

Abstract
Oxidative lesions represent the most abundant DNA lesions within the cell. In the present study, we investigated the impact of the oxidative lesions 8-oxoguanine, thymine glycol and 5-hydroxyuracil on RNA polymerase II (RNA pol II) transcription using a well-defined in vitro transcription system. We found that in a purified, reconstituted transcription system, these lesions block elongation by RNA pol II to different extents, depending on the type of lesion. Suggesting the presence of a bypass activity, the block to elongation is alleviated when transcription is carried out in HeLa cell nuclear extracts. By purifying this activity, we discovered that TFIIF could promote elongation through a thymine glycol lesion. The elongation factors Elongin and CSB, but not TFIIS, can also stimulate bypass of thymine glycol lesions, whereas Elongin, CSB and TFIIS can all enhance bypass of an 8-oxoguanine lesion. By increasing the efficiency with which RNA pol II reads through oxidative lesions, elongation factors can contribute to transcriptional mutagenesis, an activity that could have implications for the generation or progression of human diseases.
Keywords: bypass, elongation factors, oxidative DNA damage, transcription
Introduction
The integrity of cellular DNA is constantly threatened by DNA-damaging agents of both endogenous and environmental origins. Oxidative lesions represent the most common modifications within the cell, with approximately 150 000 lesions generated per cell per day (Beckman and Ames, 1997). To maintain the integrity of the genetic material, cells possess multiple DNA repair pathways, such as the nucleotide excision repair (NER) pathway, which is primarily responsible for repair of bulky helix-distorting damage, and the base excision repair (BER) pathway, which preferentially repairs non-bulky and non-helix-distorting damage (Hoeijmakers, 2001). Under some circumstances, however, these repair processes are inefficient or the load of lesions is too high, and the cell is obliged to replicate or transcribe DNA containing persistent damage.
During replication, unrepaired oxidative lesions lead to two major biological problems: miscoding, which is premutagenic, and polymerase arrest, which is cytotoxic. For example, 5-hydroxyuracil (OHdU) is not lethal, but is highly mutagenic in Escherichia coli, where it causes C to T transversions (Kreutzer and Essigmann, 1998). For 7,8-dihydro-8-oxoguanine (oxoG), a mutation frequency of 2.5–4.8% was determined in mammalian cells using a single-stranded phagemid shuttle vector system harboring a site-directed introduced oxoG (Moriya, 1993). Both, oxoG as well as OHdU are bypassed during replication (Wood et al, 1990; Shibutani et al, 1991). In contrast, 5,6-dihydroxy-5,6-dihydrothymine (thymine glycol, Tg), a non-mutagenic lesion (Hayes et al, 1988), leads to an arrest of the replicative DNA polymerase in vitro (Clark and Beardsley, 1986) and is consequently lethal in vivo (Hayes et al, 1988). To overcome such a cytotoxic status, the cell has evolved an elaborate system allowing replication past these blocking lesions. This mechanism is referred to as DNA translesion synthesis (TLS). It involves a polymerase switch, where a specialized polymerase reads through the lesion and afterwards hands over to the replicative DNA polymerase (Lehmann, 2005).
During transcription, two strategies could be envisioned to prevent acute cell death through arrested RNA polymerase II (RNA pol II) at unrepaired lesions. When encountering the damage site, elongating RNA pol II might promote transcription coupled repair (TCR), a specialized DNA repair pathway, that efficiently removes bulky lesions from the transcribed strand (Mellon et al, 1987). Whether TCR is able to remove non-bulky oxidative lesions remains controversial, since several key papers pointing in this direction have been recently retracted (Cozzarelli, 2003; Cooper et al, 2005; Le Page et al, 2005). The second strategy could be to bypass the lesion resulting in transcriptional mutagenesis (Doetsch, 2002). This pathway has been well characterized in E. coli (Bregeon et al, 2003), but there is not yet general agreement as to its importance in human cells (Tornaletti et al, 2001, 2004; Kuraoka et al, 2003; Kathe et al, 2004).
In the present study, we investigated the impact of three different oxidative lesions on RNA pol II transcription, using a well-defined in vitro transcription system. We found that a single oxidative lesion can block RNA pol II progression to a variable extent, depending on the type of the lesion. In nuclear extracts, this block is alleviated for each lesion, suggesting the presence of a bypass activity. In further experiments, we found that RNA pol II bypass activities reside in elongation factors including TFIIF, Elongin, CSB or TFIIS, which exhibit distinct specificities in their abilities to promote bypass of different oxidative lesions.
Results
An RNA pol II-associated bypass factor is present in HeLa NE
To analyze the effect of oxidative DNA damage on RNA pol II elongation, we first designed DNA substrates containing an either oxoG (DNA-oxoG), Tg (DNA-Tg) or OHdU (DNA-OHdU) lesion 488/489 nt (489 nt) downstream of the Adenovirus Major Late (AdML) promoter (Figure 1A). The presence of these nucleotide modifications at these positions abolishes an ApaLI restriction site. DNA templates with and without oxidative lesions were tested in a reconstituted transcription system (RTS), comprising the recombinant basal transcription factors TBP, TFIIB, TFIIE and TFIIF, as well as RNA pol II and TFIIH purified from HeLa cell nuclear extract (HeLa NE) (Gerard et al, 1991).
Figure 1.
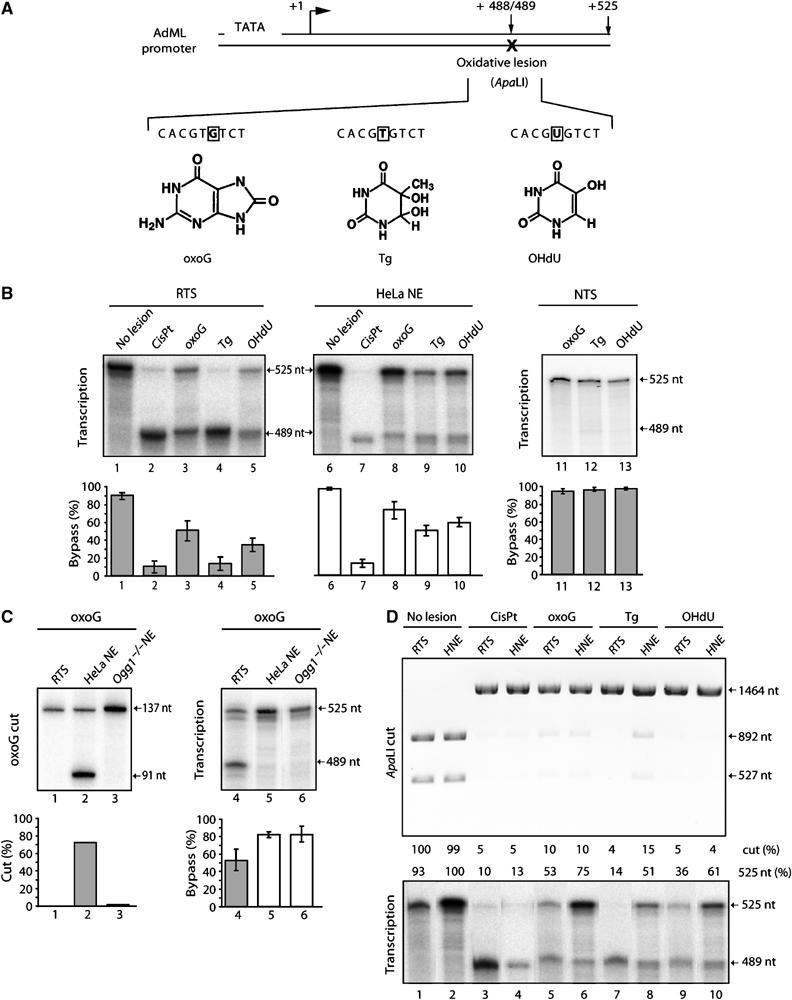
Bypass of oxidative lesions. (A) The transcription template contains a single oxoG-, Tg- or OHdU-lesion located 488/489 bp downstream of the AdMLP promoter start site. The oxidative lesion abolishes the ApaLI restriction site. (B) In vitro transcription of oxidative lesion containing template on the transcribed strand using either RTS (lanes 1–5), or HeLa NE (lanes 6–10) or on the non-transcribed strand (NTS) using RTS (lanes 11–13). Quantitative data (mean±s.d.) are derived from at least three independent experiments. Arrest (489 nt) and bypass (525 nt) transcripts are indicated. (C) Glycosylase assays (as described by Shimizu et al (2003) (lanes 1–3) and transcription assays (lanes 4–6) on an DNA-oxoG using RTS, HeLa and Ogg1−/− NE (kind gift of A Klungland). Quantitative transcription data (mean±s.d.) are derived from at least three independent experiments. (D) Repair of the damaged DNA templates as indicated at the top of the panel during the transcription reaction (lower panel). Templates (1464 bp), incubated in the presence of either RTS or HeLa NE, were next subjected to digestion by ApaLI. The presence of bands corresponding to 892 and 527 bp indicates digestion by ApaLI and thus repair of the lesion.
When no lesion was present, RNA pol II synthesized full-length, 525 nt transcripts (Figure 1B, lane 1). Transcription of DNA containing a well-characterized blocking lesion, a cisplatin 1,3-d(GTG) intrastrand crosslink (CisPt), results in the synthesis of 489 nt long RNA, suggesting that RNA synthesis stopped at the lesion (lane 2). When transcription was performed either on DNA-oxoG or DNA-OHdU templates, transcripts arrested at the lesion were synthesized in addition to the 525 nt full-length RNA, while transcription of DNA-Tg mainly produced the 489 nt RNA transcripts (lanes 3–5). This first set of experiments demonstrates that oxidative DNA modifications can block elongation by RNA pol II, albeit with different efficiencies depending on the lesion investigated.
We next carried out transcription assays in HeLa NE instead of the purified RTS. Under these conditions, we observed for each oxidative lesion studied, a two- to three-fold increase in the relative production of full-length transcripts, while CisPt still blocked elongation by RNA pol II (lanes 7–10). Note that the presence on the nontemplate strand of any oxidative DNA lesion investigated does not block elongation using RTS (lanes 11–13).
Based on these results, it appears that HeLa NE either permits the repair of oxidative DNA damage during transcription or contains factors that stimulate the bypass of these lesions. To discriminate between these hypotheses, we designed two sets of experiments. Nuclear extracts from either HeLa cells or oxoG-repair-impaired mouse embryonic fibroblasts derived from oxoG DNA glycosylase 1 (Ogg1)−/− mice (Klungland et al, 1999) were incubated with a γ-32P-ATP 5′-end labeled DNA-oxoG substrate. Only HeLa NE led to DNA-oxoG cleavage, while neither Ogg1−/− NE nor the RTS exhibited any oxoG excision activity (Figure 1C, lanes 1–3). Nevertheless, the oxoG-excision-impaired Ogg1−/− NE allowed high levels of full-length transcription (up to 90%), comparable to what we observed during transcription in HeLa NE and clearly higher than in the RTS (∼60%) (Figure 1C, lanes 4–6). This suggests that accumulation of full-length RNA transcripts is not (only) the result of repair of the DNA-oxoG, but rather due to the presence of an oxo-G bypass activity.
We next tested whether HeLa NE would repair oxidative lesions during the transcription reaction. Similar amounts of DNA-oxoG, DNA-Tg or DNA-OHdU were first transcribed in the presence of either RTS or HeLa NE and then purified and analyzed following ApaLI digestion. In the absence of a DNA lesion, we observed two restriction fragments of 892 and 572 bp (Figure 1D, lanes 1 and 2). Only a very low percentage of DNA-oxoG and DNA-OHdU templates as well as the DNA-CisPt template was cleaved by ApaLI. Importantly, no increase in the cleavage by incubation with NE in comparison to RTS was observed for these lesions, thus demonstrating that, in our assay, NE—compared to RTS—did not lead to an increased repair during transcription (lanes 3–6, 9 and 10). In contrast, cleavage of DNA-Tg by ApaLI increased slightly following incubation with HeLa NE compared to RTS, suggesting that some minor repair of Tg lesions occurred during the NE transcription reaction (lanes 7 and 8). This weak increase in repair activity (∼11%), however, was not sufficient to account entirely for the increase in full-length RNA synthesis seen with HeLa NE (∼35%). We thus conclude first that production of full-length RNA is due to RNA pol II translesion transcription rather than to the elimination of the oxidative damage and second, that HeLa NE contains at least one bypass activity.
Purification of an RNA pol II bypass factor
To isolate the protein(s) responsible for bypass stimulation, HeLa whole-cell extract was sequentially fractionated using heparin-Ultrogel, DEAE-Sephacryl, sulfopropyl-Spherodex and phenyl-5PW chromatography (Figure 2A and Materials and methods). The 0.40 M (NH4)2SO4 fraction eluted from the phenyl column contained the bypass activity and was subsequently purified on sulfopropyl-5PW and finally on heparin-5PW columns (Figure 2A). The protein fractions eluting from heparin-5PW between 0.22 and 0.35 M KCl were analyzed by SDS–PAGE and tested in a transcription assay containing the DNA-Tg template, RNA pol II, and each of the transcription factors required for promoter-specific initiation. A Tg bypass activity that supports increased production of full-length 525 nt RNA transcripts is detected in fractions 40–43 of the final Heparin column (Figure 2B, transcription, lanes 3–6). Starting from 20 × 1010 HeLa cells (∼3 g of total protein extract), we thus were able to recover after six chromatographic steps an active bypass fraction containing about 300 μg of protein. Bypass activity copurified with three major polypeptides of approximately 30, 50 and 75 kDa (Figure 2B, Coomassie staining, lanes 2–6). Analysis by mass spectrometry (MALDI-MS) and Western blot revealed that two of these polypeptides are the RAP30 and RAP74 subunits of the general transcription factor TFIIF (Figure 2B, Western blot). This factor is required for promoter-specific initiation and has been shown to stimulate the rate of transcript elongation by RNA pol II (Flores et al, 1989). The 50 kDa polypeptide corresponds to the ‘PKA and PKC Substrate In Neurons 3′ (PACSIN3) protein, a cytosolic adapter protein implicated in endocytosis and signal transduction (Modregger et al, 2000; Sumoy et al, 2001; Mori et al, 2003).
Figure 2.
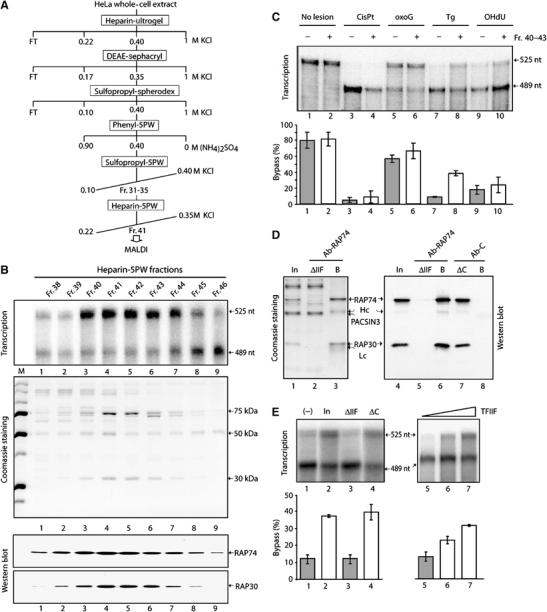
The bypass factor TFIIF. (A) Purification scheme from HeLa WCE. (B) Transcription, Coomassie staining, and TFIIF-Western blot using the fractions from the final heparin 5PW column. (C) Fr. 40–43 bypass activity towards several DNA lesions as indicated at the top of the panel. The quantification (mean±s.d.) was performed from at least three independent experiments. (D) TFIIF immunodepletion from the Pool of Fr.40–43 of the Heparin-5PW column (In), using either Ab-RAP74, an antibody raised against the larger subunit of TFIIF, or Ab-C, a control-antibody. The heavy (Hc) and light (Lc) chains are indicated. The proteins retained on the beads (B) as well as the ones in the flow through fraction (ΔIIF) were analyzed by SDS–PAGE (Coomassie staining, lanes 1–3) and Western blot (lanes 4–8) using the RAP74 and RAP30 antibodies. (E) These fractions (lanes 1–4) as well as the recombinant TFIIF (lanes 5–7) were further tested for their bypass activity in transcription. Quantitative data derived from at least three independent experiments are shown (mean±s.d., lower panels).
Since the screening for the bypass activity was performed using the DNA-Tg template, we wondered whether the purified factor also helps to bypass the other oxidative lesions. RTS was next incubated with either DNA-CisPt, -oxoG, -Tg, -OHdU or undamaged template with or without a pool of the active fractions 40–43 (Fr. 40–43) from the final heparin-5PW column. In the presence of the pool, we observed an increase in synthesis of the full-length RNA when DNA-Tg is used as a template, while there was weak, but no substantial increase when reactions contained DNA-oxoG or DNA-OHdU (Figure 2C).
To test whether the Tg bypass activity is due to TFIIF, Fr. 40–43 from the heparin-5PW column was immunodepleted with an antibody that specifically recognizes the RAP74 subunit of TFIIF (Figure 2D). Demonstrating the specificity of the immunodepletion, no additional proteins co-immunoprecipitated with TFIIF (lane 3), and TFIIF was not immunoprecipitated with a control antibody (lane 7 and 8). In addition, the immunodepletion was quantitative since no TFIIF was detectable in the TFIIF immunodepleted fraction (ΔIIF) by Coomassie staining (lane 2) or by Western blot (lane 5). When added to the RTS, the ΔIIF fraction does not stimulate bypass of the Tg lesion (Figure 2E, lanes 2 and 3). In contrast, the control-antibody treated pool of Fr. 40–43 (ΔC) showed bypass stimulation comparable to the input (lanes 1 and 4). It also has to be noted that PACSIN3, the 50 kDa polypeptide that copurified with TFIIF, remained in the ΔIIF fraction (Figure 2D, lane 2), which did not exhibit any bypass activity (Figure 2E, lane 3). Importantly, purified recombinant TFIIF increased the Tg bypass activity in a dose-dependent manner (Figure 2E, lanes 5–7). Taken together, our findings demonstrate that Tg bypass activity in our purified active fractions can be attributed to the transcription factor TFIIF.
Elongation factors act directly on the stalled ternary transcription complex
To investigate further how TFIIF promotes bypass of the Tg lesion and to determine whether other elongation factors have a similar activity, we established an in vitro transcription assay in which the damaged DNA template was immobilized on magnetic beads (Laine et al, 2006). After performing transcription using the RTS (Figure 3A, Incub I), all the basal transcription factors were removed from the immobilized damaged DNA template by extensive washes with a buffer containing 0.40 M KCl and 0.01% sarkosyl. The remaining immobilized ternary transcription complexes, which contain only the hyperphosphorylated elongating RNA pol II (RNA pol IIO) (Figure 3B, lane 3), were subsequently incubated (Incub II) with the purified recombinant elongation factors TFIIF, CSB (Troelstra et al, 1992, 1993) Elongin, (Aso et al, 1995; Kong et al, 2003) or TFIIS (Nakanishi et al, 1981) (Figure 3C). In this well-defined transcription elongation system, both the activity in Fr. 40–43 and recombinant TFIIF stimulate bypass of the Tg lesion, excluding the possibility that TFIIF acts indirectly at or prior to initiation and indicating instead that it acts directly on stalled RNA pol IIO (Figure 3D, lanes 1–3). In addition, we observe that CSB and Elongin also allow RNA pol IIO to bypass the Tg damage, leading to an increase in synthesis of the full-length 525 nt RNA (lanes 4–6). Interestingly, TFIIS, which functions by a mechanism distinct from that utilized by TFIIF, CSB, and Elongin, was not able to promote bypass of the Tg lesion by RNA pol IIO (lane 7).
Figure 3.
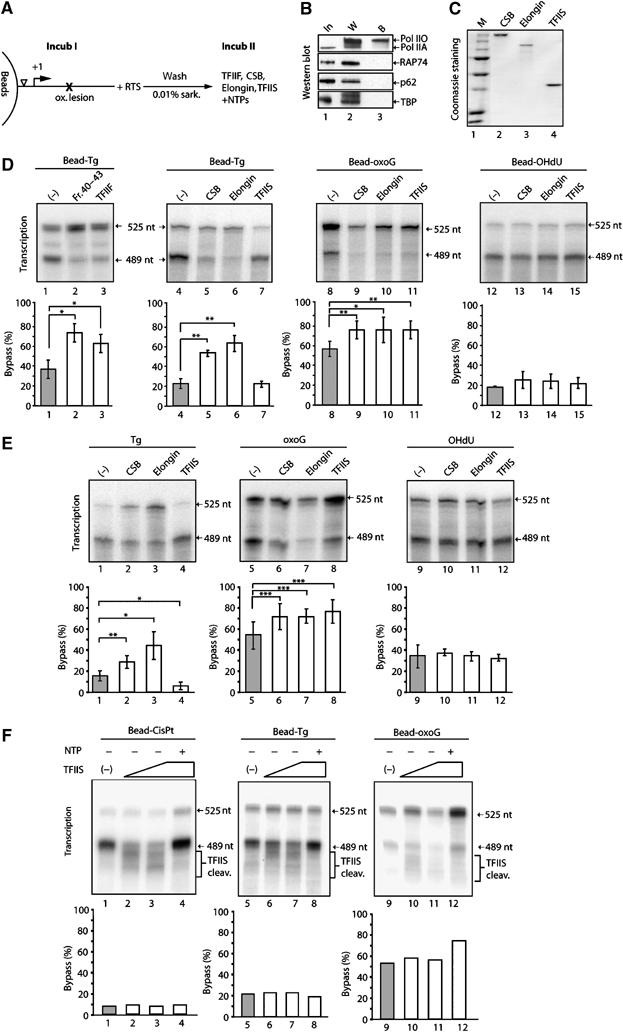
Elongation factor mediated specific bypass stimulation. (A) Scheme of transcription assay using immobilized DNA. After the first incubation (IncubI) with RTS and washes, the elongation factor such as TFIIF, CSB, Elongin or TFIIS was added for the second incubation (Incub II). (B) Western blot analysis of transcription components (In, lane 1), factors washed off (W, lane 2) or remaining bound (B, lane 3) to the immobilized damaged DNA. (C) Coomassie staining of purified recombinant CSB, Elongin and TFIIS. (D) Transcription on oxidative lesion containing DNA immobilized on magnetic beads in the presence of elongation factors (5 pmol) as indicated at the top of the panel. (E) Transcription as described above with a circular DNA template. Statistical analysis for (D) and (E) was performed by a pairwise comparison of elongation factor present versus negative control using a Student's t-test. Different degrees of significance were indicated as follows: *P<0.05, **P<0.01, ***P<0.001. (F) TFIIS-stimulated transcript cleavage (TFIIS cleav.), sensitivity of transcription complexes halted on immobilized DNA-oxoG, DNA-Tg and DNA-CisPt. After the first incubation, when indicated, TFIIS with and without NTPs was added to the reaction.
We also carried out similar experiments using immobilized DNA-oxoG and DNA-OHdU templates. RNA pol IIO stalled at an oxoG lesion can bypass the damage site when supplemented with CSB, Elongin and TFIIS (lanes 8–11). In contrast, none of these elongation factors are able to promote OHdU TLS by RNA pol IIO (lanes 12–15). Furthermore, we did not observe cooperative effects in the presence of various combinations of elongation factors; in fact, TFIIS even seems to slightly decrease the activity of Elongin, CSB and TFIIF on Tg lesions (data not shown).
Finally, we obtained similar results using nonimmobilized circular templates (Figure 3E). CSB and Elongin, but not TFIIS, could support bypass of a Tg lesion (Figure 3E, lanes 1–4); all of the elongation factors supported bypass of an oxoG lesion (lanes 5–8), and none of the factors tested could stimulate bypass of an OHdU lesion (lanes 9–12).
The above data suggest that each elongation factor exhibit its own specificity in controlling the bypass of the oxidative damage by the elongating RNA pol IIO and also demonstrate that the structure of the DNA template (linearized versus closed circular DNA), does not substantially influence the bypass reaction.
To address the question, why TFIIS mediates the bypass of oxoG but not Tg, even though they are both not complete blocks of transcription, we tested, whether RNA pol II halted in front of oxoG and Tg responds differently in terms of TFIIS-stimulate transcript cleavage. After having performed transcription on immobilized DNA-Tg, DNA-oxoG and DNA-CisPt as illustrated in Figure 3A, TFIIS was added in the absence of NTP to monitor transcript cleavage. For all lesion, the TFIIS-stimulated transcript cleavage is observed: RNA shorter than the transcript generated from lesion-stalled RNA pol II is formed as well as the of 489 nt transcript in a TFIIS-dose-dependent manner (Figure 3F, lanes 2 and 3, 6 and 7, as well as 10 and 11). These backtracked ternary complexes can be reactivated upon NTP addition (lanes 4, 8 and 12) but do not lead to increased bypass of Tg or CisPt, reinforcing the results shown before (Figure 3 D and E). Thus, the difference in RNA pol II behavior with regard to TFIIS bypass stimulation between DNA-Tg and DNA-oxoG cannot be explained by different sensitivity towards TFIIS-mediated cleavage induction. To study TFIIF's mode of action, KMnO4-footprinting experiments were performed and indicated that TFIIF slightly changes the DNA structure in the context of a stalled RNA pol II (data not shown).
Bypass of oxidative lesions might partially result in miscoding
To address whether increased bypass by RNA pol II would result in transcriptional mutagenesis, we analyzed the sequence of the resulting transcript. We generated by reverse transcription the corresponding cDNA followed by PCR amplification. The PCR products were either sequenced or digested with the restriction enzyme ApaLI. Sensitivity to ApaLI is diagnostic for mutagenesis, since transcriptional mutagenesis resulting from misincorporation opposite either the guanine oxidation product oxoG or the thymine oxidation product Tg would abolish the restriction site (see Figure 1). OHdU, which we introduced in place of a thymine in DNA-OHdU (Figure 1A), is a cytosine oxidation product (see Discussion). Consequently, transcriptional mutagenesis by incorporation of adenine opposite the OHdU would restore the ApaLI site in the corresponding cDNA.
Transcription of DNA-oxoG is mutagenic, since 8% of the RT–PCR products are insensitive to ApaLI digestion (Figure 4A, lane 3). Sequence analysis of the cDNA shows that mutagenic oxoG leads principally to adenine incorporation (Figure 4B, upper panel). Transcription on DNA-OHdU template is highly mutagenic, since OHdU pairs nearly exclusively with adenine, as demonstrated by the almost complete sensitivity of RT–PCR products to ApaLI digestion (Figure 4A, lane 5) and by sequencing (Figure 4B). In contrast, transcription of the DNA-Tg template results in less than 2% mutated transcripts (Figure 4A, lane 4, and Figure 4B). We note that addition of either CSB, Elongin, or TFIIS does not result in an obvious change of the mutagenic potential of DNA-oxoG by RNA pol II (Figure 4B, lower panels). Similarly, none of the elongation factors seems to have an influence on the mutation rate for either OHdU or Tg.
Figure 4.
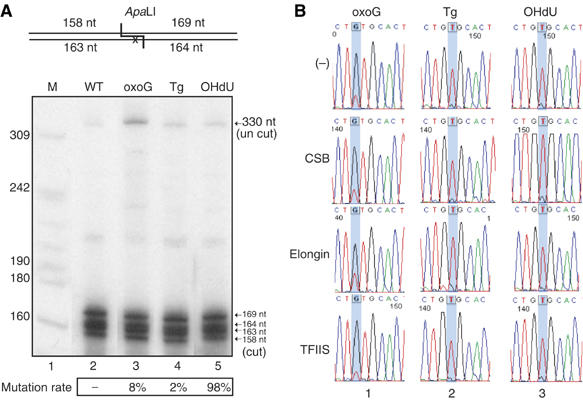
RNA transcript sequence analysis. (A) ApaLI (10 U), digestion for 3 h at 37°C of the labelled RT–PCR-product from DNA-oxoG, DNA-Tg and DNA-OHdU templates following incubation with RTS. The uncut (330 nt) and ApaLI cut (169, 164, 163 and 159 nt) DNA fragments were analyzed on a 8% denaturing polyacrylamide gel. The ratios presented at the bottom of the figure shows the percentage of mutated transcript. (B) Transcript sequence analysis by RT–PCR product sequencing. During the transcription reaction, the indicated elongation factor was present. The position of the lesion is highlighted with a blue bar.
Discussion
Cells are continuously exposed to genotoxic attack and therefore must be prepared to counteract the detrimental effects of the resulting DNA damage by either rapidly repairing the damage or activating the apoptotic response. Another option is to allow replicative or transcriptional bypass of DNA lesions at the cost of risking mutagenesis. As mentioned earlier, TLS has been particularly well studied for DNA replication, where it is facilitated by a switch between high and low fidelity DNA polymerases during bypass of the lesion (reviewed in Lehmann, 2005). During transcription, however, there is no polymerase switch to facilitate TLS.
Oxidative lesion has the ability to partially block RNA pol II elongation
In the present study, we have investigated the behavior of RNA pol II during in vitro transcription of DNA templates that contain different oxidative lesions. Using a RTS, we found that the oxidative lesions Tg, oxoG and OHdU can all block elongation by RNA pol II to a greater or lesser extent, with Tg lesions presenting the greatest impediment to elongation, while oxoG and OHdU lesions are bypassed more easily. These results are reminiscent of those obtained with DNA polymerases during replication and might be explained by the different modifications of the DNA structure specific for each type of oxidative damage (reviewed in Lukin and de Los Santos, 2006; Wallace, 2002). X-ray diffraction studies showed that the conversion of guanine to oxoG does not significantly alter the glycosidic torsion preference of the molecule and that there is no steric interaction of oxoG with the phospho-deoxyribose backbone. Similarly, OHdU in DNA adopts a nondistorted B type conformation (Thiviyanathan et al, 2005). In contrast, molecular dynamics simulations of Tg support the idea that the hydroxyl group at the 5 position results in a pseudo-axial oriented methyl group that causes distortion of the base pair 5′ to the Tg. NMR analyses of Tg containing DNA further indicate that the presence of Tg induce a significant and highly localized alteration in the structure of the DNA, in which the base of Tg and of the opposing residue are extrahelical (Kung and Bolton, 1997).
Elongation factors regulate oxidative lesion bypass
During transcription in HeLa extracts, in which it can be assumed that any activity regulating transcription should be present, we observed a higher production of full-length RNA transcripts. Our results argued that the increased efficiency of elongation on damaged DNA in these extracts was not due to either BER (Figure 1C) or TC-NER/TC-BER. Indeed, incubating either all or combinations of nucleotide excision repair factors (Laine and Egly, 2006) together with the stalled RNA pol IIO does not remove the oxidative damage (data not shown). Alternatively, stalled RNA pol IIO prevents access of the glycosylases such as Ogg1, Nth1, Neil1 or Neil2 to the damage (data not shown), demonstrating that Neil1 and Neil2 do not cut preferentially in the context of a transcription bubble, as previously speculated (Dou et al, 2003). Addition of all the factors required for TC-NER does not improve the removal of the oxidative damage by glycosylases (data not shown).
In conclusion, the absence of substantial repair during the transcription reaction suggests that activity(s) in the extracts were capable of increasing the efficiency of RNA TLS. Purification of a bypass activity from HeLa nuclear extract identified the general transcription factor TFIIF, which is capable of stimulating the rate of transcript elongation by pol II (Tan et al, 1994). Further investigation revealed that several other elongation factors, including Elongin, CSB and TFIIS, can increase the bypass of different oxidative lesions to varying extents. OxoG bypass could be stimulated by all elongation factors, Elongin, CSB and TFIIS, presumably because the presence of oxoG does not significantly alter the overall DNA structure (see above). We also observed that bypass of Tg lesions could be stimulated by TFIIF, Elongin and CSB, but not by TFIIS. Importantly, although activity(s) in HeLa NE could promote bypass of an OHdU lesion, TFIIF, Elongin, CSB as well as TFIIS could not, suggesting that additional bypass activity(s) remain to be identified and its purification is currently ongoing.
It has to be stressed that, despite some specificity, we found overlapping activities for oxidative lesion bypass stimulation. This apparent redundancy raises the question for the need of multiple bypass factors within the cell. However, we analyzed only three different oxidative lesions in our study, whereas within the cell a large variety of different oxidative lesions exist. Therefore, we could imagine that some factors that were indistinguishable in our study would show discriminable bypass activities for other lesions. This redundancy/specificity is reminiscent of the one seen with DNA-N-Glycosylases, that are responsible for the first step in BER (Parsons and Elder, 2003). Moreover, different elongation factors could act in a specific context or discrete localization. Altogether, this could explain the existence of redundant bypass activities.
How might TFIIF, Elongin, CSB and TFIIS promote transcriptional bypass of oxidative lesions? Consistent with their known abilities to act directly on the ternary transcription complex (reviewed in Sims et al, 2004), our data demonstrate that TFIIF, Elongin, CSB and TFIIS act directly on stalled RNA pol II to allow bypass of oxidative lesions.
The distinct mechanisms of action of TFIIS on the one hand and TFIIF, Elongin and CSB on the other hand may contribute to their differential abilities to reactivate RNA pol II stalled at the relatively nondistorting lesion oxoG and the highly distorting lesion Tg. During transcription, elongating RNA pol II is subject to reversible pausing or transcriptional arrest, which are believed to result from aberrant backward movement (backtracking) of RNA pol II on the DNA template and displacement of the 3′-end of the transcript from the catalytic site in a process that is spontaneously reversible (i.e. pausing) or not (i.e. arrest). TFIIF, Elongin and CSB all stimulate the rate of elongation by decreasing the frequency or duration of transient pausing by RNA pol II (reviewed in Shilatifard et al, 2003). Evidence suggests that Elongin (Takagi et al, 1995), TFIIF (RCC, JWC, BJ Elmendorf, unpublished data), and perhaps other elongation factors including CSB, suppress transient pausing by helping to lock RNA pol II into an active conformation such that the 3′-hydroxyl terminus of the nascent RNA chain is maintained in its proper position in the polymerase active site (Shilatifard et al, 2003; Sims et al, 2004). In contrast, TFIIS can reactivate arrested RNA pol II by stimulating the nascent transcript cleavage activity of the back-tracked polymerase (Izban and Luse, 1992; Reines et al, 1992; Reines, 1992). This creates a new 3′OH end that is properly aligned in the active center of the RNA pol II and can be re-extended, allowing the enzyme additional opportunities to read past the arrest site. Thus, it is tempting to speculate that TFIIF, Elongin, and CSB may be able to stimulate bypass of oxidative lesions because they prevent pausing or arrest at these sites by locking RNA pol II in an active conformation that seems resistant even to substantial distortions in the DNA template like the Tg lesion. In contrast, TFIIS does not lock RNA pol II into such a conformation but gives the RNA pol II a second chance to bypass the lesion by stimulating transcript cleavage. Theoretically, even with low probability of crossing, TFIIS-mediated cleavage and restart should allow all of the RNA pol II to bypass Tg lesion. Then, how can it be explained, that TFIIS does not promote Tg bypass even though 20% of this lesion was bypassed in the first encounter? We propose that the RNA pol II fraction that can bypass the Tg lesion is able to do so due to its association with TFIIF. RNA pol II alone would not be able to bypass the Tg lesion. In our assays, TFIIF is either absent or in limiting amounts for optimum elongation. Hence, repeated chances to resume transcription, stimulated by TFIIS, will end again at the lesion site. This is well observed with the CisPt lesion, which is also sensitive to TFIIS, but not bypassed (Figure 3F). Furthermore, we do not observe synergistic effects since TFIIF may negatively regulate TFIIS activity (Elmendorf et al, 2001).
Bypass of oxidative lesions can promote transcriptional mutagenesis
As discussed earlier, bypass of oxidative lesions occurs at the risk of transcriptional mutagenesis. Although oxoG pairs predominantly with cytosine, it can also pair with adenine during transcription (Figure 4B, Kuraoka et al, 2003). We also found that OHdU, a product of cytosine oxidation (reviewed in Wallace, 2002), is highly mutagenic as it pairs nearly exclusively with adenine. In contrast, bypass of Tg lesions is not mutagenic during in vitro transcription (Figure 4).
We also should point out that our findings demonstrate the absence of a correlation between the blocking and the miscoding properties of the studied lesions: oxoG is weakly blocking and weakly mutagenic, OHdU is weakly blocking but highly mutagenic, and Tg is strongly blocking and not mutagenic. Moreover, we showed that elongation factors do not substantially alter the miscoding properties of the oxidative lesions despite their ability to increase translesion transcription. In light of these observations, we can conclude that the mispairing potential of a given damage is an intrinsic property of the lesion and is independent from its ability to block RNA pol II.
Finally, our discoveries that transcription elongation factors can increase the efficiency with which RNA pol II is able to bypass oxidative lesions and that different elongation factors do so to different extents could also—at least partially—explain, the contradictory data found in the literature (Tornaletti et al, 2001, 2004; Kuraoka et al, 2003; Kathe et al, 2004). Thus, translesion transcription may occur to varying extents depending on the particular cell extract used, and/or on the presence of contaminating elongation factors in components of the transcription system. In addition, sequences surrounding an oxidative lesion could influence the bypass activity of RNA pol II, since it is known that the template sequence can influence pausing and/or arrest. For example, the sequence of downstream DNA can selectively alter a paused conformation of human RNA pol II (Palangat et al, 2004); this is why in the present study, the lesions were inserted in the same DNA sequence context. Moreover, we also have observed that insertion of the lesions closer to the transcription initiation site increases the efficiency of oxidative lesion bypass by RNA pol II (Table I). This could be due to some remaining influence of factors required for promoter escape and early elongation. For example, an increased portion of RNA pol II could still be associated with TFIIF closer to the transcription start site.
Table 1.
Bypass frequency for different lesions depending on the distance
| Lesiondistance | 105 bp | 489 bp |
|---|---|---|
| WT | 92±3 | 93±4 |
| CisPt | 24±7 | 10±4 |
| oxoG | 77±2 | 48±3 |
| Tg | 31±7 | 22±9 |
| OHdU | 65±2 | 39±7 |
| RNA pol II bypass of oxidative damage varies with the distance of the lesion from the transcription start site (105/489 nt). | ||
The present work sheds some light on the behavior of RNA pol II encountering an oxidative lesion and the transcriptional mutagenesis mechanism. By unraveling this elongation factor-mediated bypass of oxidative lesions, we have identified, on the one hand, a novel mechanism for dealing with oxidative lesions during RNA pol II transcription. On the other hand, we also highlight a novel function for elongation factors upon oxidative stress. Notably, these findings are consistent with the recent finding that CSB−/− cells exhibit an increased sensitivity toward oxidative stress (de Waard et al, 2004) and observations implicating other elongation factors in the response to stress conditions (Gerber et al, 2005; Mason and Struhl, 2005). Finally, we have shown that, depending on the type of lesion, such bypass can generate mutated RNA, which, in turn, may contribute to the generation and progression of human diseases (Saxowsky and Doetsch, 2006). By now, there are no reports addressing the question if transcriptional bypass can yield sufficient mutated mRNA and mutant proteins such that there are phenotypic changes. However, given these observations in bacterial systems (Viswanathan et al, 1999), it is likely that these changes also occur in mammalian systems. Answering this question will be the focus of future research.
Materials and methods
Construction of DNA templates
Circular as well as immobilized DNA damage containing templates were prepared as described (Laine et al, 2006). Briefly, for immobilized templates, a 972 bp fragment was generated from the pBSII(KS-)GAL CAX500/300 TS by FokI digestion and biotinylated by fill-in synthesis using the Klenow fragment of DNA pol I in the presence of 40 μM Bio-dUTP (Amersham Bioscience). After ethanol precipitation the DNA was digested with Bsu36I and DraIII and dephosphorylated with calf intestine alkaline phosphatase (PROMEGA). If necessary, the template was radioactively labelled using γ-32P-ATP and Polynucleotide Kinase (NEB).
In vitro transcription assays
Transcription was performed as described in Gerard et al (1991) for nonimmobilized DNA and in Laine et al (2006) for immobilized DNA, using RNA pol II and TFIIH purified from Hela cells and recombinant TBP, TFIIB, TFIIE and TFIIF with the following modifications: After a 15-min preincubation at 30°C, NTPs (General Electrics Healthcare) were added to a final concentration of 300 μM (ATP, CTP, GTP) or 50 μM (UTP) and incubated for further 20 min at 37°C. When indicated, 5 pmol elongation factors were added to the transcription reaction. Reactions were stopped by adding 5 μM alpha-amanitin and 10 U RNase-free DNaseI (Roche) to each transcription reaction and incubated for 20 min at 37°C. After addition of 50 μg Proteinase K (Sigma-Aldrich) and 200 μl STOP buffer (1% SDS, 300 μM sodium-acetate pH 5and 50 μg/ml glycogen) and incubation for 20 min at 37°C, the transcript was purified by phenol/chloroform extraction and ethanol precipitation.
RNase protection assay
The transcript was resuspended in hybridization solution containing 80% formamide and 400 mM NaCl, 50 mM PIPES pH 6.5, 1.25 mM EDTA and 1 fmol of 32P-labelled probe. This mixture was heated for 2 min at 95°C and hybridised over night at 65°C. The following day hybridisation was stopped by transferring the samples on ice. Single-strand RNA was digested by addition of 4 μg RNase A and 20 U of RNase T1 in 80 μl RPA buffer (300 mM NaCl, 10 mM Tris pH 7.5, 10 mM EDTA) and incubation for 30 min at 37°C. This step was followed by phenol/chloroform purification and ethanol precipitation. Total reaction product was loaded in sample buffer (80% formamide, 0.5% TBE, 0.02% bromphenolblue, 0.02% xylenecyanol) on 8% urea polyacrylamide gels and analyzed by autoradiography. Quantification was performed using a Typhoon phosphoimager and ImageQuant software from GE Healthcare. The rate of bypass was established by dividing the intensity of the 595 nt transcript band by the sum of the intensity of the 498 nt and the 595 nt band.
RNA probe preparation
The DNA template for the RNA probe contains nucleotides 325–523 of the transcript initiated from the AdML promoter of pBSII(KS-)GAL CAX500/300 TS and was generated by PCR using a T7 containing reverse primer, a gene-specific forward primer and pBSII(KS-)GAL CAX500/300 TS as a template. Transcription was performed using 10 U T7 RNA polymerase (Promega) and 100ng of PCR product, 500 μM ATP, UTP, GTP in a final concentration of cold 50 μM CTP and 5 μM 32αP-CTP (∼400 μCi/mmol). Reactions were performed at 37°C for 30 min followed by DNaseI digestion (5 U/100ng DNA) for additional 20 min. The reaction was stopped by adding equal volume loading dye and separated on an 8% denaturing urea gel followed by extraction from the gel.
TFIIS-stimulated transcript cleavage analysis
After the first round of transcription on immobilized DNA, washes with 400 mM KCl and 0.01% sarkosyl, and reequilibration with transcription buffer, 0.5–1.5 pmol TFIIS was added to the reaction and incubated at 30°C for 20 min before stopping the reaction as described above for the in vitro transcription.
Purification of the Tg bypass factor TFIIF
All procedures were carried out at 4°C. Heparin-Ultrogel and DEAE Spherodex were purchased from IBF (France). TSK-phenyl-5PW, TSK-Heparin-5PW and TSK-SP-5PW were from Toso Haas (Germany). The first two steps of the purification were performed as described (Gerard et al, 1991).
Step 3: Six DEAE 0.35 M fractions (6 × 400 ml) that contained bypass activity were pooled, adjusted to 60 mM KCl by dilution with buffer A (50 mM Tris/HCl pH 7.9, 0.5 mM DTT, 0.1 mM EDTA, 8.7% Glycerol) and loaded onto a 100 ml SP Spherodex column (6 × 5.5 cm2) equilibrated in buffer B (50 mM Tris pH 7.9, 50 mM KCl, 0.5 mM DTT, 0.1 mM EDTA, 8.7% Glycerol). The column was loaded over night by gravity (height difference between columns 60 cm, initial flow rate 3.3 ml/min). The elution was performed at 3 ml/min with 100 and 400 mM KCl in buffer B. Nine milliliter fractions were collected at the 400 mM step.
Step 4: The active fractions (45 ml) were pooled, dialyzed against buffer C (50 mM Tris pH 7.9, 900 mM (NH4)2SO4, 50 mM KCl, 0.5 mM DTT, 0.1 mM EDTA, 8.7% Glycerol) and loaded onto an in buffer C equilibrated analytical phenyl-5PW column (0.75 × 7.5 cm2, 0.3 ml/min). The flow-through was collected in 3 ml fractions. The activity was eluted in two steps of 400 mM and 0 mM (NH4)2SO4, which established as steep gradients due to the small volume of the column. Fractions (0.3 ml) were collected.
Step 5: Active fractions (18, 19 and 20) from 15 phenyl-5PW columns were pooled, dialyzed against buffer B and loaded onto an SP-5PW column (0.75 × 7.5 cm, 0.3 ml/min) equilibrated in buffer B. After a step of 1.5 column volumes (4.5 ml) at 100 mM KCl, the activity was eluted at 0.3 ml/min with a 4.5 ml linear gradient from 100 mM to 400 mM KCl. 0.3 ml fractions were collected.
Step 6: The bypass activity (Fr. 31–34) was dialyzed against buffer B and loaded onto a heparin-5PW column (0.75 × 7.5 cm2, 0.3 ml/min) equilibrated in buffer B. After washing with 4.5 ml of buffer B containing 225 mM KCl, the column was eluted with a 4.5 ml linear gradient from 225 to 375 mM KCl in buffer B. The activity eluted between Fr. 39 and 43. For analysis, the fraction was dialyzed in buffer B.
Purification of recombinant proteins
Elongin was prepared as described in Kong et al (2003). CSB was prepared as described in Bradsher et al (2002) and TFIIS was prepared as described in Powell and Reines (1996).
Immunodepletion of TFIIF
Fraction 40–43 from the final column (Heparin-5PW) were pooled and dialyzed against buffer D containing 50 mM Tris pH 7.9, 150 mM KCl, 0.5 mM DTT, 0.1 mM EDTA, 8.7% Glycerol and 0.05%NP40. Afterwards, 150 μl of the pool was supplemented with buffer D equilibrated RAP74 or a control antibody crosslinked to Protein A beads or left untreated (input). The next day, beads were carefully pelleted at 200 g and supernatants were removed. The beads were washed with 3 × 10 volumes in buffer D and gel loading buffer was added. Aliquots of the supernatant and the input were also subjected to gel loading buffer addition. For in vitro transcription aliquots of supernatants were dialyzed in buffer A (50 mM Tris/HCl pH 7.9, 0.5 mM DTT, 0.1 mM EDTA, 8.7% glycerol).
Reverse transcription and PCR
After the transcription reaction, the template DNA was digested for 1 h at 37°C using 10 U DNaseI per reaction. This step was followed by routine RNA extraction and reverse transcription was performed according to the manufacturer's protocol (SuperScript II Invitrogen). Briefly, RNA was resuspended in RT reaction mix containing 2 pmol gene specific primer, 1 × first strand buffer, 500 μM dNTP and 10 μl H2O, heated for 5 min at 65°C and quick frozen on ice. Afterwards, 10 mM DTT, 5 U RNasin (Promega) and 10 U Reverse Transcriptase (RT) SuperScript II (invitrogen) is added and incubated 50 min at 42°C. RT was inactivated for 15 min at 65°C. 1/10 of the RT reaction was added to a PCR reaction. 10 U PFU polymerase (NEB) was used with the following reaction conditions: 2 min 94°C, 25 cycles of 94°C for 40 s, 52°C for 40 s, 72°C for 50 s and 5 min final elongation. After purification using QIAGEN quick PCR purification kit, sequencing was performed using gene specific primer. Alternatively, PCR products were digested with 10 U ApaLI restriction enzyme at 37°C o.n. (NEB/Fermentas) followed by end labelling using 0.1 μM 32P-γ ATP and 5 U polynucleotide kinase (NEB), separation on 8% denaturing urea polyacrylamide gel and revelation using autoradioactivity.
Acknowledgments
We thank M Bjoras for the Tg-lesion containing oligonucleotide and fruitful discussion. We thank Z Burton for providing the recombinant TFIIF, D Reines and C Kane for the TFIIS containing plasmid. We also are indebted to M Argentini for mass spectrometry analysis as well as to J Gintz, C Braun and A Larnicol for their skilful technical assistance. This study has been supported by funds from ‘La Ligue contre le Cancer' (équipe labellisée, Contract No. EL2004), the ‘Ministère de l'Education National et de la Recherche', the ACI Grants (BCMS No. 03 2 535) and the Agence Nationale de la Recherche (ANR-05-MRAR-005-01). SF was granted by an EEC grant (MRTN-CT-2003-503618). This work is dedicated to Professor E Seeberg, who was a pioneer in the DNA repair field
References
- Aso T, Lane WS, Conaway JW, Conaway RC (1995) Elongin (SIII): a multisubunit regulator of elongation by RNA polymerase II. Science 269: 1439–1443 [DOI] [PubMed] [Google Scholar]
- Beckman KB, Ames BN (1997) Oxidative decay of DNA. J Biol Chem 272: 19633–19636 [DOI] [PubMed] [Google Scholar]
- Bradsher J, Auriol J, Proietti de Santis L, Iben S, Vonesch JL, Grummt I, Egly JM (2002) CSB is a component of RNA pol I transcription. Mol Cell 10: 819–829 [DOI] [PubMed] [Google Scholar]
- Bregeon D, Doddridge ZA, You HJ, Weiss B, Doetsch PW (2003) Transcriptional mutagenesis induced by uracil and 8-oxoguanine in Escherichia coli. Mol Cell 12: 959–970 [DOI] [PubMed] [Google Scholar]
- Clark JM, Beardsley GP (1986) Thymine glycol lesions terminate chain elongation by DNA polymerase I in vitro. Nucleic Acids Res 14: 737–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper PK, Nouspikel T, Clarkson SG (2005) Retraction. Science 308: 1740. [DOI] [PubMed] [Google Scholar]
- Cozzarelli NR (2003) Editorial expression of concern. Proc Natl Acad Sci USA 100: 11816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Waard H, de Wit J, Andressoo JO, van Oostrom CT, Riis B, Weimann A, Poulsen HE, van Steeg H, Hoeijmakers JH, van der Horst GT (2004) Different effects of CSA and CSB deficiency on sensitivity to oxidative DNA damage. Mol Cell Biol 24: 7941–7948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetsch PW (2002) Translesion synthesis by RNA polymerases: occurrence and biological implications for transcriptional mutagenesis. Mutat Res 510: 131–140 [DOI] [PubMed] [Google Scholar]
- Dou H, Mitra S, Hazra TK (2003) Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J Biol Chem 278: 49679–49684 [DOI] [PubMed] [Google Scholar]
- Elmendorf BJ, Shilatifard A, Yan Q, Conaway JW, Conaway RC (2001) Transcription factors TFIIF, ELL, and Elongin negatively regulate SII-induced nascent transcript cleavage by non-arrested RNA polymerase II elongation intermediates. J Biol Chem 276: 23109–23114 [DOI] [PubMed] [Google Scholar]
- Flores O, Maldonado E, Reinberg D (1989) Factors involved in specific transcription by mammalian RNA polymerase II. Factors IIE and IIF independently interact with RNA polymerase II. J Biol Chem 264: 8913–8921 [PubMed] [Google Scholar]
- Gerard M, Fischer L, Moncollin V, Chipoulet JM, Chambon P, Egly JM (1991) Purification and interaction properties of the human RNA polymerase B(II) general transcription factor BTF2. J Biol Chem 266: 20940–20945 [PubMed] [Google Scholar]
- Gerber M, Tenney K, Conaway JW, Conaway RC, Eissenberg JC, Shilatifard A (2005) Regulation of heat shock gene expression by RNA polymerase II elongation factor, Elongin A. J Biol Chem 280: 4017–4020 [DOI] [PubMed] [Google Scholar]
- Hayes RC, Petrullo LA, Huang HM, Wallace SS, LeClerc JE (1988) Oxidative damage in DNA. Lack of mutagenicity by thymine glycol lesions. J Mol Biol 201: 239–246 [DOI] [PubMed] [Google Scholar]
- Hoeijmakers JH (2001) Genome maintenance mechanisms for preventing cancer. Nature 411: 366–374 [DOI] [PubMed] [Google Scholar]
- Izban MG, Luse DS (1992) The RNA polymerase II ternary complex cleaves the nascent transcript in a 3′ → 5′ direction in the presence of elongation factor SII. Genes Dev 6: 1342–1356 [DOI] [PubMed] [Google Scholar]
- Kathe SD, Shen GP, Wallace SS (2004) Single-stranded breaks in DNA but not oxidative DNA base damages block transcriptional elongation by RNA polymerase II in HeLa cell nuclear extracts. J Biol Chem 279: 18511–18520 [DOI] [PubMed] [Google Scholar]
- Klungland A, Rosewell I, Hollenbach S, Larsen E, Daly G, Epe B, Seeberg E, Lindahl T, Barnes DE (1999) Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc Natl Acad Sci USA 96: 13300–13305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong SE, Shilatifard A, Conaway RC, Conaway JW (2003) Preparation and assay of RNA polymerase II elongation factors elongin and ELL. Methods Enzymol 371: 276–283 [DOI] [PubMed] [Google Scholar]
- Kreutzer DA, Essigmann JM (1998) Oxidized, deaminated cytosines are a source of C → T transitions in vivo. Proc Natl Acad Sci USA 95: 3578–3582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung HC, Bolton PH (1997) Structure of a duplex DNA containing a thymine glycol residue in solution. J Biol Chem 272: 9227–9236 [DOI] [PubMed] [Google Scholar]
- Kuraoka I, Endou M, Yamaguchi Y, Wada T, Handa H, Tanaka K (2003) Effects of endogenous DNA base lesions on transcription elongation by mammalian RNA polymerase II. Implications for transcription-coupled DNA repair and transcriptional mutagenesis. J Biol Chem 278: 7294–7299 [DOI] [PubMed] [Google Scholar]
- Laine JP, Egly JM (2006) Initiation of DNA repair mediated by a stalled RNA polymerase IIO. EMBO J 25: 387–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine JP, Mocquet V, Egly JM (2006) TFIIH enzymatic activities in transcription and nucleotide excision repair. Methods Enzymol 408: 246–263 [DOI] [PubMed] [Google Scholar]
- Le Page F, Kwoh EE, Avrutskaya A, Gentil A, Leadon SA, Sarasin A, Cooper PK (2005) Transcription-coupled repair of 8-oxoguanine: requirement for XPG, TFIIH, and CSB and implications for Cockayne syndrome. Cell 123: 711. [DOI] [PubMed] [Google Scholar]
- Lehmann AR (2005) Replication of damaged DNA by translesion synthesis in human cells. FEBS Lett 579: 873–876 [DOI] [PubMed] [Google Scholar]
- Lukin M, de Los Santos C (2006) NMR structures of damaged DNA. Chem Rev 106: 607–686 [DOI] [PubMed] [Google Scholar]
- Mason PB, Struhl K (2005) Distinction and relationship between elongation rate and processivity of RNA polymerase II in vivo. Mol Cell 17: 831–840 [DOI] [PubMed] [Google Scholar]
- Mellon I, Spivak G, Hanawalt PC (1987) Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell 51: 241–249 [DOI] [PubMed] [Google Scholar]
- Modregger J, Ritter B, Witter B, Paulsson M, Plomann M (2000) All three PACSIN isoforms bind to endocytic proteins and inhibit endocytosis. J Cell Sci 113 (Part 24): 4511–4521 [DOI] [PubMed] [Google Scholar]
- Mori S, Tanaka M, Nanba D, Nishiwaki E, Ishiguro H, Higashiyama S, Matsuura N (2003) PACSIN3 binds ADAM12/meltrin alpha and up-regulates ectodomain shedding of heparin-binding epidermal growth factor-like growth factor. J Biol Chem 278: 46029–46034 [DOI] [PubMed] [Google Scholar]
- Moriya M (1993) Single-stranded shuttle phagemid for mutagenesis studies in mammalian cells: 8-oxoguanine in DNA induces targeted G.C → T.A transversions in simian kidney cells. Proc Natl Acad Sci USA 90: 1122–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi Y, Mitsuhashi Y, Sekimizu K, Yokoi H, Tanaka Y, Horikoshi M, Natori S (1981) Characterization of three proteins stimulating RNA polymerase II. FEBS Lett 130: 69–72 [DOI] [PubMed] [Google Scholar]
- Palangat M, Hittinger CT, Landick R (2004) Downstream DNA selectively affects a paused conformation of human RNA polymerase II. J Mol Biol 341: 429–442 [DOI] [PubMed] [Google Scholar]
- Parsons JL, Elder RH (2003) DNA N-glycosylase deficient mice: a tale of redundancy. Mutat Res 531: 165–175 [DOI] [PubMed] [Google Scholar]
- Powell W, Reines D (1996) Mutations in the second largest subunit of RNA polymerase II cause 6-azauracil sensitivity in yeast and increased transcriptional arrest in vitro. J Biol Chem 271: 6866–6873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reines D (1992) Elongation factor-dependent transcript shortening by template-engaged RNA polymerase II. J Biol Chem 267: 3795–3800 [PMC free article] [PubMed] [Google Scholar]
- Reines D, Ghanouni P, Li QQ, Mote J Jr (1992) The RNA polymerase II elongation complex. Factor-dependent transcription elongation involves nascent RNA cleavage. J Biol Chem 267: 15516–15522 [PMC free article] [PubMed] [Google Scholar]
- Saxowsky TT, Doetsch PW (2006) RNA polymerase encounters with DNA damage: transcription-coupled repair or transcriptional mutagenesis? Chem Rev 106: 474–488 [DOI] [PubMed] [Google Scholar]
- Shibutani S, Takeshita M, Grollman AP (1991) Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature 349: 431–434 [DOI] [PubMed] [Google Scholar]
- Shilatifard A, Conaway RC, Conaway JW (2003) The RNA polymerase II elongation complex. Annu Rev Biochem 72: 693–715 [DOI] [PubMed] [Google Scholar]
- Shimizu Y, Iwai S, Hanaoka F, Sugasawa K (2003) Xeroderma pigmentosum group C protein interacts physically and functionally with thymine DNA glycosylase. EMBO J 22: 164–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims RJ III, Belotserkovskaya R, Reinberg D (2004) Elongation by RNA polymerase II: the short and long of it. Genes Dev 18: 2437–2468 [DOI] [PubMed] [Google Scholar]
- Sumoy L, Pluvinet R, Andreu N, Estivill X, Escarceller M (2001) PACSIN 3 is a novel SH3 domain cytoplasmic adapter protein of the pacsin-syndapin-FAP52 gene family. Gene 262: 199–205 [DOI] [PubMed] [Google Scholar]
- Takagi Y, Conaway JW, Conaway RC (1995) A novel activity associated with RNA polymerase II elongation factor SIII. SIII directs promoter-independent transcription initiation by RNA polymerase II in the absence of initiation factors. J Biol Chem 270: 24300–24305 [DOI] [PubMed] [Google Scholar]
- Tan S, Aso T, Conaway RC, Conaway JW (1994) Roles for both the RAP30 and RAP74 subunits of transcription factor IIF in transcription initiation and elongation by RNA polymerase II. J Biol Chem 269: 25684–25691 [PubMed] [Google Scholar]
- Thiviyanathan V, Somasunderam A, Volk DE, Gorenstein DG (2005) 5-hydroxyuracil can form stable base pairs with all four bases in a DNA duplex. Chem Commun (Camb) 3: 400–402 [DOI] [PubMed] [Google Scholar]
- Tornaletti S, Maeda LS, Kolodner RD, Hanawalt PC (2004) Effect of 8-oxoguanine on transcription elongation by T7 RNA polymerase and mammalian RNA polymerase II. DNA Repair (Amst) 3: 483–494 [DOI] [PubMed] [Google Scholar]
- Tornaletti S, Maeda LS, Lloyd DR, Reines D, Hanawalt PC (2001) Effect of thymine glycol on transcription elongation by T7 RNA polymerase and mammalian RNA polymerase II. J Biol Chem 276: 45367–45371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troelstra C, Hesen W, Bootsma D, Hoeijmakers JH (1993) Structure and expression of the excision repair gene ERCC6, involved in the human disorder Cockayne's syndrome group B. Nucleic Acids Res 21: 419–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troelstra C, van Gool A, de Wit J, Vermeulen W, Bootsma D, Hoeijmakers JH (1992) ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne's syndrome and preferential repair of active genes. Cell 71: 939–953 [DOI] [PubMed] [Google Scholar]
- Viswanathan A, You HJ, Doetsch PW (1999) Phenotypic change caused by transcriptional bypass of uracil in nondividing cells. Science 284: 159–162 [DOI] [PubMed] [Google Scholar]
- Wallace SS (2002) Biological consequences of free radical-damaged DNA bases. Free Radical Biol Med 33: 1–14 [DOI] [PubMed] [Google Scholar]
- Wood ML, Dizdaroglu M, Gajewski E, Essigmann JM (1990) Mechanistic studies of ionizing radiation and oxidative mutagenesis: genetic effects of a single 8-hydroxyguanine (7-hydro-8-oxoguanine) residue inserted at a unique site in a viral genome. Biochemistry 29: 7024–7032 [DOI] [PubMed] [Google Scholar]