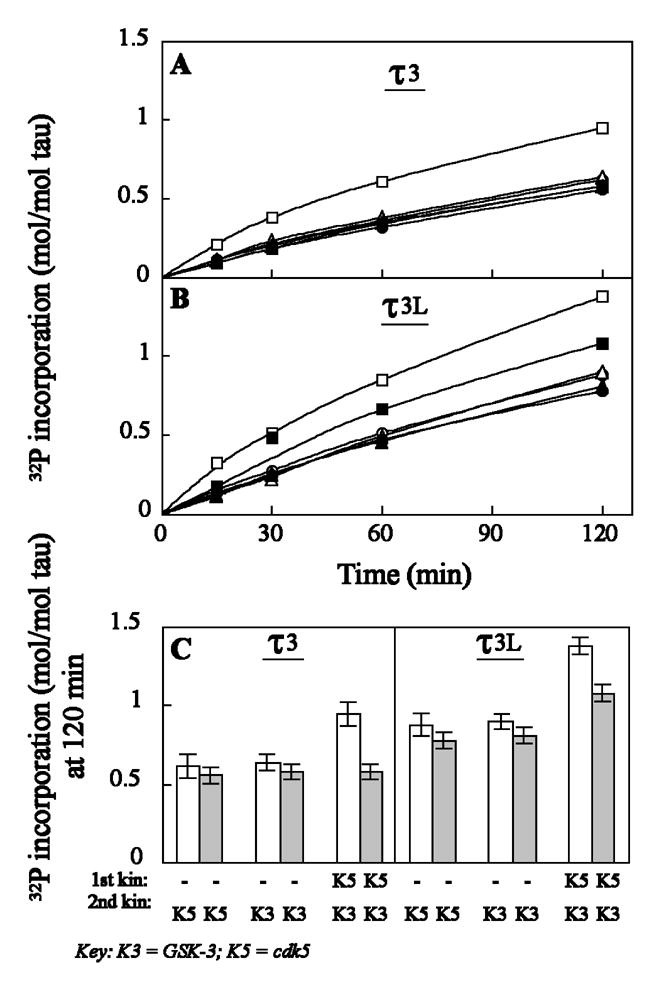
Phosphorylation of free and microtubule-bound tau isoforms (τ3 and τ3L) by a combination of cdk5 (K5) and GSK-3 (K3). τ3 (A) and τ3L (b) as free (open symbols) or microtubule-bound (closed symbols) incubated in the absence (○, •, ▵ and ▴) or presence (□, ▪) of cdk5 as described in Materials and Methods. After 120 min at 30°C, the phosphorylated taus (with non-radioactive ATP) were further phosphorylated by [γ-32P]ATP and cdk5 (○, •) or GSK-3 (▵, ▴, □ and ▪) for another 120 min at 30°C. Aliquots of the phosphorylation mixture were removed at different times, the reaction was stopped by heating at 95°C for 5 min and denatured tubulin removed by centrifugation (10,000 x g, 10 min). The heat-stable phosphorylated tau in the supernatant was spotted on filter paper and 32P incorporation was analyzed as described in Materials and Methods; (□, ▪) incubated with cdk5, followed by GSK-3; (▵, ▴) incubated without kinase, followed by GSK-3; (○, •) incubated without kinase followed by cdk5. (C) 32P-incorporation by second kinase in 120 min at 30°C. Open bars, free taus; closed bars, MT-bound taus.
Binding of different phosphorylated species of tau isoforms to microtubules
Since the phosphorylation of tau at only some and not all sites might be of physiological significance, we investigated whether the upregulation of the phosphorylation of tau due to N-terminal inserts inhibited its binding to MT. We examined the effect of phosphorylation by cdk5, GSK-3 or by combination of cdk5 and GSK-3 on the binding of tau to MT using τ3, τ4, τ3L, and τ4L. For all the four tau isoforms, the kinase combination cdk5 followed by GSK-3 was most effective for causing inhibition of tau binding to MT (Fig. 3A). In contrast, no significant increase in inhibition was observed when tau phosphorylated by GSK-3 was subsequently phosphorylated by cdk5 (compare column K3K5 with column K3 in each panel). For both τ3L and τ4L, phosphorylation by cdk5 followed by GSK-3 combination resulted in ~80% inhibition of binding of phosphorylated tau to MT, whereas for both τ3 and τ4, this inhibition was ~40% (Table 1). Thus, the presence of N-terminal inserts markedly reduced the binding of tau to MT when it was phosphorylated by a combination of cdk5 and GSK-3. Unlike the amino-terminal inserts, the presence of the fourth microtubule binding repeat did not significantly affect on phosphorylation by cdk5 and GSK-3 the binding of tau to MT.
Fig. 3.
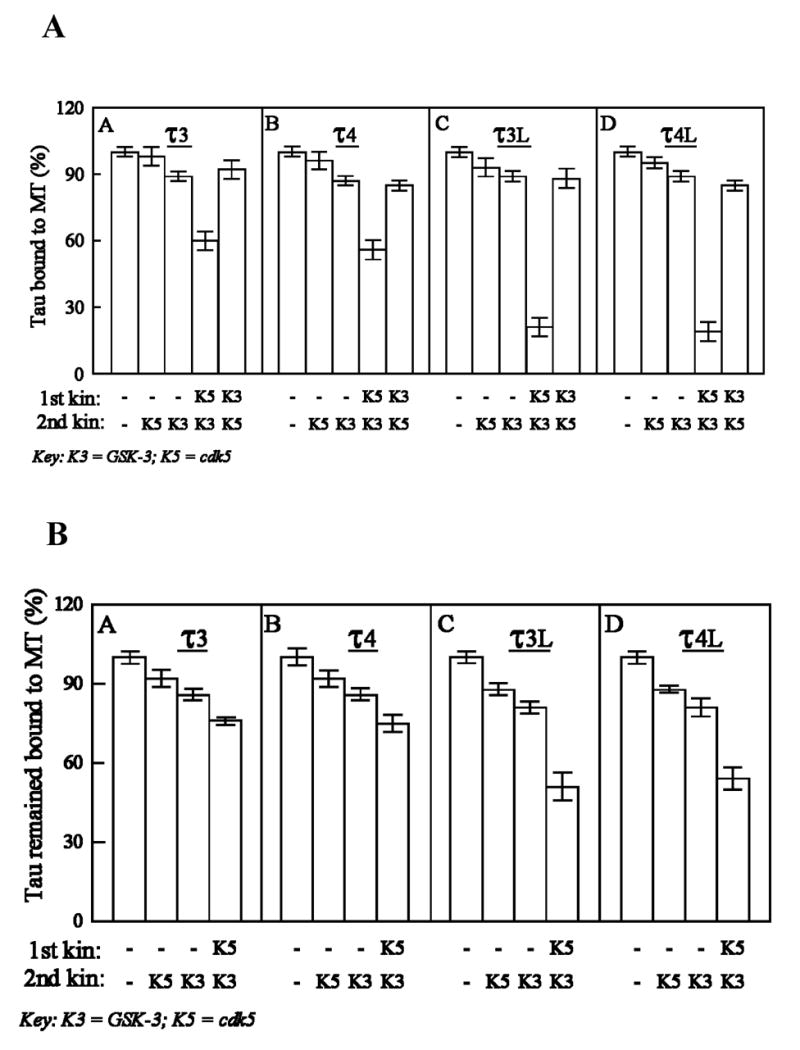
(A) Comparison of the binding of different phosphorylated species of tau isoforms to microtubules, and (B) release of microtubule-bound tau isoforms after phosphorylation by cdk5 and GSK-3. τ3 (A), τ4 (B), τ3L (C) or τ4L (D) was phosphorylated by cdk5 and or GSK-3, and its binding to taxol-stabilized MT was determined (A). Alternatively, taxol-stabilized MT were phosphorylated as above and the release of tau from MT was determined (B). Tau levels were assayed by immunoblots developed with phosphorylation-independent tau antibody 92e, and probed with 125I-labeled secondary antibody. Immunoreactive bands were quantitated. All values are expressed as a percentage of the binding observed with non-phosphorylated-τ (100%). Data (mean ± SD) are from four separate experiments.
Table 1.
Effect of phosphorylation by cdk5, GSK-3 or cdk5 plus GSK-3 on the binding of different free or MT-bound tau isoforms to microtubules
| τ isoform used | Kinase used (1st, 2nd) | τ bound to MT (%)(phosphorylation of τ followed by binding to MT) | τ remained bound to MT (%)(phosphorylation of MT-bound τ) |
|---|---|---|---|
| τ3 | – | 100 ± 2 | 100 ± 2 |
| –, cdk5 | 98 ± 4 | 92 ± 3 | |
| –, GSK-3 | 89 ± 2 | 86 ± 2 | |
| cdk5, GSK-3 | 60 ± 4* | 76 ± 1* | |
| τ4 | – | 100 ± 2 | 100 ± 3 |
| –, cdk5 | 96 ± 2 | 92 ± 3 | |
| –, GSK-3 | 87 ± 2 | 86 ± 2 | |
| cdk5, GSK-3 | 56 ± 4* | 75 ± 3* | |
| τ3L | – | 100 ± 2 | 100 ± 3 |
| –, cdk5 | 93 ± 3 | 88 ± 2 | |
| –, GSK-3 | 89 ± 2 | 81 ± 2 | |
| cdk5, GSK-3 | 21 ± 3** | 51 ± 5** | |
| τ4L | – | 100 ± 3 | 100 ± 2 |
| –, cdk5 | 95 ± 2 | 88 ± 2 | |
| –, GSK-3 | 89 ± 2 | 81 ± 3 | |
| cdk5, GSK-3 | 19 ± 4** | 54 ± 4** |
p<0.05;
p<0.01
Next, we examined whether the N-terminal inserts or the fourth repeat regulate the phosphorylation of the MT-bound tau that affects tau’s association to MT. For this purpose, we first bound tau to taxol-stabilized microtubules (tubulin/tau molar ratio 15:1, well below the saturation level) and then different MT-bound tau species were phosphorylated by cdk5 and GSK-3, acting singly or in combination. The released tau remained in the supernatant, whereas the MT-bound tau was recovered in the pellet. The MT-bound tau identically treated but without kinase(s) served as a control. We found that ~8% tau was released when MT-bound τ3, τ4, τ3L and τ4L were incubated in phosphorylation buffer in the absence of any kinase. When the MT-bound tau isoforms were phosphorylated by the combination of cdk5 and GSK-3, as a result of this phosphorylation 24%, 25%, 49% and 46% of τ3, τ4, τ3L and τ4L, respectively, were released, compared to the MT-bound taus which were not phosphorylated (100%; Fig. 3B). A comparison of these results to those in Fig. 3A revealed that when phosphorylated by cdk5 plus GSK-3, MT-bound taus behave differently from the free tau in associating to MT; the MT-bound was ~30% (τ3, τ4) to ~300% (τ3L, τ4L) more associated to MT than the corresponding free taus when phosphorylated with cdk5 followed by GSK-3 (see Table 1). The fourth repeat had no significant effect on the release of tau from MT produced by phosphorylation by cdk5 and GSK-3 (compare τ3 with τ4 and τ3L with τ4L in Table 1).
Phosphorylation of tau at Thr231, Ser235 and Ser262 by cdk5, GSK-3α and GSK-3β
While both Thr231 and Ser235 are canonical sites for proline-dependent protein kinases and, thus, are expected to be phosphorylated by the proline-dependent protein kinases cdk5 and GSK-3, the phosphorylation of tau at Ser262, a non-proline-dependent site, by these kinases has been controversial [29–35]. Thus, before ascribing the effect of the phosphorylation of tau to Thr231, Ser235 or Ser262 by cdk5 and GSK-3, we investigated which of these three sites could be phosphorylated by the two kinases. For these studies, we employed recombinant enzymes and as well each kinase purified by immunoprecipitation with different specific antibodies. We found that GSK-3α and neither GSK-3β nor cdk5 could phosphorylate tau at Ser262 (Fig. 4A,B). Both PKA and CaMKII, used as positive controls, as expected, phosphorylated τ4L at Ser262.
Fig. 4.

In vitro phosphorylation of recombinant human brain τ4L at Ser262 (A, B), Thr231 or Ser235 (C), by different protein kinases. τ4L was phosphorylated for 2 hr at 30°C, followed by Western blots of the samples developed with a rabbit antibody pS262 (Biosource Intl.) to tau phosphorylated at Ser262 (A, B); mAb M4 = a mouse monoclonal antibody to tau phosphorylated at Thr231/Ser235 (C); a.pT231 = a rabbit antibody to tau phosphorylated at Thr231 (Biosource Intl.); and a.pSer235 = a rabbit antibody to tau phosphorylated at Ser235 (Biosource Intl.). Cont = τ4L treated identically but without any kinase; rc = recombinant; IP = immunoprecipitated; UBI = antibody from Upstate Biotechnology, Inc.; TL = antibody from Transduction Labs; 127d = a rabbit antibody to GSK-3β [10]; cdk5 rc = recombinant cdk5 plus p25. cdk5IP in (B) and a.pT231 staining of Cont. in (C) are non-specific.
cdk5 phosphorylated tau at Ser235 more robustly than GSK-3α; the phosphorylation of tau at Ser235 by GSK-3β was not detected (Fig. 4C). However, phosphorylation of tau by cdk5 not only markedly enhanced the phosphorylation at Ser235 but also resulted in a robust phosphorylation of Thr231 by GSK-3, especially the GSK-3β.
Phosphorylation of free and MT-bound taus at different Alzheimer abnormal phosphorylation sites
To determine the specificity of the differences in the phosphorylation of tau at Thr-231/Ser235 and Ser262/Ser356 between the free and the MT-bound taus, we studied the phosphorylation of these taus at several other abnormal sites. With the aid of site-specific phosphorylation-dependent antibodies, we examined the phosphorylation of eight sites (Ser46, Thr231, Ser235, Ser262, Ser356, Ser396, Ser404 and Ser422 in τ3L and τ4L, and all the above sites except Ser46 in τ3 and τ4, because τ3 and τ4 lack Ser46, which is in the N-terminal inserts) which are known to be phosphorylated in PHF-tau [7, 36]. The antibody 102c binds to dephosphorylated Ser46, M4 binds to P-Thr231 and P-Ser235, 12E8 binds to P-Ser262 and P-Ser356, PHF-1 binds to P-Ser396 and P-Ser404, and anti-P-Ser422 antibody binds to P-Ser422. Quantitative data from immunoblots revealed that the extent of phosphorylation of Ser46 in both free and MT-bound taus (τ3L and τ4L) were similar, which indicates that this N-terminal site in both free and MT-bound taus is equally susceptible to phosphorylation (Fig. 5). Thr231/Ser235 and Ser262/Ser356 were phosphorylated in free tau to the extent of ~1.5-fold higher in all the four isoforms studied compared to the corresponding MT-bound taus, indicating that the proline-rich and the repeat regions might be either involved in the binding of tau to MT or present as less favorable conformation for phosphorylation in MT-bound tau. Both free and MT-bound taus with two N-terminal inserts (τ3L and τ4L) were phosphorylated at Thr231/Ser235 and Ser262/Ser356 by cdk5 and GSK-3 markedly more than taus lacking these inserts (τ3 and τ4). However, no such differences in the extent of phosphorylation of Ser396/Ser404 and Ser422 in free or MT-bound taus were found. These findings suggested that C-terminal sites were equally accessible for phosphorylation in both free and MT-bound taus.
Fig. 5.
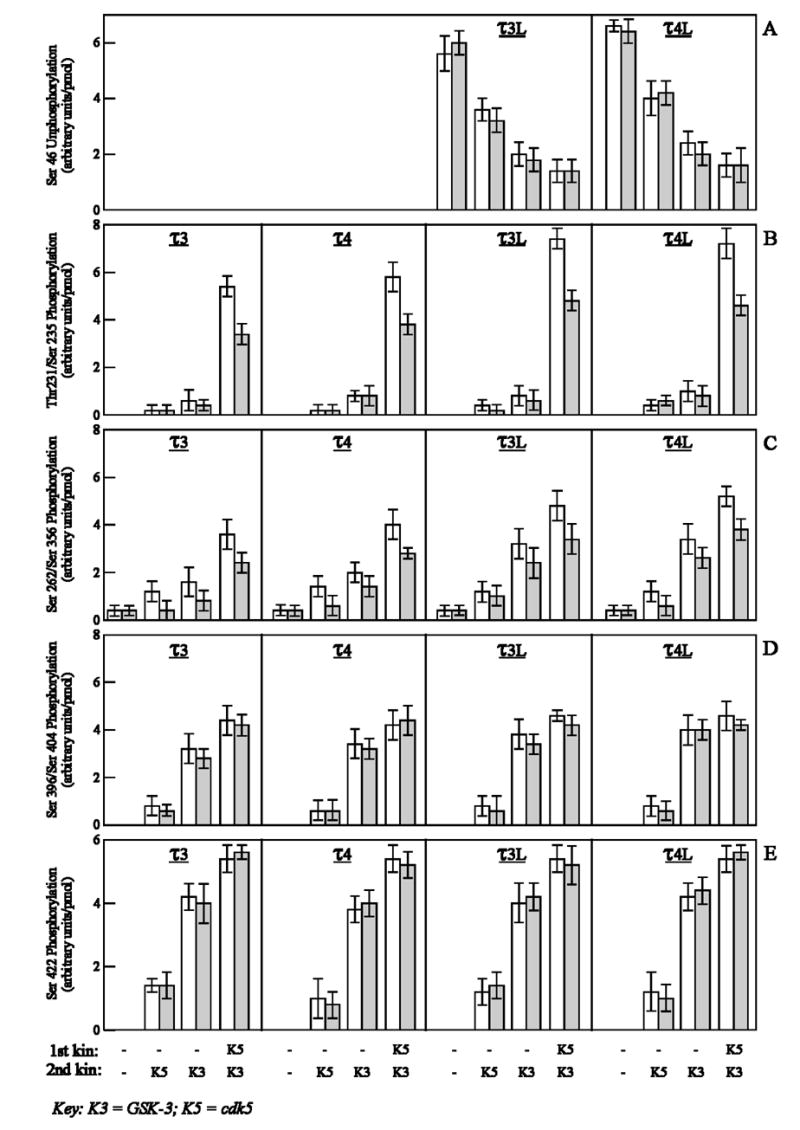
Phosphorylation of Ser46, Thr231/Ser235, Ser262/Ser356, Ser396/Ser404 and Ser422 in free and microtubule-bound tau isoforms by cdk5, GSK-3 and cdk5 plus GSK-3. τ3, τ4, τ3L, or τ4L (4 μM) were incubated in the phosphorylation reaction mixture as free (open bars) or microtubule-bound (closed bars) in the absence (−) or presence of cdk5 (K5). After 2 hr at 30°C, the phosphorylated taus were further phosphorylated by cdk5 (K5) or GSK-3 (K3) for 2 hr at 30°C. The reactions were terminated by heating with SDS-PAGE sample buffer at 95°C for 5 min. The different phosphorylated tau species were immunoblotted with 102c (A), M4 (B), 12E8 (C), PHF-1 (D) and anti-PSer422 (E) antibodies, and probed with 125I-labeled secondary antibodies. Data (mean ± SD) are from four separate experiments.
The confirmation of the above findings was provided by using three mutants of τ4L, namely τ4L (Ala231), τ4L (Ala235) and τ4L (Ala262) in which Thr231, Ser235 and Ser262 were mutated to Ala231, Ala235 and Ala262, respectively. As in Fig. 4, we first bound each mutant tau to taxol-stabilized microtubules and then the different MT-bound tau species were phosphorylated by cdk5, GSK-3 or cdk5 followed by GSK-3. As a result of phosphorylation, a part of the bound tau was released in the supernatant, whereas the rest of it (still bound to MT) was recovered in the pellet. The percent of mutant and wild-type taus that remained bound to MT were compared (Fig. 6). After phosphorylation by cdk5 plus GSK-3, the MT-bound Ala231 mutant tau remained bound to a ~25% higher extent compared to MT-bound wild tau. Similarly, when phosphorylated by a combination of cdk5 plus GSK-3, the MT-bound tau mutants Ala262 and Ala235 were bound to MT to ~13% and ~2% higher extents, respectively, compared to MT-bound wild-type tau. Thus, the phosphorylation of Thr231 and Ser262 in MT-bound tau contributed ~25% and ~13% inhibition in binding to MT. Previously, we have shown that, in free tau, phosphorylation of Thr231 and Ser262 contribute almost equally (~30%) towards inhibition of binding of tau to MT [16]. Together, these results suggest that, as a whole, the phosphorylation of Thr231 contributes more than the phosphorylation of Ser262 in the inhibition of tau binding to microtubules.
Fig. 6.
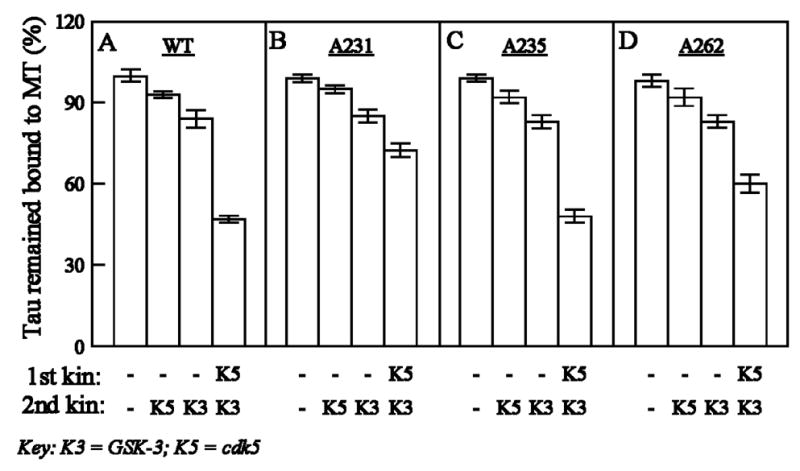
Comparison of the release of microtubule-bound wild-type τ4L and different τ4L mutants after phosphorylation by cdk5 and GSK-3. τ4L (A), τ4L(Ala231) (B), τ4L(Ala235) (C) and τ4L(Ala262) (D) were bound to taxol-stabilized microtubules and then phosphorylated by the first and second kinases as in Fig. 4. The reactions were stopped and the levels of wild-type and mutant taus that remained bound to MT assessed as in Fig. 4. All values are expressed as a percentage of the binding observed with MT-bound non-phosphorylated-τ4L (100%). Data (mean ± SD) are from four different experiments.
4. Discussion
Abnormal hyperphosphorylation of tau is a key biochemical abnormality that leads to neuronal degeneration, the formation of neurofibrillary tangles and the dementia syndrome (for review see [8]). Thus, it is critical to understand the regulation of the phosphorylation of tau. The hyperphosphorylation of tau appears to be controlled at both enzyme and substrate levels. The tau hyperphosphorylation at enzyme level could occur by activation of one or more tau kinases and decline in the activities of one or more tau phosphatases. Furthermore, the activities of both kinases and phosphatases can be modulated at substrate (tau) level. This could occur due to difference in the prior phosphorylation states of tau and as well as controlled by tau’s structure. Previous studies had focused primarily on the levels of the activities of tau kinases and tau phosphatases in AD brain and on the effect of phosphorylation of tau by one kinase on the subsequent phosphorylation by a second kinase (see ref. [8]).
The present study demonstrates that the phosphorylation of tau can also be regulated at the substrate level by tau’s structure. We found that tau isoforms with the two amino terminal inserts (τ4L, τ3L) were more favorable substrates than tau isoforms without the inserts (τ4, τ3) for phosphorylation by cdk5 followed by GSK-3 at Thr231 and Ser262, the two major sites involved in the binding of tau to microtubules. The combined action of these two protein kinases, which is believed to be involved in the abnormal hyperphosphorylation of tau in AD [26, 28, 37], caused considerably more inhibition (~80%) of binding to microtubules of tau isoforms with two amino terminal inserts than tau isoforms lacking these inserts (~40%) (Table 1). Previous studies have also been consistent with the presence of one or both amino terminal inserts making tau a more favorable substrate for phosphorylation by protein kinases [38]. The presence of both the extra microtubule binding repeat (4R taus) and as well as the amino terminal inserts make tau a more favorable substrate for sequestration by the AD abnormally hyperphosphorylated tau [39] and its self-assembly into tangles of PHF [40].
The present study showed that the phosphorylation of sites in the proline-rich region (Thr231/Ser235) and the microtubule binding repeats (Ser262/Ser356) but neither in the amino terminal (Ser46) nor carboxy terminal tail (Ser396/Ser404, Ser422) regions were decreased in MT-bound tau as compared to free tau. This decreased phosphorylation of the MT-bound τ4L and τ3L, which is consistent with the retardation of phosphorylation of MT-bound τ4L at Ser262 by MARK110 kinase reported previously [41], also resulted in about one third (~30%) lesser release of these proteins from MT than the binding of these tau isoforms to MT after phosphorylation by cdk5 plus GSK-3. We found that in MT-bound tau, cdk5 prephosphorylated tau and unmodified tau were phosphorylated to the same extent by GSK-3. Thus, Thr231/Ser235, which on phosphorylation by cdk5 become more accessible for subsequent phosphorylation by GSK-3 in free tau with N-terminal inserts, are protected in MT-bound tau.
The role of Thr231 and Ser262 phosphorylations in the inhibition of the binding of tau to MT was confirmed by studies in which these sites were mutated to Ala. While on phosphorylation by ckd5 plus GSK-3 MT-bound τ4L remained bound to MT to the extent of ~50%, binding of Ala231 and Ala262 mutant taus to MT was increased by ~25% and ~13%, respectively. Hence, in MT-bound tau, both of these sites appeared to be involved in controlling tau binding to MT. It is important to note that, in MT-bound tau, Thr231 contributes more (~25%) than Ser262 (~13%) in the inhibition of binding of tau to MT when phosphorylated, whereas in a previous study [16], phosphorylation of free tau at Thr231 and Ser262 was found to contribute ~26% and ~33%, respectively, of the overall inhibition of tau binding to microtubules. Thus, free tau and MT-bound tau may attain different conformations upon phosphorylation which, in turn, bind differently to microtubules.
It appears that not all sites which have been reported to be phosphorylated in PHF-tau directly affect the binding of tau to microtubules. We found that the sites Ser46, Ser396, Ser404 and Ser422 are phosphorylated to the same extent in both free and microtubule-bound tau. On the contrary, Thr231/Ser235 and Ser262/Ser356 are phosphorylated to a higher extent in free tau compared to MT-bound tau. These results suggest that both the proline-rich region and repeat regions are directly involved in the binding of tau to microtubules, whereas the N-terminal and C-terminal regions exert only indirect effect to this binding. These findings are consistent with those of Goode et al. [42], who showed that functional interaction between proline-rich and repeat regions of tau enhanced microtubule binding and assembly. Phosphorylation of tau at Ser396 and the subsequent reduction of its binding to MT reported previously [43] is probably an example of indirect involvement of the C-terminal region in the binding of tau to MT.
Ser262 is the only site abnormally hyperphosphorylated in the microtubule binding repeat region of tau in AD. This site is believed to play a major role in regulating the binding of tau to MT [41]. GSK-3 and cdk5 have been implicated as two major protein kinases involved in the abnormal hyperphosphorylation of tau in AD. The phosphorylation of tau at Ser262 by these two protein kinases, however, has been controversial [27, 29–35]. The present study, employing both recombinant cdk5 and GSK-3 and these protein kinases purified by immunoprecipitation, have demonstrated that GSK-3α and neither GSK-3β nor cdk5 could phosphorylate Ser262 in tau. The GSK-3 isolated from brain is a mixture of GSK-3α and GSK-3β. The phosphorylation of tau at Ser262 by GSK-3 isolated from brain reported previously [29, 32–34] was most probably due to GSK-3α and not GSK-3β in the preparations. The present study suggests a possible specific role of GSK-3α in the phosphorylation of tau at Ser262.
The isoform composition of tau is altered in different neurodegenerative disorders that are characterized by tau pathology, and in every one of these diseases, the tau is abnormally hyperphosphorylated (see ref. [8]). Furthermore, several missense or deletion mutations, especially in the microtubule binding regions of tau, and the segregation of these mutations with the disease in the hereditary frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17) have been observed [44]. In light of the present study, which shows the substrate regulation of the phosphorylation of tau at sites that affect its biological activity, we speculate the involvement of this process in the pathogenesis of Alzheimer disease and related tauopathies in which the isoforms of the composition of tau is altered.
Acknowledgments
We would like to thank Y. Ihara (University of Tokyo) for mAb M4 and Ezzat El-Akkad and T. Zaidi for purification of recombinant taus. We are also grateful to Janet Murphy for secretarial assistance. These studies were supported in part by the New York State Office of Mental Retardation and Developmental Disabilities, National Institutes of Health grants AG08776, AG05892, AG019158, NS18105, Howard Hughes Medical Institute (Grant #75195-54701, MN), Human Frontier Science Program Organization (Grant #93/93B, MN), and Slovak Academy of Sciences.
Footnotes
E-mail: < iqbalk@worldnet.att.net >
References
- 1.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. 1986;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alonso A, Zaidi T, Grundke-Iqbal I, Iqbal K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:5562–5566. doi: 10.1073/pnas.91.12.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alonso A, Grundke-Iqbal I, Barra HS, Iqbal K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc Natl Acad Sci USA. 1997;94:298–303. doi: 10.1073/pnas.94.1.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alonso Adel C, Li B, Grundke-Iqbal I, Iqbal K. Polymerization of hyperphosphorylated tau into filaments eliminates its inhibitory activity. Proc Natl Acad Sci U S A. 2006;103:8864–8869. doi: 10.1073/pnas.0603214103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang JZ, Gong CX, Zaidi T, Grundke-Iqbal I, Iqbal K. Dephosphorylation of Alzheimer paired helical filaments by protein phosphatase-2A and -2B. J Biol Chem. 1995;270:4854–4860. doi: 10.1074/jbc.270.9.4854. [DOI] [PubMed] [Google Scholar]
- 6.Wang JZ, Grundke-Iqbal I, Iqbal K. Restoration of biological activity of Alzheimer abnormally phosphorylated tau by dephosphorylation with protein phosphatase-2A, -2B and -1. Brain Res Mol Brain Res. 1996;38:200–208. doi: 10.1016/0169-328x(95)00316-k. [DOI] [PubMed] [Google Scholar]
- 7.Hanger DP, Betts JC, Loviny TL, Blackstock WP, Anderton BH. New phosphorylation sites identified in hyperphosphorylated tau (paired helical filament-tau) from Alzheimer’s disease brain using nanoelectrospray mass spectrometry. J Neurochem. 1998;71:2465–2476. doi: 10.1046/j.1471-4159.1998.71062465.x. [DOI] [PubMed] [Google Scholar]
- 8.Iqbal K, Alonso Adel C, Chen S, Chohan MO, El-Akkad E, Gong CX, Khatoon S, Li B, Liu F, Rahman A, Tanimukai H, Grundke-Iqbal I. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta. 2005;1739:198–210. doi: 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 9.Yamaguchi H, Ishiguro K, Uchida T, Takashima A, Lemere CA, Imahori K. Preferential labeling of Alzheimer neurofibrillary tangles with antisera for tau protein kinase (TPK) I/glycogen synthase kinase-3 beta and cyclin-dependent kinase 5, a component of TPK II. Acta Neuropathol (Berl) 1996;92:232–241. doi: 10.1007/s004010050513. [DOI] [PubMed] [Google Scholar]
- 10.Pei JJ, Tanaka T, Tung YC, Braak E, Iqbal K, Grundke-Iqbal I. Distribution, levels, and activity of glycogen synthase kinase-3 in the Alzheimer disease brain. J Neuropathol Exp Neurol. 1997;56:70–78. doi: 10.1097/00005072-199701000-00007. [DOI] [PubMed] [Google Scholar]
- 11.Pei JJ, Grundke-Iqbal I, Iqbal K, Bogdanovic N, Winblad B, Cowburn RF. Accumulation of cyclin-dependent kinase 5 (cdk5) in neurons with early stages of Alzheimer’s disease neurofibrillary degeneration. Brain Res. 1998;797:267–277. doi: 10.1016/s0006-8993(98)00296-0. [DOI] [PubMed] [Google Scholar]
- 12.Pei JJ, Braak E, Braak H, Grundke-Iqbal I, Iqbal K, Winblad B, Cowburn RF. Distribution of active glycogen synthase kinase 3beta (GSK-3beta) in brains staged for Alzheimer disease neurofibrillary changes. J Neuropathol Exp Neurol. 1999;58:1010–1019. doi: 10.1097/00005072-199909000-00011. [DOI] [PubMed] [Google Scholar]
- 13.Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003;40:471–483. doi: 10.1016/s0896-6273(03)00627-5. [DOI] [PubMed] [Google Scholar]
- 14.Engel T, Hernandez F, Avila J, Lucas JJ. Full reversal of Alzheimer’s disease-like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase-3. J Neurosci. 2006;26:5083–5090. doi: 10.1523/JNEUROSCI.0604-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho JH, Johnson GV. Primed phosphorylation of tau at Thr231 by glycogen synthase kinase 3beta (GSK3beta) plays a critical role in regulating tau’s ability to bind and stabilize microtubules. J Neurochem. 2004;88:349–358. doi: 10.1111/j.1471-4159.2004.02155.x. [DOI] [PubMed] [Google Scholar]
- 16.Sengupta A, Kabat J, Novak M, Wu Q, Grundke-Iqbal I, Iqbal K. Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch Biochem Biophys. 1998;357:299–309. doi: 10.1006/abbi.1998.0813. [DOI] [PubMed] [Google Scholar]
- 17.Li T, Hawkes C, Qureshi HY, Kar S, Paudel HK. Cyclin-dependent protein kinase 5 primes microtubule-associated protein tau site-specifically for glycogen synthase kinase 3beta. Biochemistry. 2006;45:3134–3145. doi: 10.1021/bi051635j. [DOI] [PubMed] [Google Scholar]
- 18.Plattner F, Angelo M, Giese KP. The roles of cyclin-dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J Biol Chem. 2006 doi: 10.1074/jbc.M603469200. [DOI] [PubMed] [Google Scholar]
- 19.Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 20.Hasegawa M, Watanabe A, Takio K, Suzuki M, Arai T, Titani K, Ihara Y. Characterization of two distinct monoclonal antibodies to paired helical filaments: further evidence for fetal-type phosphorylation of the tau in paired helical filaments. J Neurochem. 1993;60:2068–2077. doi: 10.1111/j.1471-4159.1993.tb03491.x. [DOI] [PubMed] [Google Scholar]
- 21.Seubert P, Mawal-Dewan M, Barbour R, Jakes R, Goedert M, Johnson GV, Litersky JM, Schenk D, Lieberburg I, Trojanowski JQ, et al. Detection of phosphorylated Ser262 in fetal tau, adult tau, and paired helical filament tau. J Biol Chem. 1995;270:18917–18922. doi: 10.1074/jbc.270.32.18917. [DOI] [PubMed] [Google Scholar]
- 22.Greenberg SG, Davies P. A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci USA. 1990;87:5827–5831. doi: 10.1073/pnas.87.15.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grundke-Iqbal I, Vorbrodt AW, Iqbal K, Tung YC, Wang GP, Wisniewski HM. Microtubule-associated polypeptides tau are altered in Alzheimer paired helical filaments. Brain Res. 1988;464:43–52. doi: 10.1016/0169-328x(88)90017-4. [DOI] [PubMed] [Google Scholar]
- 24.Iqbal K, Grundke-Iqbal I, Smith AJ, George L, Tung YC, Zaidi T. Identification and localization of a tau peptide to paired helical filaments of Alzheimer disease. Proc Natl Acad Sci USA. 1989;86:5646–5650. doi: 10.1073/pnas.86.14.5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tanaka T, Zhong J, Iqbal K, Trenkner E, Grundke-Iqbal I. The regulation of phosphorylation of tau in SY5Y neuroblastoma cells: the role of protein phosphatases. FEBS Lett. 1998;426:248–254. doi: 10.1016/s0014-5793(98)00346-9. [DOI] [PubMed] [Google Scholar]
- 26.Singh TJ, Haque N, Grundke-Iqbal I, Iqbal K. Rapid Alzheimer-like phosphorylation of tau by the synergistic actions of non-proline-dependent protein kinases and GSK-3. FEBS Lett. 1995;358:267–272. doi: 10.1016/0014-5793(94)01445-7. [DOI] [PubMed] [Google Scholar]
- 27.Qi Z, Huang QQ, Lee KY, Lew J, Wang JH. Reconstitution of neuronal Cdc2-like kinase from bacteria-expressed Cdk5 and an active fragment of the brain-specific activator. Kinase activation in the absence of Cdk5 phosphorylation. J Biol Chem. 1995;270:10847–10854. doi: 10.1074/jbc.270.18.10847. [DOI] [PubMed] [Google Scholar]
- 28.Sengupta A, Wu Q, Grundke-Iqbal I, Iqbal K, Singh TJ. Potentiation of GSK-3-catalyzed Alzheimer-like phosphorylation of human tau by cdk5. Mol Cell Biochem. 1997;167:99–105. doi: 10.1023/a:1006883924775. [DOI] [PubMed] [Google Scholar]
- 29.Singh TJ, Wang JZ, Novak M, Kontzekova E, Grundke-Iqbal I, Iqbal K. Calcium/calmodulin-dependent protein kinase II phosphorylates tau at Ser-262 but only partially inhibits its binding to microtubules. FEBS Lett. 1996;387:145–148. doi: 10.1016/0014-5793(96)00485-1. [DOI] [PubMed] [Google Scholar]
- 30.Taniguchi S, Fujita Y, Hayashi S, Kakita A, Takahashi H, Murayama S, Saido TC, Hisanaga S, Iwatsubo T, Hasegawa M. Calpain-mediated degradation of p35 to p25 in postmortem human and rat brains. FEBS Lett. 2001;489:46–50. doi: 10.1016/s0014-5793(00)02431-5. [DOI] [PubMed] [Google Scholar]
- 31.Paudel HK, Lew J, Ali Z, Wang JH. Brain proline-directed protein kinase phosphorylates tau on sites that are abnormally phosphorylated in tau associated with Alzheimer’s paired helical filaments. J Biol Chem. 1993;268:23512–23518. [PubMed] [Google Scholar]
- 32.Yang SD, Yu JS, Shiah SG, Huang JJ. Protein kinase FA/glycogen synthase kinase-3 alpha after heparin potentiation phosphorylates tau on sites abnormally phosphorylated in Alzheimer’s disease brain. J Neurochem. 1994;63:1416–1425. doi: 10.1046/j.1471-4159.1994.63041416.x. [DOI] [PubMed] [Google Scholar]
- 33.Song JS, Yang SD. Tau protein kinase I/GSK-3 beta/kinase FA in heparin phosphorylates tau on Ser199, Thr231, Ser235, Ser262, Ser369, and Ser400 sites phosphorylated in Alzheimer disease brain. J Protein Chem. 1995;14:95–105. doi: 10.1007/BF01888367. [DOI] [PubMed] [Google Scholar]
- 34.Moreno FJ, Medina M, Perez M, Montejo de Garcini E, Avila J. Glycogen synthase kinase 3 phosphorylates recombinant human tau protein at serine-262 in the presence of heparin (or tubulin) FEBS Lett. 1995;372:65–68. doi: 10.1016/0014-5793(95)00934-2. [DOI] [PubMed] [Google Scholar]
- 35.Godemann R, Biernat J, Mandelkow E, Mandelkow EM. Phosphorylation of tau protein by recombinant GSK-3beta: pronounced phosphorylation at select Ser/Thr-Pro motifs but no phosphorylation at Ser262 in the repeat domain. FEBS Lett. 1999;454:157–164. doi: 10.1016/s0014-5793(99)00741-3. [DOI] [PubMed] [Google Scholar]
- 36.Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Yoshida H, Titani K, Ihara Y. Proline-directed and non-proline-directed phosphorylation of PHF-tau. J Biol Chem. 1995;270:823–829. doi: 10.1074/jbc.270.2.823. [DOI] [PubMed] [Google Scholar]
- 37.Arioka M, Tsukamoto M, Ishiguro K, Kato R, Sato K, Imahori K, Uchida T. Tau protein kinase II is involved in the regulation of the normal phosphorylation state of tau protein. J Neurochem. 1993;60:461–468. doi: 10.1111/j.1471-4159.1993.tb03173.x. [DOI] [PubMed] [Google Scholar]
- 38.Singh TJ, Grundke-Iqbal I, Iqbal K. Differential phosphorylation of human tau isoforms containing three repeats by several protein kinases. Arch Biochem Biophys. 1996;328:43–50. doi: 10.1006/abbi.1996.0140. [DOI] [PubMed] [Google Scholar]
- 39.Alonso A, Zaidi T, Novak M, Barra HS, Grundke-Iqbal I, Iqbal K. Interaction of tau isoforms with Alzheimer’s disease abnormally hyperphosphorylated tau and in vitro phosphorylation into the disease-like protein. J Biol Chem. 2001;276:37967–37973. doi: 10.1074/jbc.M105365200. [DOI] [PubMed] [Google Scholar]
- 40.Alonso A, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci U S A. 2001;98:6923–6928. doi: 10.1073/pnas.121119298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Drewes G, Trinczek B, Illenberger S, Biernat J, Schmitt-Ulms G, Meyer HE, Mandelkow EM, Mandelkow E. Microtubule-associated protein/microtubule affinity-regulating kinase (p110mark). A novel protein kinase that regulates tau-microtubule interactions and dynamic instability by phosphorylation at the Alzheimer-specific site serine 262. J Biol Chem. 1995;270:7679–7688. doi: 10.1074/jbc.270.13.7679. [DOI] [PubMed] [Google Scholar]
- 42.Goode BL, Denis PE, Panda D, Radeke MJ, Miller HP, Wilson L, Feinstein SC. Functional interactions between the proline-rich and repeat regions of tau enhance microtubule binding and assembly. Mol Biol Cell. 1997;8:353–365. doi: 10.1091/mbc.8.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bramblett GT, Goedert M, Jakes R, Merrick SE, Trojanowski JQ, Lee VM. Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron. 1993;10:1089–1099. doi: 10.1016/0896-6273(93)90057-x. [DOI] [PubMed] [Google Scholar]
- 44.Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–825. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]