

Abstract
NBS1 forms a complex with MRE11 and RAD50 (MRN) that is proposed to act on the upstream of two repair pathways of DNA double-strand break (DSB), homologous repair (HR) and non-homologous end joining (NHEJ). However, the function of Nbs1 in these processes has not fully been elucidated in mammals due to the lethal phenotype of cells and mice lacking Nbs1. Here, we have constructed mouse Nbs1-null embryonic fibroblasts and embryonic stem cells, through the Cre-loxP and sequential gene targeting techniques. We show that cells lacking Nbs1 display reduced HR of the single DSB in chromosomally integrated substrate, affecting both homology-directed repair (HDR) and single-stranded annealing pathways, and, surprisingly, increased NHEJ-mediated sequence deletion. Moreover, focus formation at DSBs and chromatin recruitment of the Nbs1 partners Rad50 and Mre11 as well as Rad51 and Brca1 are attenuated in these cells, whereas the NHEJ molecule Ku70 binding to chromatin is not affected. These data provide a novel insight into the function of MRN in the branching of DSB repair pathways.
Keywords: DNA double-strand break, homology-directed repair, NBS1, nonhomologous end-joining, single-stranded annealing
Introduction
The NBS1 protein (hypomorphically mutated in Nijmegen Breakage Syndrome; NBS) forms a complex (MRN) with Mre11 (mutated in A–T-like disorder, A-TLD) and Rad50 in mammalian cells (D'Amours and Jackson, 2002). The functional or physical interactions between the MRN complex with ATM, ATR, MDC1, H2AX, BRCA1 and FANCs suggest how MRN may coordinate checkpoint molecules and the double-strand break (DSB) repair machinery (Shiloh, 2003; Lavin, 2004). Thus, NBS cells also show checkpoint defects in cell cycle progression (Shiloh, 2003). NBS1 has been shown to directly activate ATM and to stimulate the kinase activity of ATM towards its substrates in vitro (Lee and Paull, 2004, 2005; Falck et al, 2005). Consistent with such a role, activation of ATM targets is impaired in human NBS cells or mouse Nbs1-deficient B cells (Carson et al, 2003; Uziel et al, 2003; Difilippantonio et al, 2005).
DNA DSBs repair is carried out by two major pathways: homologous repair (HR) and non-homologous end joining (NHEJ) (West, 2003). HR is conducted by Rad52 epistasis group molecules, including Rad51, Rad54, Rad59, XRCC2/3 and BRCA1/2 (Symington, 2002), while NHEJ repair is carried out by the Ku complex consisting of the Ku70/Ku80 heterodimer and the catalytic subunit of DNA-PK (DNA-PKcs), which recruit XRCC4, DNA Ligase IV and artemis (Weterings and van Gent, 2004). However, how a cell chooses to undergo repair by either pathway remains largely unexplained. It is proposed that cell cycle phase is one factor that modulates the choice of repair pathway (Lieber et al, 2003). The repair of DSBs in G0/G1 cells is carried out mainly by NHEJ because of the limited supply of homology template, whereas HR would become the major form of repair in S/G2 cells when sister chromatids are available. Other factors may also influence the use of pathways. For example, the CDK1 inhibitor represses HR in budding yeast arrested in G2 phase but results in a compensatory increase in NHEJ (Ira et al, 2004). Different from other DSB molecules, MRN is proposed to act at the branching point upstream of both HR and NHEJ. However, there is conflicting evidence on the role of MRN in HR and NHEJ. In budding yeast Saccharomyces cerevisiae, the Mre11–Rad50–Xrs2 complex is required for both HR and NHEJ, whereas in fission yeast Schizosaccharomyces pombe, the Rad32(Mre11)–Rad50 complex is dispensable for NHEJ (Manolis et al, 2001; Tomita et al, 2003; see review by Khanna and Jackson, 2001). Finally, although Nbs1-deficient chicken DT-40 cells showed defects in homology-directed repair (HDR) (Tauchi et al, 2002), the precise role of Nbs1 in mammalian cells and in particular repair steps have not been studied.
Although the function of mammalian NBS1 has been extensively studied using biochemical approaches and using cell lines derived from NBS patients who carry NBS1 hypomorphic mutations (Maser et al, 2001), the biological role of the entire Nbs1 protein in the repair of DSBs remains elusive. One of the reasons is that null mutation of Nbs1 and members of the MRN complex in mammals cause a lethal phenotype at the cellular or mouse level (Xiao and Weaver, 1997; Luo et al, 1999; Zhu et al, 2001; Dumon-Jones et al, 2003). We and others have applied Cre-loxP technology to overcome the lethality and to delete Nbs1 in specific tissues of mice (Demuth et al, 2004; Frappart et al, 2005; Kracker et al, 2005; Reina-San-Martin et al, 2005) and studied the biological function of Nbs1 in lymphoid development and in neurogenesis. In the present study, we engineered an Nbs1-null cellular system by the constitutive (gene targeting) and inducible (via Cre-loxP technology) deletion of the gene in embryonic stem (ES) cells and fibroblasts (MEFs). Using these cells, we have investigated the role of Nbs1 in the regulation of DSB repair pathways.
Results
Generation of inducible Nbs1-null mouse embryonic fibroblast cells
In order to study the function of the entire Nbs1 protein in DNA repair in mammals, we constructed MEFs with an inducible Nbs1 deletion by disrupting exon 6 of the Nbs1 gene (the murine homolog of NBS1) using the Cre-loxP technology (Supplementary Figure S1A). We isolated MEFs from E13.5 fetuses of Nbs1f6/f6 and Nbs1neo/f6 genotypes (Dumon-Jones et al, 2003; Frappart et al, 2005) and immortalized them according to the 3T3 protocol. These MEFs were then stably transfected with the Cre-ER vector expressing Cre recombinase (CER clones) or an empty vector (PSG clones) (Figure 1A). The inducibility of the disruption of the Nbs1 locus by 4-hydroxytamoxifen (OHT) in CER clones was analyzed by Southern blotting and RT–PCR (Figure 1B). Nbs1 deletion was further confirmed by Western blotting (Figure 1B, lower panel). Genetically, the Nbs1neo and Nbs1Δ6 alleles are null because homozygous mutant embryos (Nbs1neo/neo; Nbs1Δ6/Δ6; Nbs1neo/Δ6) died at the peri-implantation stage (Dumon-Jones et al, 2003; Frappart et al, 2005). These molecular and genetic data confirm that these Nbs1Δ6/Δ6 and Nbs1neo/Δ6 CER cells are Nbs1-null mutants and thus designated as ‘inducible' Nbs1-null MEF cells.
Figure 1.
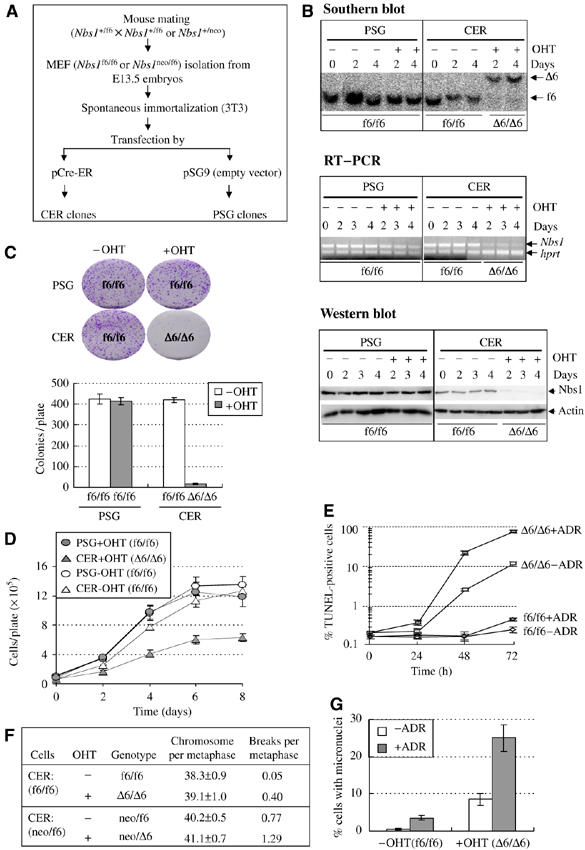
Generation and characterization of inducible Nbs1 deletion in mouse 3T3 MEFs. (A) Protocol used for generation of ‘inducible' Nbs1-null MEFs. (B) Southern blot, RT–PCR and Western blot analysis of Nbs1 deletion after OHT treatment in ‘inducible' Nbs1-null CER MEFs. The ‘f6' and ‘Δ6' alleles are indicated in the Southern blot. The genotype of each sample after OHT treatment is indicated at the bottom of the gels. Hprt served as an internal control for RT–PCR analysis. Actin is the loading control for Western blotting. (C) Colony formation assay of Nbs1 deletion in MEFs. Representative plates (upper panel) and quantification (lower panel) of colony formation assay are shown. Bars represent the mean of six clones of each sample from two independent experiments. (D) The proliferation curve of Nbs1-proficient (PSG±OHT and CER−OHT, f6/f6) and null (CER+OHT, Δ6/Δ6) MEFs. Curves represent the mean of duplicate samples from two independent experiments. (E) TUNEL analysis of inducible Nbs1-null MEFs (Δ6/Δ6) after treatment with adriamycin (+ADR) or not (−ADR) for 3 h (Time 0). TUNEL staining was performed at the indicated time points after drug removal. (F) Cytogenetic analysis of Nbs1f6/f6 or Nbs1neo/f6 CER cells after OHT-mediated Nbs1 deletion. Forty metaphases of each cell line were analyzed. (G) Spontaneous and ADR-induced micronucleus formation in the absence of Nbs1. Bars represent the mean of three clones of each genotype from two experiments.
Characterization of DNA damage response of inducible Nbs1-null MEF cells
We first examined the effect of Nbs1 null on DNA damage response and chromosome stability. To this end, we applied inducible Nbs1-null MEF cells to study the effect of Nbs1 depletion on cell proliferation, survival and chromosome stability. After depletion of Nbs1 by OHT treatment, CER clones (Δ6/Δ6) completely lost clonogenic formation ability (Figure 1C). To test whether this phenotype was due to proliferation and/or apoptosis, we analyzed the proliferation profile as well as apoptosis. As expected, Nbs1 deletion in these cells resulted in defective proliferation (Figure 1D) and increased TUNEL-positive populations, which were further enhanced by DNA damaging agent adriamycin (ADR) (Δ6/Δ6+ADR) (Figure 1E). Moreover, cytogenetic analysis revealed that inducible Nbs1 deletion in MEF CER cells (Δ6/Δ6, neo/Δ6; after OHT treatment) caused a high degree of spontaneous chromosomal aberrations and micronuclei (Figure 1F and G). The findings support the proposed role of Nbs1 in cell viability and DNA damage response.
Impaired HDR of DSBs in MEFs after induced Nbs1 deletion
To investigate repair pathways of DSBs, we constructed PSG (without Cre) and inducible Nbs1-null CER (Cre) cells in which a single copy of the HDR reporter substrate DRGFP was integrated into the genome (Figure 2A). To characterize the HR pathway, we used seven CER clones that carried a single intact copy of DRGFP and were treated with or without OHT. After expression of the I-SceI endonuclease that induces a single DSB in the substrate, HDR activity can be monitored by flow cytometry for GFP-positive cells (Pierce et al, 1999). We found that GFP-positive populations were significantly lower in all seven inducible Nbs1-null CER cells (Δ6/Δ6) compared to Nbs1-proficient MEF cells (f6/f6; Figure 2B). In contrast, almost none of GFP-positive MEFs were detected among mock-transfected cells (Figure 2B, upper panel; data not shown).
Figure 2.
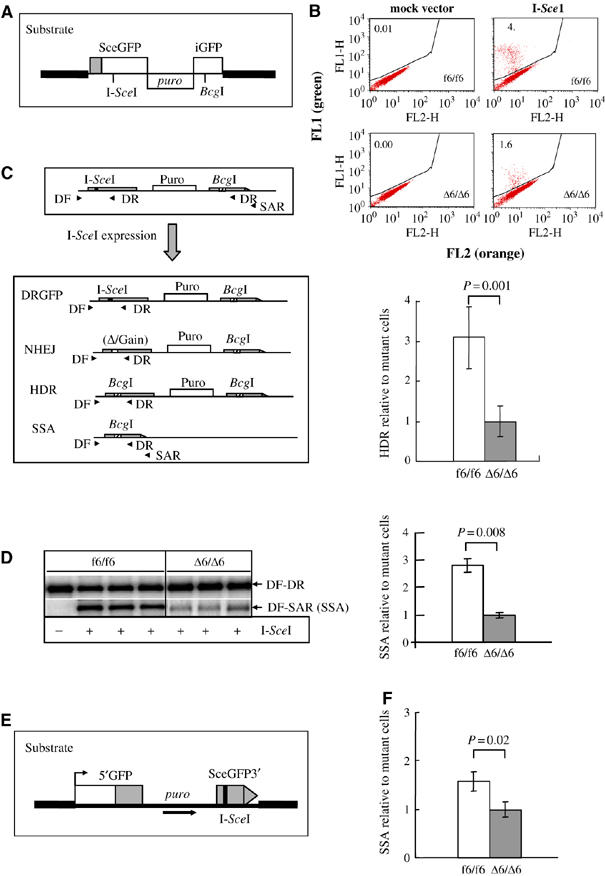
Nbs1 depletion compromised both HDR and SSA in inducible Nbs1-null MEFs. (A) The HDR assay substrate DRGFP. (B) Nbs1-proficient or -null cells carrying a single intact copy of substrate DRGFP were analyzed by flow cytometry. Upper panel: representative flow cytometric analysis for HDR in MEFs after I-SceI expression. The GFP-positive population, reflecting HDR repair events, is separated from GFP-negative population. FL1, green fluorescence; FL2, orange fluorescence. Lower panel: The HDR events were scored as the percentage of GFP-positive populations after I-SceI expression. The percentage of GFP-positive cells in inducible (Δ6/Δ6) Nbs1-null mutants was set as 1. Bars represent the mean of seven MEF clones of each genotype from two independent experiments. (C) Strategy for analysis of SSA products in the DRGFP substrate. Four putative products surrounding the original genomic region of the I-SceI site: HDR, SSA, NHEJ and uncut DRGFP are shown. SSA was quantified by the products from primers DF and SAR, compared to that of the total PCR products from primers DF and DR (including HDR, SSA, DRGFP and NHEJ products). (D) Representative PCR-Southern blotting image (left panel) and quantification of SSA products (right panel) after I-SceI expression. The density of SSA products (DF-SAR) was normalized to the total PCR products (DF-DR) within the same sample. The SSA value of inducible (Δ6/Δ6) Nbs1-null mutants was set as 1. Bars represent the mean of three MEF clones of each genotype from two independent experiments. (E) SSA assay substrate SAGFP was stably integrated into MEFs and SSA was analyzed by flow cytometry after I-SceI transfection. (F) Summary of SSA in Nbs1-null (Δ6/Δ6) and -proficient (f6/f6) MEF cells. The SSA events were scored by the percentage of GFP-positive populations. The percentage of GFP-positive cells in Nbs1-null mutants was set as 1. Bars represent the mean of seven MEF clones of each genotype from two independent experiments. P-values by t-test are indicated in (B, D, F).
To monitor the transfection efficiency, we transfected a GFP-expressing plasmid and found similar populations of these inducible Nbs1-null and -proficient MEFs that were GFP-positive (data not shown), suggesting no difference in the uptake of the I-SceI-expressing plasmid in both genotypes. Thus, the HDR deficiency observed in inducible Nbs1-null MEFs was solely due to the depletion of the Nbs1 protein. Taken together, these findings indicate that Nbs1 is required for efficient HDR of DSBs in mammalian cells.
Decreased single-stranded annealing of DSBs in inducible Nbs1-null MEFs
To determine the specific step of HR pathways in which Nbs1 is involved, we analyzed single-stranded annealing (SSA) in MEFs after induced Nbs1 deletion. To this end, using the primers DF and SAR (Figure 2C), we amplified SSA products from inducible Nbs1-null CER cells after I-SceI transfection, and compared them to total PCR products (including HDR, SSA, NHEJ and uncut substrate DRGFP) generated using primers DF and DR (see Figure 2C). In the HDR reporter system used here, either HR (HDR and SSA) or NHEJ can repair I-SceI-induced DSBs (Figure 2C; see Yang et al, 2005). Using these assays, we found that SSA-generated products were significantly decreased (about 2.5-fold) in inducible Nbs1-null CER MEFs (Δ6/Δ6) compared to Nbs1-proficient control clones (f6/f6) (Figure 2D).
To test the potential contribution of LTGC (long-tract gene conversion) that can be amplified by primers DF and SAR to the authentic SSA events, we grew cells in the presence of puromycin after DSBs repair, which would eliminate cells undergoing SSA that caused the loss of the puromycin resistance gene. Primers (DF and SAR) then would solely amplify the regions around the BcgI site generated via LTGC (Figure 2C; Nakanishi et al, 2005; Yang et al, 2005). This assay did not detect any PCR products (data not shown), ruling out a significant contribution of the LTGC event to the SSA products shown in Figure 2D.
To exclude the possibility of experimental bias, we next constructed new Nbs1 CER cells containing a single intact copy of a specific SSA reporter substrate SAGFP (Figure 2E; Stark et al, 2004). After induction of Nbs1 deletion, in the presence of I-SceI, numbers of SSA-generated GFP-positive cells were reduced significantly among Nbs1-null MEFs (Δ6/Δ6), compared to wild-type cells (f6/f6) (Figure 2F). In control experiments, without I-SceI expression, almost no GFP-positive cells were detected for both genotypes (data not shown). Thus, the absence of Nbs1 affects the SSA step, consistent with the proposed role of the MRN complex in the resection of DNA DSBs.
Imprecise NHEJ is increased in inducible Nbs1-null MEF cells
We next used two independent approaches to analyze the involvement of the mammalian MRN complex in the NHEJ pathway after I-SceI induced DSBs. First, we amplified the genomic region surrounding the I-SceI site in DRGFP using PCR primers (DF and DR; see Figure 2C). The NHEJ products were significantly increased in inducible Nbs1-null MEF cells (Δ6/Δ6), compared to Nbs1-proficient controls (f6/f6) (Figure 3A). After subcloning into the pGEM-T vector, direct sequencing of these NHEJ products from 31 wild-type and 35 Nbs1-null plasmid clones revealed deletions ranging from 10 to 25 bp around the original I-SceI site (data not shown). However, no such deletion was detectable in mock controls without I-SceI expression (Figure 3A; data not shown).
Figure 3.
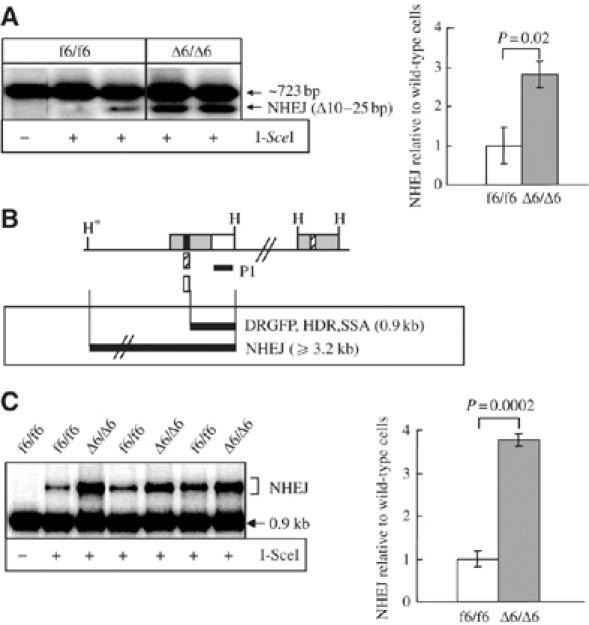
Increased NHEJ in MEF cells after induction of Nbs1 deletion. (A) PCR-Southern blot analysis of NHEJ. Representative gel (left panel) and quantification of NHEJ deletion products (right panel) after I-SceI expression. The PCR primers are shown in Figure 2C. The NHEJ frequency was examined by normalizing the density of NHEJ deletion products (Δ10–25 bp) to that of the 723-bp PCR product within the same sample. The NHEJ value of Nbs1-proficient cells (f6/f6) was set as 1. (B) Southern blot strategy for NHEJ analysis. NHEJ products are resistant to double digestion with I-SceI and BcgI enzymes. Note: the first HindIII site (star) is located outside the DRGFP substrate. Black filled box, I-SceI site arisen from uncut DRGFP; striped box, BcgI site retrieved from HDR and SSA products; empty box, NHEJ products resistant to both I-SceI and BcgI digestion. (C) Southern blot analysis of NHEJ using the strategy shown in (B). Representative Southern blot image (left panel) and quantification of total NHEJ products (right panel) in the DRGFP substrate after I-SceI expression. The NHEJ efficiency was examined by normalizing the density of NHEJ products (resistant to both I-SceI and BcgI) to that of cleaved product (0.9 kb, sensitive to both I-SceI and BcgI) within the same sample. The NHEJ value of Nbs1-proficient cells (f6/f6) was set as 1. Bars in (A, C) represent the mean of three MEF clones of each genotype from two independent experiments. P-values by t-test in (A, C) are indicated.
To confirm these findings, we adopted another approach to examine directly the total NHEJ events by digesting the genomic DNA of mock or I-SceI-transfected cells with I-SceI, followed by BcgI and HindIII enzymes (Figure 3B). The repair of a DSB in the DRGFP substrate via HDR, SSA, or imprecise NHEJ pathways generates I-SceI-site loss leading to a resistance to I-SceI digestion. HDR and SSA generate a BcgI site that replaces the I-SceI site in the substrate, leading to sensitivity to BcgI digestion (Figure 3B; Yang et al, 2005). Therefore, the uncleaved products resistant to both I-SceI and BcgI enzymes arose solely through NHEJ. The relative amount of I-SceI/BcgI-resistant DNA, indicative of this process, was significantly increased in inducible Nbs1-null CER MEFs (Δ6/Δ6), as we found about three-fold increase of NHEJ products in these cells compared to the Nbs1-proficient counterpart (f6/f6) (Figure 3C). In mock-transfected cells, genomic DNA was completely cleaved by I-SceI (Figure 3C; data not shown). Together, these results suggest that Nbs1 represses the NHEJ pathway.
Generation and characterization of constitutive Nbs1-null mouse ES and MEF cells
To verify DNA repair defects of inducible Nbs1-null MEFs, we also generated ES and MEF cells containing ‘constitutive' Nbs1 deletion following the strategy outlined in Figure 4A and Supplementary Figure S1B. By sequential gene targeting in ES cells (Supplementary Figure S1B), one homozygous Nbs1neo/hyg mutant clone was obtained. To obtain an independent Nbs1-null ES cell line, we isolated blastocysts (E3.5 embryos) from intercrosses of heterozygous Nbs1+/neo (Dumon-Jones et al, 2003) and Nbs1+/f6 (Frappart et al, 2005) mice, from which Nbs1neo/f6 ES clones were established. Following transfection of a Cre-expressing plasmid, the Nbs1neo/Δ6 mutant ES cells were identified by Southern blotting (data not shown). Western blot analysis did not detect the full-length nor truncated form of the Nbs1 protein in these ES cells (Figure 4B). We next isolated MEFs from E13.5 chimeric fetuses that were generated by injection of Nbs1neo/hyg ES cells into wild-type blastocysts. The Nbs1neo/hyg MEFs were selected against G418 to eliminate wild-type cells before immortalization (Figure 4A). Consistent with their ES cell origin, these immortalized Nbs1-null MEF cells (Nbs1neo/hyg) were devoid of the Nbs1 protein (Figure 4C) and thus these ES and MEF cells (Nbs1neo/hyg, Nbs1neo/Δ6) were designated as ‘constitutive' Nbs1-null mutants.
Figure 4.
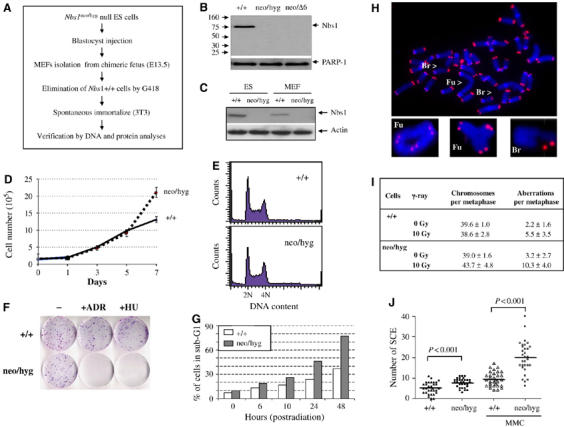
Generation and characterization of mouse ES cells and MEFs carrying constitutive Nbs1 deletion. (A) Protocol used to establish constitutive Nbs1-deleted MEFs from chimeric fetuses. (B) Western blot analysis of Nbs1-null ES cells. PARP-1 is a loading control. (C) Western blot analysis of Nbs1-null ES cells and 3T3 MEFs. Actin serves as a loading control. (D) Proliferation curve of constitutive Nbs1-null MEF cells. Curves represent the mean of triplicate samples from two independent experiments. (E) FACs analysis of cell cycle distribution of Nbs1-null ES cells. (F) Colony formation assay was performed to examine viability of constitutive Nbs1 deleted MEFs after ADR (0.2 μg/ml) or HU (2 mM) treatment. (G) Histogram shows the percentage of ES cells in sub-G1 phase extracted from FACs analysis at the indicated time points after 10 Gy of IR. Representative images of FISH analysis (H) and quantification (I) of wild type (+/+) and Nbs1-null (neo/hyg) ES cells subjected or not to 10 Gy of IR. Br: breaks; Fu: fusions. Between 16 and 19 metaphases from each treatment were analyzed and the results are shown as the mean±standard deviation. (J) SCE analysis of constitutive Nbs1-null MEFs with or without MMC treatment. At least 31 metaphases of each sample were scored and the t-test was applied for statistical analysis.
We next examined proliferation of these Nbs1-null cells and found no difference in proliferation and normal cell cycle distribution compared to wild-type controls (Figure 4D and E). The proliferation potential of Nbs1-null ES cells was further tested using chimeric embryo assays by injecting Nbs1-null ES cells into wild-type blastocysts. The presence of mutant alleles (neo and hygro) in a given tissue was quantified and compared to the wild-type allele in the same tissue by image analysis of the Southern blot. It was noted that Nbs1-null ES cells could differentiate into various cell types and participate in the formation of various tissues although the capacity varied (Supplementary Figure S2).
To test the DNA damage response of these mutant cells, we performed the colony formation assay using constitutive Nbs1neo/hyg MEFs. After treatment with ADR and hydoxyurea (HU), constitutive Nbs1-null MEFs completely lost their clonogenic capacity (Figure 4F). In addition, flow cytometric analysis revealed that Nbs1-null ES cells showed similar apoptosis (sub-G1) in spontaneous cell population compared to wild-type controls. However, after 10 Gy of ionizing radiation (IR), more Nbs1-null ES cells accumulated in the sub-G1 phase, indicating an increased apoptosis (Figure 4G). Cytogenetic analysis revealed that Nbs1-null ES cells contained high levels of chromosome breaks and fusions after 10 Gy of IR although without IR treatment their basal levels of chromosome aberrations were similar to those of wild-type counterparts (Figure 4H and I). We also found a higher rate of sister chromatid exchange (SCE) in the presence and absence of mitomycin C (MMC) in constitutive Nbs1-deficient MEFs compared to Nbs1-proficient controls (Figure 4J).
Repair defects of Nbs1-null cells are independent of the cell proliferation status
To avoid the argument that the DSB repair defects seen in ‘inducible' Nbs1 deleted MEFs is secondary to the impact of Nbs1 deletion-induced cell proliferation arrest (e.g., Figure 1C and D), we took advantage of proliferating constitutive Nbs1-null cells for investigation of the role of Nbs1 in the repair pathways. To characterize the DSB pathway, we established six Nbs1-null and six wild-type ES clones containing a single intact substrate DRGFP. After expression of the I-SceI, we found that GFP-positive populations were significantly lower in constitutive Nbs1-null ES cells (neo/hyg) than in controls (+/+) (Figure 5A). To test the SSA pathway, we used two different assays (see Figure 2C and E) and found that SSA-generated products were significantly less in constitutive Nbs1-deleted ES cells (neo/hyg), compared to their Nbs1-proficient counterparts (+/+) (Figure 5B and C). These defects were corrected by the reintroduction of mouse Nbs1 cDNA into these mutant cells (Figure 5D–F). In these experiments, we used Fanca null cells as controls because we demonstrated recently that these cells are defective in both HDR and SSA (Nakanishi et al, 2005; Yang et al, 2005).
Figure 5.
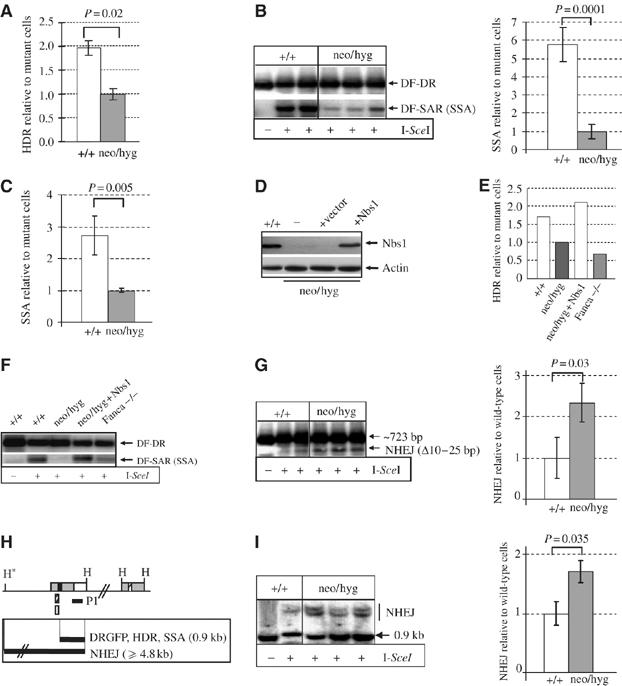
DSB repair defects in constitutive Nbs1-null ES cells. (A) The HDR events after I-SceI expression were analyzed by flow cytometry (see Figure 2B) and scored as the percentage of GFP-positive populations. The percentage of GFP-positive cells in Nbs1-null (neo/hyg) mutants was set as 1. Bars represent the mean of six ES clones of each genotype from two independent experiments. (B) Representative PCR-Southern blot image (left panel) and quantification of SSA products (right panel) in the HDR substrate DRGFP. The density of SSA products (DF-SAR) was normalized to the total PCR products (DF-DR) within the same sample. The SSA value of constitutive Nbs1-deleted mutants (neo/hyg) was set as 1. Bars represent the mean of three ES clones of each genotype from two independent experiments. (C) SSA analysis of Nbs1-null (neo/hyg) and -proficient (+/+) ES cells (see Figure 2E). The percentage of GFP-positive cells (SSA events) in Nbs1-null mutants was set as 1. Bars represent the mean of six ES clones of each genotype from two independent experiments. (D) Western blot analysis of transient ectopic expression of mouse Nbs1 cDNA in constitutive Nbs1-null ES cells harboring DRGFP. Defects in HDR (E) and SSA (F) are rescued by transient ectopic expression of mouse Nbs1 cDNA. One of two independent experiments are shown for (E) and (F). Fanconi anemia-A mutant fibroblasts (Fanca−/−; Yang et al, 2005) were used as a negative control. (G) PCR-Southern blot analysis of NHEJ activity in proliferating Nbs1-null ES cells as described in Figure 2C. Representative Southern blot image (left panel) and quantification of NHEJ deletion products (right panel). The NHEJ value of Nbs1-proficient cells (+/+) was set as 1. (H) Southern blot strategy for NHEJ analysis. Note: NHEJ products resistant to both I-SceI and BcgI digestions are ⩾4.8 kb. (I) Representative Southern blotting image (left panel) and quantification of total NHEJ products (right panel). The NHEJ efficiency was examined as described in Figure 3C. The NHEJ value of Nbs1-proficient cells (+/+) was set as 1. Bars in (G, I) represent the mean of three ES clones of each genotype from two independent experiments. The t-test was applied for statistical analysis.
We also analyzed the NHEJ activity in constitutive Nbs1-null cells by two approaches. We first analyzed DSB repair products in DRGFP in ES cells by PCR-Southern blotting. After transfection of I-SceI, we amplified the genomic region surrounding the I-SceI site in the substrate using PCR primers (DF and DR; see Figure 2C). The PCR products generated by NHEJ deletion were significantly increased in proliferating Nbs1-null ES cells (neo/hyg), compared to wild-type controls (+/+) (Figure 5G). In the second approach, we used the same strategy as shown in Figure 3B and C. Similarly, imprecise NHEJ pathways generate I-SceI-site loss leading to a resistance to I-SceI digestion, which can be visualized by uncleaved products (⩾4.8 kb) (Figure 5H). The relative amount of I-SceI/BcgI-resistant DNA, indicative of this process, was significantly increased in proliferating constitutive Nbs1-null ES cells (neo/hyg), compared to that of their wild-type counterparts (+/+) (Figure 5I). In mock-transfected cells, genomic DNA was completely cleaved by I-SceI in vitro (Figure 5I). Taken together, these data are consistent with the results obtained using inducible Nbs1-deleted MEF cells, confirming that the observed DSB repair phenotype is not a consequence of the cell cycle arrest, but rather intrinsic to the Nbs1 function.
Deletion of Nbs1 compromises recruitment of MRN and HR repair proteins to DSBs
We further examined the effect of Nbs1 null on the recruitment of its partners Mre11 and Rad50 to DNA breaks. As expected, ADR and HU treatment resulted in Rad50 foci in Nbs1-proficient MEFs (f6/f6; Supplementary Figure S3, left panel). However, Rad50 focus formation was completely abolished in both inducible (Δ6/Δ6) and constitutive (neo/hyg) Nbs1-null MEFs (Supplementary Figure S3, left panel). Similarly, Mre11 foci were also abolished in Nbs1-null cells, and instead, Mre11 was dispersed throughout the cytoplasm and also in the nucleus (Supplementary Figure S3, right panels). To investigate the molecular basis of the reduced overall HR and increased NHEJ in Nbs1-null cells, we next studied the impact of Nbs1-null mutation on focus formation of two key players in HR, Rad51 and Brca1. After DNA damage by ADR, Rad51 focus-positive cells was significantly less in Nbs1-null genotype (Δ6/Δ6) compared to Nbs1-proficient cells (f6/f6) (Figure 6A). We also found fewer Brca1 focus-containing cells among Nbs1-null MEFs (Δ6/Δ6) compared to Nbs1-proficient (f6/f6) cells (Figure 6B). Focus formation of these two DNA repair molecules in response to DNA damage was not altered in PSG control MEFs in the presence or absence of OHT (data not shown), ruling out any interference of OHT in focus formation. To rule out potential influence of Nbs1 deletion-induced cell cycle arrest (see Figure 1E) on the low number of Rad51 and Brca1 foci, we performed these experiments with proliferating Nbs1-null MEFs (neo/hyg) (see Figure 4D) and found similar reduction of these foci in the absence of Nbs1 (Figure 6A and B).
Figure 6.
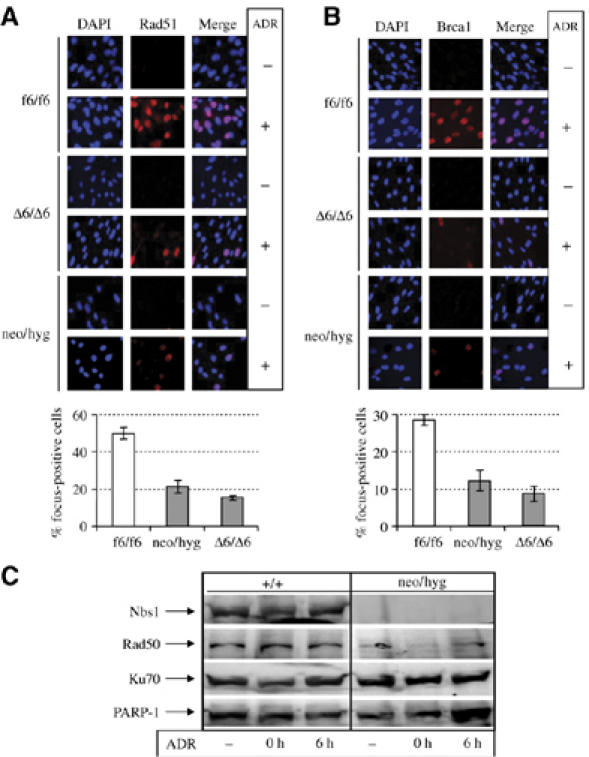
Defective focus formation of DSB repair molecules in MEF cells lacking Nbs1. Immunofluorescence analysis of Rad51 (A) and Brca1 (B) focus formation in inducible (Δ6/Δ6) and constitutive (neo/hyg) Nbs1-null MEFs after treatment with 0.2 μg/ml ADR for 3 h. Lower panels of (A) and (B) are quantifications of DNA damage-induced foci. Bars represent means±s.d. are shown for duplicate samples from two independent experiments. For each sample at least 500 nuclei were scored. (C) Western blot analysis of the chromatin-enriched fraction of MRN and Ku70 proteins. 0 h, 0 h after removal of ADR; 6 h, 6 h after removal of ADR. PARP-1 is served as a control.
Nbs1 is required for the chromatin loading of MRN, but dispensable for Ku70
To further characterize the recruitment of MRN to DSBs, we performed chromatin fractionation analysis. Although the basal level of total Rad50 was unaltered in Nbs1-null MEF cells (data not shown), Rad50 was almost absent in chromatin-enriched fractions of Nbs1-null cells (neo/hyg) with or without DNA damage (Figure 6C). The trace amount of Rad50 seen in the chromatin-bound fractions correlated with immunofluorescence images (see Supplementary Figure S3), which may be due to the slow turnover of this protein in the nucleus. In contrast to the MRN molecules, the chromatin-bound form of the NHEJ molecule Ku70 (Figure 6C) was apparently unaffected in the absence of Nbs1, which may correlate with NHEJ activities seen in these cells (see Figures 3 and 5). These results, together with focus formation experiments, suggest that Nbs1 is essential for MRN stability and recruitment of HR molecules to DNA breaks; however, its absence does not affect the recruitment of NHEJ molecules to DSBs.
Discussion
Using the Cre-LoxP technology, we have generated mouse MEFs null for Nbs1. These cells have allowed us to demonstrate that Nbs1 (possibly through MRN) modulates DNA DSB repair pathways in mammalian cells by promoting HR while repressing NHEJ. These results are apparently not secondary to the effects of Nbs1 deletion-induced cell cycle arrest, since these phenotypes were also seen in proliferating constitutive Nbs1-null cells. More importantly, ectopic expression of mouse Nbs1 cDNA in constitutive Nbs1-null cells can correct these DSB defects.
It might be surprising that we could obtain cells null for Nbs1 because complete deletion of any member of the MRN complex in mammals is lethal both at embryonic and cellular levels (Xiao and Weaver, 1997; Luo et al, 1999; Zhu et al, 2001; Dumon-Jones et al, 2003). Although p53 deficiency can rescue a lethal phenotype of mice lacking DSB repair molecules (e.g., see Lim and Hasty, 1996; Hakem et al, 1997; Ludwig et al, 1997; Gao et al, 2000), p53-null mutation did not rescue the embryonic lethality of Nbs1 null mice (our unpublished data) and the p53 pathway is functional in our Nbs1-null cells (data not shown). These observations suggest that the viability of our Nbs1-null cells is p53-independent and other genetic factors must be involved. Nevertheless, using inducible Nbs1 deletion in MEFs, we have demonstrated an indispensable role of Nbs1 in cell viability (Figure 1C and D), consistent with the general notion that Nbs1 is essential for cell life. Despite unknown factors overcoming the lethality of constitutive Nbs1-null cells, these cells are hypersensitive to DNA-damaging treatments and reproduce many phenotypes similar to those observed in human NBS cells. Thus, the Nbs1-null cellular system is a valuable reagent, given the current absence of MRN-null mutants in mammalian cells, and should greatly facilitate the dissection of the early DNA damage response.
The most intriguing result is that Nbs1 appears to modulate the branching of DNA DSB repair pathways because, in the cells lacking Nbs1, the HR pathway (i.e., HDR and SSA) is severely impaired, whereas imprecise NHEJ events are increased. The current study suggests a different mechanism operated by MRN in both HDR and NHEJ pathways in mammalian cells. Several possibilities could explain how Nbs1/MRN modulates the repair pathways. The MRN complex harbors exonuclease and DNA end-processing activity that are required for the DSB resection into ssDNA, a common step to both HDR and SSA (D'Amours and Jackson, 2002; Shiloh, 2003). Moreover, the lack of Nbs1 abrogates the loading of HR molecules (e.g., Rad51 and Brca1) to DSBs and thereby compromises HR, consistent with the fact that BRCA1 and FancD2 physically interact with Nbs1. In this regard, it is interesting to note that although Rad51 is not involved in SSA, cells mutated in BRCA1 and FANCs (-A/-C/-G/-D2) exhibit defects of both HDR and SSA (Stark et al, 2004; Nakanishi et al, 2005; Yang et al, 2005). Interestingly, Nbs1-null cells exhibit a similar defect in HDR and SSA as Fanca−/− cells (see Figure 5E and F), suggesting that the FA complex and MRN are epistatic and may participate in a common repair pathway in dealing with certain type of DNA damage. However, it might be surprising that Nbs1-null cells contain elevated MMC-induced SCEs while showing low HDR and less Rad51 foci, because IR-induced SCEs in S. cerevisiae is an HR-dependent event abolished when RAD51 is inactivated (Fasullo et al, 2001). One possible explanation for this apparent paradox is the nature of DNA breaks generated by different approaches. In the current study, HDR and Rad51 foci were analyzed from ‘frank' DSBs induced by I-SceI in the DRGFP substrate and by ADR treatment, respectively. However, SCEs were analyzed by using a classical DNA interstrand crosslink (ICL) agent MMC, which does not introduce ‘frank' DSBs. Probably, MMC collapses replication forks and initiates recombination with different requirements as compared to ‘frank' DSBs. In this regard, many mutant cells that are hypersensitive to ICLs are not clearly IR-sensitive (e.g. FA cells). On the other hand, it is conceivable that loss of MRN may compromise DNA strand resection and thereby favor the Ku-mediated NHEJ activity. Finally, Nbs1 could destabilize or antagonize NHEJ molecules (e.g., Ku70) at DSBs thereby inhibiting NHEJ activities. In support of this hypothesis is the observation that Nbs1-mediated recruitment of HR molecules (Rad51 and Brca1) to DSBs correlates with the disappearance of Ku, which favors disengagement of NHEJ activities at DSBs (Kim et al, 2005).
However, our results seem to be contradictory to the recent study using a transient reporter assay to show that NBS skin fibroblasts contain a low capacity for HDR/SSA and micro-homology directed NHEJ (Howlett et al, 2006). Discrepancy of these findings may be due to different systems used: while we used a chromosomally integrated substrate where a defined DSB is applied, Howlett et al. used episomal linearized plasmid assays where cytoplasmic components may influence the activity. In addition, the NHEJ defect was implied in human NBS cells as well as ATLD cells as judged by impaired γ-H2AX focus formation (Riballo et al, 2004). However, since MRN is required for ATM activation and γ-H2AX is a substrate of ATM, the reduced γ-H2AX foci in these human NBS and in ATLD cells may not be scored reliably as DSB rejoining defects. Moreover, our results are surprising given the fact that cells carrying either hypomorphic NBS1 mutation (Lahdesmaki et al, 2004) or complete deletion (Kracker et al, 2005; Reina-San-Martin et al, 2005) have defects in class-switch recombination (CSR), a process that requires NHEJ activities. Despite this observation, V(D)J recombination, another NHEJ-dependent process, is apparently not affected in human NBS cells (Harfst et al, 2000). CSR and V(D)J are region- and cell type-specific recombination events in lymphoid cells. Our NHEJ data were obtained from fibroblasts and ES cells in which lymphoid specific factors are not present. Thus, we cannot rule out the specificity of MRN in CSR (and V(D)J) recombination.
It is well established that HR repair is predominant in S/G2 whereas NHEJ is more frequently used in G0/G1 due to the lack of homology sequences in these phases. It might not have been surprising to see increased NHEJ in Nbs1-null cells while HR is suppressed. For example, it has been shown that suppression of NHEJ in mammalian cells by disruption of Ku70, Xrcc4 and DNA-PKcs results in increased levels of HDR, as Ku antagonizes DNA strand resection and thereby inhibits the HDR pathway (Pierce et al, 2001; Stark et al, 2004). However, the increased NHEJ in our study is not secondary to an Nbs1-null-induced proliferation arrest in G0/G1 because we observed these phenotypes also in proliferating Nbs1-null cells (see Figure 5). Moreover, since our experimental system provides the homology sequence right next to the DSB site (see Figure 2A), it avoids the consideration that the limited availability of homology sequence itself could promote NHEJ. Nonetheless, we cannot formally rule out the possibility that when HR is reduced, the substrates may be processed through the NHEJ pathway. Since the proliferation and the cell cycle distribution of constitutive Nbs1-null cells are similar to wild-type controls (Figure 4D and E) and the reintroduction of Nbs1 cDNA corrected DNA repair defects, it seems that the increased NHEJ activity is cell cycle-independent and is not limited by the substrate. These data thus suggest that MRN acts at the branching point upstream of both DSB repair pathways, that is, HR and NHEJ, which thus makes the MRN complex a good candidate that plays an instrumental role in the cellular decision about the use of these pathways, that is, Rad52 epistasis proteins or the Ku complex.
The current study demonstrates, for the first time in mammalian cells completely lacking the Nbs1 protein, that while this protein (possibly via the MRN complex) represses the NHEJ activity, Nbs1 promotes the processing of DNA DSBs, thereby facilitating recruitments of HR molecules (e.g., Brca1 and Rad51) to DNA breaks to conduct HR. Therefore, mammalian cells have evolved a delicate repair mechanism modulated through the MRN complex: by promoting HR, cells can maximize their capacity to repair lethal DSBs with a high fidelity and, by repressing NHEJ, they minimize the generation of ‘dangerous liaisons' between broken chromosomes.
Materials and methods
Generation of inducible Nbs1-null MEFs (Δ6/Δ6)
The construction of inducible Nbs1-null MEFs by the Cre-loxP technique was carried out as described previously (Herceg et al, 2001). We isolated MEFs from E13.5 embryos derived from crosses of Nbs1+/f6 mice (Frappart et al, 2005) with Nbs1+/f6 or Nbs1+/neo mice (Dumon-Jones et al, 2003) and immortalized them by the 3T3 protocol. Nbs1f6/f6 or Nbs1f6/neo MEF cell lines were co-transfected with a pCre-ER plasmid (referred to as CER) or an empty pSG9 plasmid (no Cre gene) (referred to as PSG), with a PGK-Hyg plasmid containing the hygromycin-resistance gene. To induce Nbs1 deletion, Nbs1f6/f6 or Nbs1f6/neo CER cells were incubated 2 days with 1 μM 4-hydroxytamoxifen (OHT) (Sigma, St Louis, USA) that triggers the Cre-mediated deletion of the Nbs1 exon 6 flanked by two loxP sites to generate Nbs1Δ6/Δ6 or Nbs1neo/Δ6 cells, respectively.
Establishment of constitutive Nbs1-null cells (neo/hyg)
To generate ES cells mutant for Nbs1, we designed strategies depicted in Supplementary Figure S1B. The first strategy used a ‘conventional' gene targeting approach to disrupt both alleles of exon 6 by sequential electroporation of the targeting vectors pTV-neo and pTV-hyg into D3 ES cells. The second strategy was to isolate Nbs1neo/f6 ES cells from E3.5 blastocysts derived from intercrosses of Nbs1+/f6 and Nbs1+/neo mice. Following transfection of Cre recombinase, the loxP-flanked exon 6 was deleted. To establish constitutive Nbs1-null MEFs, primary embryonic fibroblasts were isolated from E13.5 chimeric embryos and the G418-resistant fibroblasts (Nbs1neo/hyg) were immortalized according to the 3T3 protocol (see Figure 4A).
RT–PCR assay
Total RNA was extracted using Tri-Reagent (Sigma), and reversely transcribed into cDNA following the instruction in the kit. To analyze the expression of Nbs1 transcript, Nbs1 cDNA was amplified by PCR using primers Nbn658F (5′-ctaagaaacagcctccagata-3′; corresponding to exon 5) and Nbn905R (5′-attcctacatcaacaacgcag-3′; corresponding to exon 7), yielding 248-bp PCR products. For the internal control, the primer pair P3 (5′-ccatcacattgtggccctctgtgtgctca-3′) and P4 (5′-gttaaagttgagagatcatctccacca-3′), corresponding to hprt exon 3 and 4, respectively, amplifies a 200-bp PCR fragment.
Cytogenetic analysis
To facilitate visualization and identification of chromosome aberrations, we applied fluorescence in situ hybridization using a telomere-specific probe Cy3-PNA (Applied Biosystems, Courtaboeuf, France). Cells were collected for metaphase preparation as described previously (Tong et al, 2001). For examination of SCE, MEFs were cultured in the presence or absence of 30 nM MMC (Sigma) and were collected for preparation of chromosome spreads (Wang et al, 1997). After staining with 4% Giemsa, SCEs were scored.
Cell survival and proliferation assays
For the colony formation assay, MEFs were plated in duplicate at 1000 cells/10-cm dish. PSG or CER cells were cultured in the presence or absence of 1 μM OHT for 2 days and then for a further 10 days before fixing in methanol/acetic acid and staining with Giemsa. Colonies per dish were counted and expressed as the percentage of untreated controls. To test DNA damage response of constitutive Nbs1neo/hyg cells, cells were treated for 12 h with either 0.2 μg/ml ADR (Sigma) or 2 mM hydroxyurea (HU, Sigma) before the colony formation assay. To analyze apoptosis in cultured cells, TUNEL-positive cells were determined by in situ immunostaining with FITC-conjugated BrdU antibody (Roche, Meylan, France). For the proliferation assay, 1.5 × 105 cells were plated into one well of a six-well plate in triplicate and the cell number was determined at the indicated time points.
Cell cycle analysis
Cells were treated with or without 10 Gy of γ-radiation (137Cesium) and collected at the indicated time points. For FACS analysis, the percentage of cells in each phase of the cell cycle was measured by a FACScalibur apparatus equipped with Cellquest software (Becton-Dickinson) and ModFit software (Verity Software House Inc., Topsham, USA).
Construction of stable cell lines for DSB repair assay
Cell lines containing a single intact copy of the HDR reporter substrate DRGFP in a random locus (MEFs) or in the hprt locus (ES) were generated as described previously (Yang et al, 2005). Cells containing the SSA reporter substrate SAGFP were constructed as described (Stark et al, 2004). To analyze DSB repair, I-SceI-expressing plasmid pCBASce, a mock plasmid pCAGGS and the GFP-expressing plasmid pCAGGS-EGFP were electroporated into cells. To perform genetic rescue experiments, the mouse Nbs1 cDNA from pSK-mNbs1 (kindly provided by Dr Martin Digweed) was subcloned into expression vector pCAGGS, which then was transfected into Nbs1-null ES cells harboring DRGFP.
HDR and SSA analysis
Flow cytometric analysis was performed on cells carrying a single intact copy of the DRGFP (for HDR) or SAGFP (for SSA) reporter 2 days after electroporation with I-SceI-expressing plasmid (Stark et al, 2004; Yang et al, 2005). To analyze SSA in the HDR substrate DRGFP by PCR/Southern blotting, primers DF (ggttcggcttctggcgtg) and SAR (cacaggaaacagctatgacc) were used to generate SSA products and the primers DF and DR (gtagccttcgggcatggcgg) for total PCR products. After Southern blotting, the density of SSA products was quantified by PhosphorImage and normalized to the total PCR products within the same sample.
NHEJ analysis
The first assay was based on PCR and Southern blotting as described previously (Yang et al, 2005). The efficiency of sequence deletion via NHEJ was quantified by the density of smaller DNA bands compared to that of the 723-bp PCR products within each sample, using PhosphorImager. For the second approach, the genomic DNA of mock or I-SceI-transfected cells was extracted and digested with I-SceI, followed by BcgI and HindIII enzymes. After Southern blotting, blots were probed with a DNA fragment corresponding to the C-terminus of SceGFP. The total NHEJ efficiency was quantified by the density of DNA bands resistant to I-SceI and BcgI cleavage, normalized to that of uncut DNA within each sample, using PhosphorImage.
Chromatin fractionation
Chromatin fractionation was carried out as described previously (Mendez and Stillman, 2000) with modifications. For DNA damage treatment, cells were incubated with 0.2 μg/ml ADR for 3 h. Briefly, 3 × 106 cells were washed with PBS and resuspended in 200 μl of buffer A (10 mM HEPES at pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM DTT, 10 mM NaF, 1 mM Na3VO4, 1 mM β-glycerolphosphate, protease inhibitors). Triton X-100 was added to a final concentration of 0.1%. Nuclei were lysed in 200 μl of buffer B (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT, 10 mM NaF, 1 mM Na3VO4, 1 mM β-glycerolphosphate, protease inhibitors) and chromatin fractions were then separated from soluble nuclear proteins and sheared by sonication.
Immunoblotting
Fractionized proteins or proteins extracted from cells in RIPA buffer (20 mM HEPES pH 7.6, 20% glycerol, 0.5 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA pH 8.0, 0.5% NP-40, 1 mM DTT, 1 mM PMSF, 5 μg/ml leupeptin, 2 μg/ml aprotinin, 1 mM β-glycerolphosphate, 1 mM Na3VO4 and 10 mM NaF) were resolved by SDS–PAGE, blotted and stained with antibodies in TBST containing 5% non-fat dried milk (NFDM), followed by incubation with horseradish peroxidase-conjugated secondary antibodies and detected by the ECL reagents (Amersham Biosciences, Buckinghamshire, UK). The following antibodies were used: rabbit anti-Nbs1 serum (1:3000, Oncogene, San Diego, USA); rabbit anti-Nbs1 serum (1:2000; Dumon-Jones et al, 2003); mouse anti-Rad50 (1:1000, Upstate, Dundee, UK); mouse anti-PARP-1 (1:2000, R&D systems, Minneapolis, USA); rabbit anti-Ku70 (1:5000, Serotec, Oxford, UK); mouse anti-actin serum (1:10 000, Santa Cruz, Heidelberg, Germany).
Focus formation and cell imaging
Immunofluorescence staining of focus formation was performed as described previously (Yang et al, 2004) with modification. Briefly, MEFs were treated with or without 0.2 μg/ml ADR and washed once in PBS with 1 mM β-glycerolphosphate, 1 mM Na3VO4 and 10 mM NaF, 1 mM PMSF, and further treated with a hypotonic lysis solution (10 mM Tris–HCl pH 7.4, 2.5 mM MgCl2, 1 mM PMSF, 1 mM β-glycerolphosphate, 1 mM Na3VO4, 10 mM NaF, and 0.5% NP-40) for 8 min on ice. Subsequently, cells were fixed in ice-cold acetone–methanol (1:1) for 30 min on ice. The slides were then incubated with appropriate primary antibody in TBST containing 5% NFDM and mounted in Vectashield mounting medium with DAPI (Vector Laboratories, Burlingame, CA, USA). A cell with at least five distinct foci in the nucleus was scored as focus-positive. The following antibodies were used: rabbit anti-Rad51 antibody (1:200, kindly proved by Dr Steve West); rabbit anti-Mre11 (1:200, Novus); mouse anti-Rad50 (1:200, Upstate); rabbit anti-Brca1 (1:200, kindly provided by Dr Chu-Xia Deng).
Supplementary Material
Supplementary Figures
Legends to Supplementary Figures
Acknowledgments
We thank Maria Jasin for pDRGFP, pCBASce and pSAGFP plasmids, Steve West for rabbit anti-Rad51 antibody and Chu-Xia Deng for rabbit anti-Brca1 antibody. We also thank Christine Caux for flow cytometric analysis, Dominique Galendo for her excellent assistance in the maintenance of the animal colonies, Cyrille Cuenin and Kathrin Eberhardt for their excellent technical support. Further thanks are due to Janet Hall for her critical reading of the manuscript and John Cheney for the editing of the manuscript. We thank Tomas Lindahl for his support of the study. We are also grateful to many members of ZQW's laboratory for the helpful discussions. This work was supported in part by the Association pour la Recherche sur le Cancer (ARC), France and the Association for International Cancer Research (AICR), UK.
References
- Carson CT, Schwartz RA, Stracker TH, Lilley CE, Lee DV, Weitzman MD (2003) The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J 22: 6610–6620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amours D, Jackson SP (2002) The Mre11 complex: at the crossroads of DNA repair and checkpoint signalling. Nat Rev Mol Cell Biol 3: 317–327 [DOI] [PubMed] [Google Scholar]
- Demuth I, Frappart PO, Hildebrand G, Melchers A, Lobitz S, Stockl L, Varon R, Herceg Z, Sperling K, Wang ZQ, Digweed M (2004) An inducible null mutant murine model of Nijmegen breakage syndrome proves the essential function of NBS1 in chromosomal stability and cell viability. Hum Mol Genet 13: 2385–2397 [DOI] [PubMed] [Google Scholar]
- Difilippantonio S, Celeste A, Fernandez-Capetillo O, Chen HT, Reina San Martin B, Van Laethem F, Yang YP, Petukhova GV, Eckhaus M, Feigenbaum L, Manova K, Kruhlak M, Camerini-Otero RD, Sharan S, Nussenzweig M, Nussenzweig A (2005) Role of Nbs1 in the activation of the Atm kinase revealed in humanized mouse models. Nat Cell Biol 7: 675–685 [DOI] [PubMed] [Google Scholar]
- Dumon-Jones V, Frappart PO, Tong WM, Sajithlal G, Hulla W, Schmid G, Herceg Z, Digweed M, Wang ZQ (2003) Nbn heterozygosity renders mice susceptible to tumor formation and ionizing radiation-induced tumorigenesis. Cancer Res 63: 7263–7269 [PubMed] [Google Scholar]
- Falck J, Coates J, Jackson SP (2005) Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434: 605–611 [DOI] [PubMed] [Google Scholar]
- Fasullo M, Giallanza P, Dong Z, Cera C, Bennett T (2001) Saccharomyces cerevisiae rad51 mutants are defective in DNA damage-associated sister chromatid exchanges but exhibit increased rates of homology-directed translocations. Genetics 158: 959–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frappart PO, Tong WM, Demuth I, Radovanovic I, Herceg Z, Aguzzi A, Digweed M, Wang ZQ (2005) An essential function for NBS1 in the prevention of ataxia and cerebellar defects. Nat Med 11: 538–544 [DOI] [PubMed] [Google Scholar]
- Gao Y, Ferguson DO, Xie W, Manis JP, Sekiguchi J, Frank KM, Chaudhuri J, Horner J, DePinho RA, Alt FW (2000) Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature 404: 897–900 [DOI] [PubMed] [Google Scholar]
- Hakem R, de la Pompa JL, Elia A, Potter J, Mak TW (1997) Partial rescue of Brca1 (5–6) early embryonic lethality by p53 or p21 null mutation. Nat Genet 16: 298–302 [DOI] [PubMed] [Google Scholar]
- Harfst E, Cooper S, Neubauer S, Distel L, Grawunder U (2000) Normal V(D)J recombination in cells from patients with Nijmegen breakage syndrome. Mol Immunol 37: 915–929 [DOI] [PubMed] [Google Scholar]
- Herceg Z, Hulla W, Gell D, Cuenin C, Lleonart M, Jackson S, Wang ZQ (2001) Disruption of Trrap causes early embryonic lethality and defects in cell cycle progression. Nat Genet 29: 206–211 [DOI] [PubMed] [Google Scholar]
- Howlett NG, Scuric Z, D'Andrea AD, Schiestl RH (2006) Impaired DNA double strand break repair in cells from Nijmegen breakage syndrome patients. DNA Repair (Amst) 5: 251–257 [DOI] [PubMed] [Google Scholar]
- Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S, Carotenuto W, Liberi G, Bressan D, Wan L, Hollingsworth NM, Haber JE, Foiani M (2004) DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 431: 1011–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna KK, Jackson SP (2001) DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet 27: 247–254 [DOI] [PubMed] [Google Scholar]
- Kim JS, Krasieva TB, Kurumizaka H, Chen DJ, Taylor AM, Yokomori K (2005) Independent and sequential recruitment of NHEJ and HR factors to DNA damage sites in mammalian cells. J Cell Biol 170: 341–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kracker S, Bergmann Y, Demuth I, Frappart PO, Hildebrand G, Christine R, Wang ZQ, Sperling K, Digweed M, Radbruch A (2005) Nibrin functions in Ig class-switch recombination. Proc Natl Acad Sci USA 102: 1584–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahdesmaki A, Taylor AM, Chrzanowska KH, Pan-Hammarstrom Q (2004) Delineation of the role of the Mre11 complex in class switch recombination. J Biol Chem 279: 16479–16487 [DOI] [PubMed] [Google Scholar]
- Lavin MF (2004) The Mre11 complex and ATM: a two-way functional interaction in recognising and signaling DNA double strand breaks. DNA Repair (Amst) 3: 1515–1520 [DOI] [PubMed] [Google Scholar]
- Lee JH, Paull TT (2004) Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 304: 93–96 [DOI] [PubMed] [Google Scholar]
- Lee JH, Paull TT (2005) ATM activation by DNA double-strand breaks through the Mre11–Rad50–Nbs1 complex. Science 308: 551–554 [DOI] [PubMed] [Google Scholar]
- Lieber MR, Ma Y, Pannicke U, Schwarz K (2003) Mechanism and regulation of human non-homologous DNA end-joining. Nat Rev Mol Cell Biol 4: 712–720 [DOI] [PubMed] [Google Scholar]
- Lim DS, Hasty P (1996) A mutation in mouse rad51 results in an early embryonic lethal that is suppressed by a mutation in p53. Mol Cell Biol 16: 7133–7143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig T, Chapman DL, Papaioannou VE, Efstratiadis A (1997) Targeted mutations of breast cancer susceptibility gene homologs in mice: lethal phenotypes of Brca1, Brca2, Brca1/Brca2, Brca1/p53, and Brca2/p53 nullizygous embryos. Genes Dev 11: 1226–1241 [DOI] [PubMed] [Google Scholar]
- Luo G, Yao MS, Bender CF, Mills M, Bladl AR, Bradley A, Petrini JH (1999) Disruption of mRad50 causes embryonic stem cell lethality, abnormal embryonic development, and sensitivity to ionizing radiation. Proc Natl Acad Sci USA 96: 7376–7381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolis KG, Nimmo ER, Hartsuiker E, Carr AM, Jeggo PA, Allshire RC (2001) Novel functional requirements for non-homologous DNA end joining in Schizosaccharomyces pombe. EMBO J 20: 210–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maser RS, Zinkel R, Petrini JH (2001) An alternative mode of translation permits production of a variant NBS1 protein from the common Nijmegen breakage syndrome allele. Nat Genet 27: 417–421 [DOI] [PubMed] [Google Scholar]
- Mendez J, Stillman B (2000) Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol Cell Biol 20: 8602–8612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi K, Yang YG, Pierce AJ, Taniguchi T, Digweed M, D'Andrea AD, Wang ZQ, Jasin M (2005) Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. Proc Natl Acad Sci USA 102: 1110–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce AJ, Hu P, Han M, Ellis N, Jasin M (2001) Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev 15: 3237–3242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce AJ, Johnson RD, Thompson LH, Jasin M (1999) XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev 13: 2633–2638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reina-San-Martin B, Nussenzweig MC, Nussenzweig A, Difilippantonio S (2005) Genomic instability, endoreduplication, and diminished Ig class-switch recombination in B cells lacking Nbs1. Proc Natl Acad Sci USA 102: 1590–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riballo E, Kuhne M, Rief N, Doherty A, Smith GC, Recio MJ, Reis C, Dahm K, Fricke A, Krempler A, Parker AR, Jackson SP, Gennery A, Jeggo PA, Lobrich M (2004) A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol Cell 16: 715–724 [DOI] [PubMed] [Google Scholar]
- Shiloh Y (2003) ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 3: 155–168 [DOI] [PubMed] [Google Scholar]
- Stark JM, Pierce AJ, Oh J, Pastink A, Jasin M (2004) Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol Cell Biol 24: 9305–9316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS (2002) Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol Mol Biol Rev 66: 630–670, table of contents [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauchi H, Kobayashi J, Morishima K, van Gent DC, Shiraishi T, Verkaik NS, vanHeems D, Ito E, Nakamura A, Sonoda E, Takata M, Takeda S, Matsuura S, Komatsu K (2002) Nbs1 is essential for DNA repair by homologous recombination in higher vertebrate cells. Nature 420: 93–98 [DOI] [PubMed] [Google Scholar]
- Tomita K, Matsuura A, Caspari T, Carr AM, Akamatsu Y, Iwasaki H, Mizuno K, Ohta K, Uritani M, Ushimaru T, Yoshinaga K, Ueno M (2003) Competition between the Rad50 complex and the Ku heterodimer reveals a role for Exo1 in processing double-strand breaks but not telomeres. Mol Cell Biol 23: 5186–5197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong WM, Hande MP, Lansdorp PM, Wang ZQ (2001) DNA strand break-sensing molecule poly(ADP-Ribose) polymerase cooperates with p53 in telomere function, chromosome stability, and tumor suppression. Mol Cell Biol 21: 4046–4054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y (2003) Requirement of the MRN complex for ATM activation by DNA damage. EMBO J 22: 5612–5621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZQ, Stingl L, Morrison C, Jantsch M, Los M, Schulze-Osthoff K, Wagner EF (1997) PARP is important for genomic stability but dispensable in apoptosis. Genes Dev 11: 2347–2358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West SC (2003) Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol 4: 435–445 [DOI] [PubMed] [Google Scholar]
- Weterings E, van Gent DC (2004) The mechanism of non-homologous end-joining: a synopsis of synapsis. DNA Repair (Amst) 3: 1425–1435 [DOI] [PubMed] [Google Scholar]
- Xiao Y, Weaver DT (1997) Conditional gene targeted deletion by Cre recombinase demonstrates the requirement for the double-strand break repair Mre11 protein in murine embryonic stem cells. Nucleic Acids Res 25: 2985–2991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YG, Cortes U, Patnaik S, Jasin M, Wang ZQ (2004) Ablation of PARP-1 does not interfere with the repair of DNA double-strand breaks, but compromises the reactivation of stalled replication forks. Oncogene 23: 3872–3882 [DOI] [PubMed] [Google Scholar]
- Yang YG, Herceg Z, Nakanishi K, Demuth I, Piccoli C, Michelon J, Hildebrand G, Jasin M, Digweed M, Wang ZQ (2005) The Fanconi anemia group A protein modulates homologous repair of DNA double-strand breaks in mammalian cells. Carcinogenesis 26: 1731–1740 [DOI] [PubMed] [Google Scholar]
- Zhu J, Petersen S, Tessarollo L, Nussenzweig A (2001) Targeted disruption of the Nijmegen breakage syndrome gene NBS1 leads to early embryonic lethality in mice. Curr Biol 11: 105–109 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures
Legends to Supplementary Figures