

Abstract
Enlargeosomes are small cytoplasmic vesicles that undergo rapid, Ca2+-dependent exo/endocytosis. The role of the cytoskeleton in these processes was unknown. In PC12-27 cells, microtubule disassembly had little effect on enlargeosomes, whereas microfilament disassembly increased markedly both their resting and stimulated exocytosis, and inhibited their endocytosis. Even at rest enlargeosomes are coated at their cytosolic surface by an actin-associated protein, annexin2, bound by a dual, Ca2+-dependent and Ca2+-independent mechanism. In contrast, the other enlargeosome marker, desmoyokin/Ahnak, is transported across the organelle membrane, apparently by an ABC transporter, and binds to its lumenal face. Annexin2-GFP expression revealed that, upon stimulation, the slow and random enlargeosome movement increases markedly and becomes oriented toward the plasma membrane. After annexin2 downregulation enlargeosome exocytosis induced by both [Ca2+]i rise and cytoskeleton disruption is inhibited, and the NGF-induced differentiation is blocked. Binding of annexin2 to the enlargeosome membrane, the most extensive ever reported (>50% annexin2 bound to ∼3% of total membrane area), seems therefore to participate in the regulation of their exocytosis.
Keywords: annexin2, cytoskeleton, enlargeosome, exocytosis, membrane traffic
Introduction
Enlargeosomes are small cytoplasmic vesicles, expressed by many, but not all cultured and tissue cells (Borgonovo et al, 2002), that previous patch clamp (Kasai et al, 1999; Cocucci et al, 2004) and immunocytochemical (Borgonovo et al, 2002; Cerny et al, 2004; Cocucci et al, 2004) studies have shown to undergo rapid (1 s), [Ca2+]i-dependent discharge by a tetanus toxin-insensitive exocytosis. This process does not seem to serve for secretion, but for the enlargement of the cell surface. By the use of a monoclonal antibody (mAb) against desmoyokin/Ahnak (d/A), a large (∼600 kDa) peripheral protein attached to the lumenal face of the organelle membrane, we were able to map the distribution of the enlargeosomes in the proximity of the plasma membrane; to reveal the appearance of d/A-positive surface patches upon exocytosis; and to follow their ensuing internalization by a peculiar form of endocytosis (Borgonovo et al, 2002; Cocucci et al, 2004). However, the interactions of enlargeosomes with the surrounding structures of the cells remained completely unknown.
By analogy with other exocytic systems, a structure expected to participate in operational and regulatory processes of enlargeosome traffic and discharge is the cytoskeleton. Working primarily in PC12-27 cells, a defective clone of the neurosecretory PC12 line very rich in enlargeosomes, we have found that both the enlargeosome exocytosis and the ensuing endocytosis are controlled by actin microfilaments. Further studies in PC12-27 cells and also in HeLa, BSC1 and wild-type (wt) PC12 have revealed a peculiar property of the organelles, their surface coating with annexin2 (anx2), a soluble cytoskeleton-binding protein involved in variety of functions (Gerke et al, 2005). By the study of anx2, we have revealed the movements of enlargeosomes, at rest, after stimulation and upon microfilament disassembly; provided evidence about the d/A transport, from the cytosol into the enlargeosomes; identified the anx2 region participating in its binding to the enlargeosome membrane; and demonstrated that anx2 is needed for enlargeosome exocytosis to take place.
Results
Actin filaments, a barrier to enlargeosome surface traffic
Our first studies were focused on the effects of treatments, known to affect the structure and function of microtubules or microfilaments, on the distribution of d/A, the enlargeosome marker. PC12-27 cells, which at rest appear almost completely surface-negative for d/A (Figure 1A), exhibit a dose-dependent rise of their surface immunolabelling after stimulation with the Ca2+ ionophore, ionomycin (0.1–5 μM; 5 min) (Figure 1B; Cocucci et al, 2004). After 60 min treatment with nocodazole (30 μM), microtubules exhibited the expected disruption, yet the surface d/A immunostaining of the cells, both before and after stimulation, was not appreciably different from controls (Figure 1C and D; quantification in N).
Figure 1.
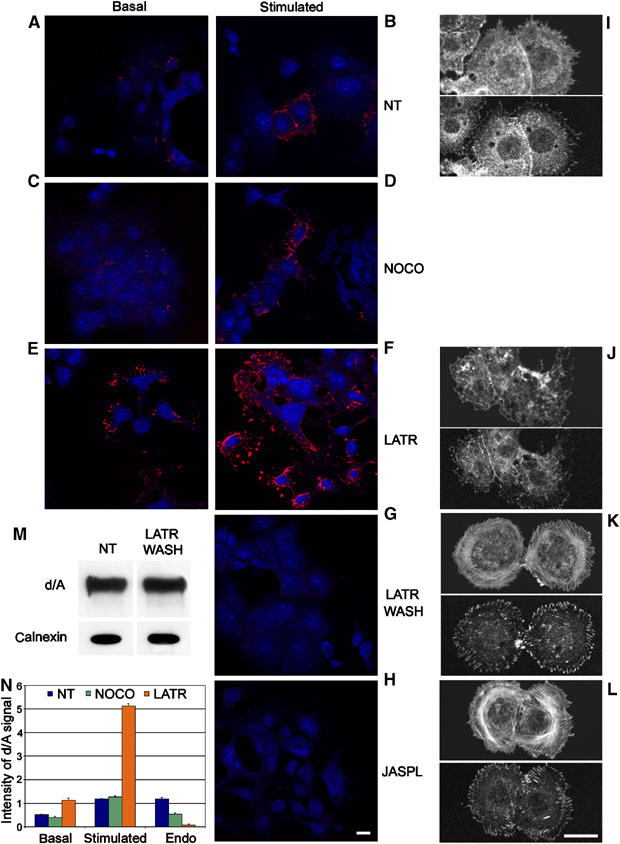
Basal and stimulated exocytosis of enlargeosomes is unaffected by microtubule depolymerization and increased by microfilament disruption. (A–H) Surface immunolabelling for d/A (red) of PC12-27 cells exposed or not to various treatments, fixed while at rest (left column) or after 5 min stimulation with 1 μM ionomycin (central column). The distribution of the cells is revealed by their own blue autofluorescence. (A, B) Non-treated cells (NT); (C, D) cells treated with 30 μM nocodazole (NOCO) for 1 h; (E, F) cells treated with 5 μM latrunculinA (LATR) for 15 min; (G) cells treated with 5 μM latrunculinA for 15 min, then reincubated for 24 h without treatments (LATR WASH); (H) cells treated with 100 nM jasplakinolide (JASPL) for 1 h. (I–L) The state of the actin cytoskeleton in cells untreated and treated as in (F–H) is illustrated. Notice the extensive disruption induced by latrunculinA (J), affecting specifically stress fibers and focal adhesions, revealed by phalloidin staining (upper part of each panel), and by immunolabelling of paxillin (lower part of each panel), respectively, and the strengthened stress fibers and cortical actin after long wash-out of latrunculinA (K), and treatment with jasplakinolide (L). The bar in (H), valid also for (A–G), and the bar in (L), valid also for (I–K), corresponds to 10 μm. (M) Western blots of d/A and the ER membrane protein, calnexin, of cells non-treated (NT) or treated with latrunculinA, and reincubated for 24 h (LATR WASH), such as those illustrated in (G, K). (N) The morphometric averages of d/A immunofluorescence data of (A, C and E) (basal), (B, D and F) (stimulated with 1 μM ionomycin) and Figure 2E–G (endocytosis occurring during stimulation with 5 μM ionomycin), calculated from five fields of 10 cells each ±s.d. and expressed as average intensity per cell.
LatrunculinA (5 μM, 15 min), which sequesters actin monomers, induced in PC12-27 cells marked flattening, finger-like protrusions and disappearance of F-actin bundles (stress fibers) and of paxillin-positive focal adhesion associated with the end of stress fibers (Figure 1, compare I to J), yet, the plasma membrane continuity was not affected since non-permeabilized cells were not stained with fluorescein–phalloidin (not shown). Many latrunculinA-treated PC12-27 cells were surface-positive for d/A already at rest (Figure 1E). Stimulation with ionomycin induced further increase (Figure 1F), up to ∼5 fold those observed after ionomycin alone (1 μM) or latrunculinA alone (Figure 1N). At higher ionomycin concentrations, the differences between cells treated or not with latrunculinA tended to disappear (not shown).
Reinforcement of the actin cytoskeleton induced marked inhibition of d/A surface accumulation. After 24 h re-incubation in plain medium, the PC12-27 cells treated with latrunculinA exhibited a compact cytoskeleton with increased stress fibers and focal adhesions (Figure 1K). Neither the d/A expression (Figure 1M) nor the whole-cell d/A immunolabelling (not shown) were changed, yet no surface appearance of d/A was observed after ionomycin (5 min, 1–5 μM; Figure 1G). Similar results (Figure 1H) were obtained with PC12-27 treated with 100 nM jasplakinolide (120 min), an inhibitor of actin depolymerization (Figure 1L).
Both exo and endocytosis are affected by cytoskeletal alterations
The styryl dye, FM1-43, which increases markedly its fluorescence when dissolved into membranes, was used in PC12-27 to reveal the cumulative area of surface-exposed membranes, including those exocytosed and internalized by endocytosis. Figure 2A and B compares the time course of FM1-43 fluorescence in cells exposed or not to 5 μM latrunculinA before 1 μM ionomycin. The overall rise induced by ionomycin after latrunculinA pretreatment was of 2–2.5-fold that induced by ionomycin only, not of five-fold as the d/A surface accumulation (Figure 1N). To investigate whether this difference was due to inhibition of endocytosis, PC12-27 cells, exposed or not to the high dose of ionomycin (5 μM, 5 min) in the presence of the anti-d/A mAb, were washed with incubation medium supplemented or not with 300 mM NaCl. Since d/A is a peripheral protein, its pool exposed to the cell surface was removed by the high ionic strength wash (Figure 2, compare C to D), whereas that already endocytosed was unaffected (Figure 2E). Compared to controls, the endocytosed immunolabelling was significantly lower and no longer visible in the cells treated with nocodazole (30 μM) or latrunculinA (5 μM), respectively (Figure 2F and G; quantification in Figure 1N). In contrast, endocytosis of transferrin was unaffected (not shown). Therefore, in nocodazole-treated cells, enlargeosome exocytosis is unaffected and endocytosis partially inhibited; in latrunculinA-treated cells, stimulation of exocytosis accounts for only a fraction of the ionomycin-induced d/A surface overaccumulation, while the rest is due to the large inhibition of endocytosis (Figure 2H).
Figure 2.
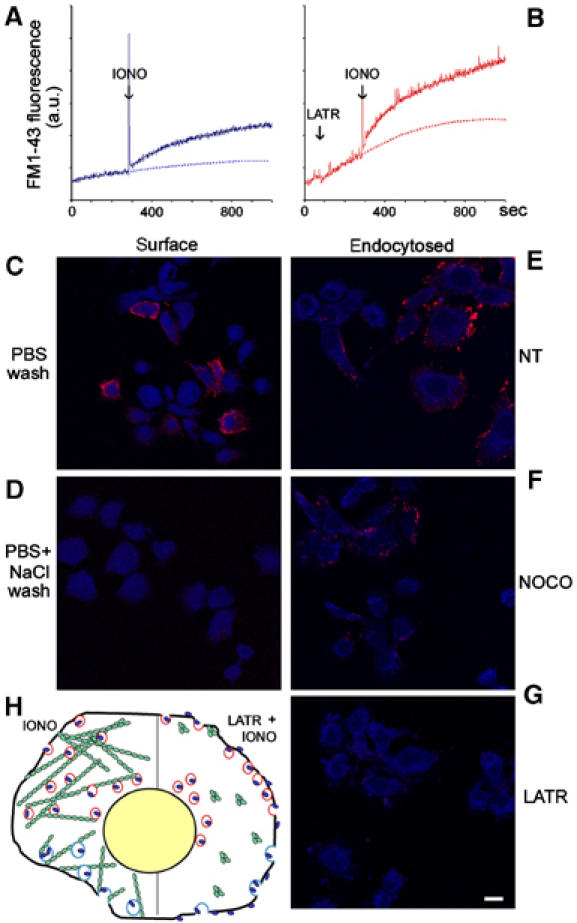
d/A endocytosis is blocked by microfilament disruption and reduced by microtubule depolymerization. (A, B) The increase of FM1-43 fluorescence in PC12-27 cell suspensions treated or not with 5 μM latrunculinA (LATR) and then stimulated with 1 μM ionomycin (IONO). (C–G) Cells stimulated for 5 min with 5 μM ionomycin in the presence of the anti-d/A mAb, then washed either with PBS (C) or PBS with 300 mM NaCl (D–G), fixed and decorated with the TRITC-conjugated secondary Ab, either without (surface, left column) or after permeabilization (endocytosed, right column). The cells of (F) had been treated with nocodazole (NOCO, 30 μM for 1 h), and those of (G) with latrunculinA (LATR, 5 μM, 15 min). The distribution of the cells is revealed by their own blue autofluorescence. The bar in (G), valid also for (C–F), corresponds to 10 μm. (H) A schematic drawing summarizing the results illustrated in Figures 1 and 2. Red circles are enlargeosomes containing d/A (blue dots), which appears at the cell surface upon ionomycin-induced exocytosis, and blue circles are d/A specific endocytic vesicles. The green signs are actin, polymerized into fibrils to the left and depolymerized by latrunculinA to the right, with ensuing stimulation of exocytosis and inhibition of endocytosis.
Anx2 is associated with enlargeosomes
To identify the protein(s) mediating the enlargeosome/cytoskeleton interaction, we analyzed by 2D PAGE the particulate subcellular fraction mostly enriched in d/A (Figure 3C), isolated from PC12-27 by sucrose gradient centrifugation (Figure 3A and B), and compared it to the corresponding fraction from wt PC12 (Figure 3D), a cell line that expresses neither the marker nor the enlargeosomes (Borgonovo et al, 2002). Among the 22 sequenced spots present only or predominantly in the PC12-27 fraction, one was anx2 (36 kDa; red-encircled in Figure 3C), a member of the cytosolic anx family known to bind both membranes and actin (Rescher et al, 2004; Gerke et al, 2005). Anx2 was not seen in the fraction of wt PC12 (Figure 3D), which expresses much lower levels of the protein diffuse in the cytoplasm (Figures 3G and 4D). Immunofluorescence shows the abundant anx2 of PC12-27 both diffuse and concentrated in small puncta coinciding with d/A (Figures 3E and 4F), and with its tetramer-associated protein, S100A10 (not shown; Gerke et al, 2005). Such a distribution was unaffected by latrunculinA (Figure 3F). Co-localization of anx2-positive puncta with endosome markers, EEA1 and transferrin receptor (TfR), and with calnexin (ER), giantin (Golgi complex) and LAMP1 (lysosomes), was negligible (not shown). The dual distribution of anx2 differs from that of annexin1 (anx1), mostly diffuse to the cytoplasm (Figure 3H).
Figure 3.
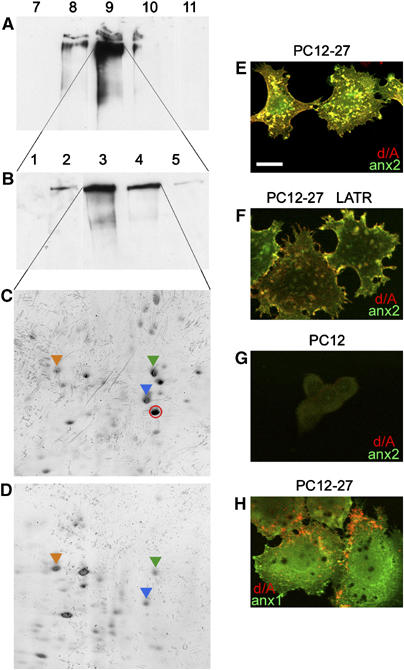
PC12-27 and wt PC12 cells: 1D and 2D Western blots of subcellular fractions, and dual immunofluorescence for d/A with anx2 or anx1. (A, B) The distribution of d/A in the particulate fractions of a density gradient loaded with the PC12-27 postnuclear supernatant (A), and of a subsequent velocity gradient loaded with fraction 9 of the first (B); (C) the 2D protein pattern from the fractions 3 and 4 of (B). (D) The pattern from the corresponding two fractions, however, from wt PC12, isolated by the same gradient of (B). The red encircled spot in (C) corresponds to anx2. It lacks in (D) because most anx2 of wt PC12 is localized in the cell sap, and not bound to membranes (Figure 4D). The additional indicated spots correspond to: HSP71 (orange arrowhead), tubulin (green arrowhead), actin (blue arrowhead). (E, F) The expression and distribution of d/A (red) and anx2 (green) in PC12-27, not treated or treated with latrunculinA (LATR); (G) The same proteins in wt PC12; (H) d/A (red) and anx1 (green) in PC12-27 cells. The bar in (E), valid also for the other panels, corresponds to 10 μm.
Figure 4.
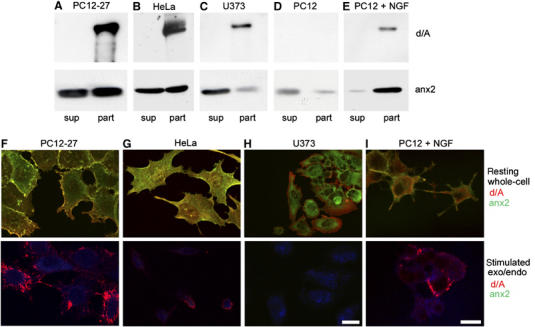
Western blot analysis of the final supernatant (sup) and particulate (part) subcellular fractions isolated from various cell types; whole cell (upper row) and surface (lower row) immunofluorescence labelling of the same cells. (A–E) The distribution of d/A and anx2 in the particles and final supernatants from cells rich (PC12-27, HeLa and NGF-differentiated wt PC12) or free (U373 and growing wt PC12) of enlargeosomes. (F–I) The immunofluorescence of d/A (red) and anx2 (green) in the indicated types of cells, immunolabelled after detergent permeabilization, to illustrate the distribution of the organelles while at rest (upper row); without permeabilization, but upon 5 min with 5 μM ionomycin, to illustrate the stimulation-induced appearance of antigens to the cell surface (bottom row). The distribution of the cells is revealed by their own blue autofluorescence. The bar in (H, lower panel), valid for (F–H), and the bar in (I, lower panel), valid also for (I, upper panel), correspond to 10 μm.
The particulate and cytosolic distribution of anx2 in PC12-27 was confirmed by subcellular fractionation showing ∼60% recovery (together with 100% d/A) in the post-nuclear, particulate fraction, and ∼40% in the final supernatant (Figure 4A). A similar dual recovery was found in HeLa (Figure 4B) and BSC1 (not shown) cells, also rich of enlargeosomes (Borgonovo et al, 2002; Cerny et al, 2004), but not in U373 cells where d/A and anx2 were recovered separately (Figure 4C). In undifferentiated wt PC12, which lack d/A, the scarce anx2 was recovered mostly in the supernatant (Figure 4D). After NGF-induced differentiation, however, the appearance of d/A and enlargeosomes (Borgonovo et al, 2002) were accompanied by a considerable increase of anx2 (Fox et al, 1991; Jacovina et al, 2001; Vaudry et al, 2002), and by a switch of its recovery, from supernatant to particulate (Figure 4E).
To illustrate the differential distribution of anx2 in relation to the expression of enlargeosomes and their exocytosis, two series of parallel fluorescence experiments were carried out (Figure 4F–I), the first showing the immunolabelling d/A and anx2 in cells permeabilized by a detergent (whole-cell labelling, upper row), and the second looking for the surface appearance of d/A in response to ionomycin (lower row). PC12-27, HeLa (Figure 4F and G) and BSC1 (not shown) cells, as well as wt PC12 cells differentiated with NGF (Figure 4I), all rich of enlargeosomes, exhibited the dual distribution of anx2, diffuse and in puncta coinciding with d/A, as well as the exo-endocytosis of d/A induced by ionomycin. In contrast, in U373 cells (Figure 4H), d/A was spread to the peripheral cytoplasm and anx2 to the perinuclear area, and no d/A appeared at the surface upon stimulation. In all cells exposed to ionomycin, anx2 failed to appear at the surface, thus excluding its association with the lumenal face of the enlargeosome membrane (Figure 4F–I, lower panels).
Anx2 is bound to the cytosolic face of the enlargeosome membrane
In various cell types, anx2 has been reported to translocate across the plasma membrane and to be released upon stimulation (Gerke et al, 2005). In PC12-27, however, the media bathing cells exposed for 15 min to 5 μM latrunculinA or ionomycin revealed just trace amounts or no anx2 (Supplementary Figure 1). Its role of enlargeosome cargo could thus be excluded.
In order to investigate further the problem, we used living PC12-27 cells permeabilized not with detergents, but with streptolysin-O (SLO), which induces small holes only in the plasma membrane. Incubation of the SLO-permeabilized cells in a medium containing 10 μM [Ca2+] (SLO/Ca2+) induced release of neither d/A nor anx2 (Figure 5A and C), while with 0.01 μM [Ca2+]/pH 7.4 (SLO/EGTA), only anx2 was largely released to the medium (Figure 5A and D), confirming the localization of the two proteins at the cytosolic (anx2) and lumenal (d/A) faces of the enlargeosome membrane (Figure 5B). Some decrease of the cellular d/A was observed only at 0.01 μM [Ca2+]/pH 11 (SLO/EGTA+Na2CO3) (Figure 5A and E), which releases also proteins segregated within membrane-bound organelles. Integral plasma membrane proteins, such as Na+/K+ ATPase (Figure 5A), were recovered only with the cells.
Figure 5.
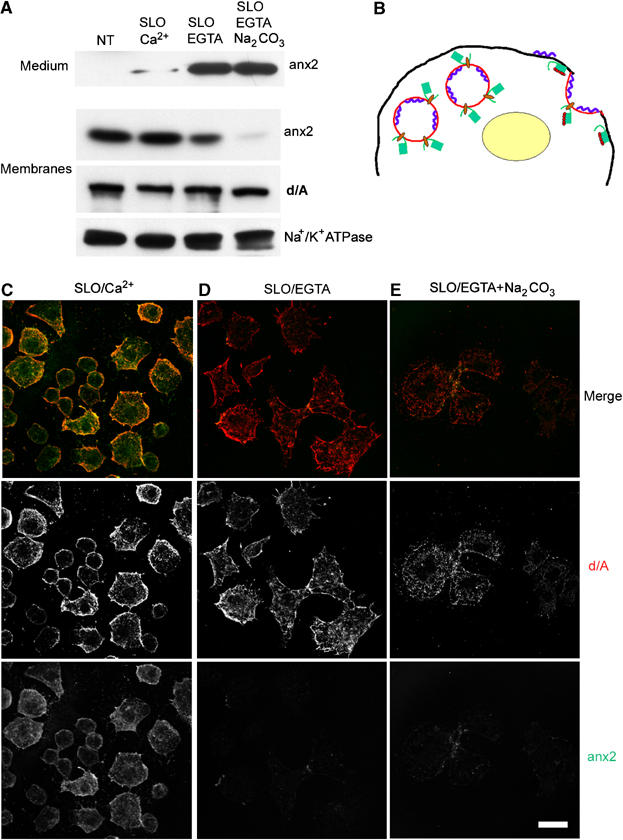
Recovery of d/A and anx2 in PC12-27 cells permeabilized with streptolysin-O, and in their incubation medium. (A) The recovery in the medium and in the particulate (membranes) fractions from PC12-27 cells non-treated (NT) or permeabilized on ice with streptolysin-O (SLO) and then incubated for 30 min in K+ glutamate buffer containing either 10 (Ca2+) or 0.01 μM free Ca2+ at pH 7.4 or 11 (EGTA and EGTA/Na2CO3, respectively). (B) A schematic drawing of enlargeosomes in the cytoplasm and after exocytosis, with d/A (blue ribbon) and anx2 (green box) bound to the opposite faces of the organelle and redistributed to the plasma membrane upon exocytosis. Binding of Ca2+ (red dots) to the core domain of anx2 is needed for the interaction of the protein to the membrane lipids. (C–E) The immunofluorescence of d/A (middle row) and anx2 (bottom row) of cells treated as in (A). The merge images are in the top row. The bar in bottom (E), valid also for (C, D), corresponds to 10 μm.
d/A may be transported into the enlargeosomes by an ABC transporter
The d/A mRNA does not include a signal sequence (Shtivelman and Bishop, 1993). Most likely, therefore, its synthesis takes place on free polysomes, with subsequent transport across the enlargeosome membrane, possibly by an ABC transporter (Herget and Tampe, 2006). Supplementary Figure 2 shows that various ABCC and ABCB transporters are expressed by both wt PC12 and PC12-27. In addition, ABCB1 is present only in the latter. We investigated whether drugs known to inhibit distinct classes of ABCs (Wang et al, 2003) were able to affect the intracellular distribution of d/A. Probenecid, MK571 and diisothiocyanatostilbene 2,2′-disulfonic acid disodium salt hydrate (DIDS) (active on all ABCC; on ABCC1 and ABCC2; and on ABCA, D, E and F, respectively) were inactive (not shown). In contrast, verapamil (50 μM) and glybenclamide (100 μM), two blockers of ABCB and ABCC, induced a slow redistribution of d/A from the enlargeosome puncta to the cytosol. Figure 6 summarizes results with verapamil (50 μM). The drug-induced cell level changes neither of d/A nor of anx2 (Figure 6A). However, the apparent co-localization of d/A with anx2 was completely dissociated after 16 h of treatment, with anx2 maintaining a particulate distribution and d/A diffuse throughout the cytoplasm (Figure 6B, right). Figure 6C shows that ionomycin-induced surface appearance of d/A (left) no longer occurred in cells treated for 16 h with verapamil (right); Figure 6D shows that the verapamil wash-out resulted in the redistribution of d/A, from the diffuse (left) to the punctate pattern, appreciable after 4 h (center) and complete after 24 h (right), when the d/A/annexin2 co-localization was also re-established (not shown).
Figure 6.
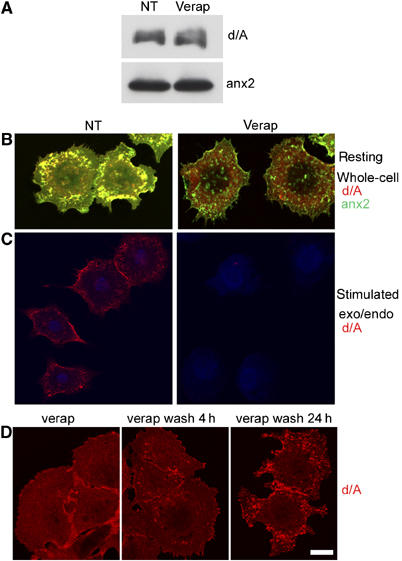
Verapamil-induced redistribution of d/A from the enlargeosome lumen to the cytosol and vice versa. (A) Expression of both d/A and anx2 is unaffected by 16 h treatment of PC12-27 cells with 50 μM verapamil (verap), a blocker of the ABCB and ABCC transporters. (B) Untreated (NT) cells exhibiting the typical d/A punctate distribution coinciding with anx2 (left, see also Figures 3B and 4F), and diffuse to the cytosol in cells treated with verapamil as in (A). In contrast, the distribution of anx2 remains punctate (right). (C) Exocytic surface appearance of d/A induced by a 5-min stimulation with 5 μM ionomycin (left, see also Figures 1B and 4F, lower panel), no longer occurs in the cells treated with verapamil as in (A) (right). (D) Recovery of d/A, from the diffuse pattern in the cytosol induced by 16 h of verapamil (left) to the punctate distribution, already appreciable in cells washed and reincubated for 4 h (center), and complete in those reincubated for 24 h (right).
Anx2 is needed for enlargeosome exocytosis
To investigate the role of anx2 in enlargeosome exocytosis, PC12-27 cells were transfected, at 48 h distance, with two anti-anx2 siRNAs sequence mixtures, which induced a considerable decrease of anx2, with no effect on the expression of d/A and other proteins, such as calnexin (an ER membrane protein) and tubulin (Figure 7A). Transfected cells were exposed for 5 min to the anti-d/A mAb administered either with or without 5 μM ionomycin (IONO and NT, respectively). Figure 7B, top row, shows that, in anx2 downregulated cells, the recovery of d/A exo/endocytosed by the cells in response to ionomycin was largely decreased. This decrease correlated well with the ∼60% average decrease of the anx2 level (Figure 7A and B, bottom rows, and 7C). In contrast, the level of calnexin, not involved in enlargeosome exocytosis, was unchanged (Figure 7B, middle row).
Figure 7.
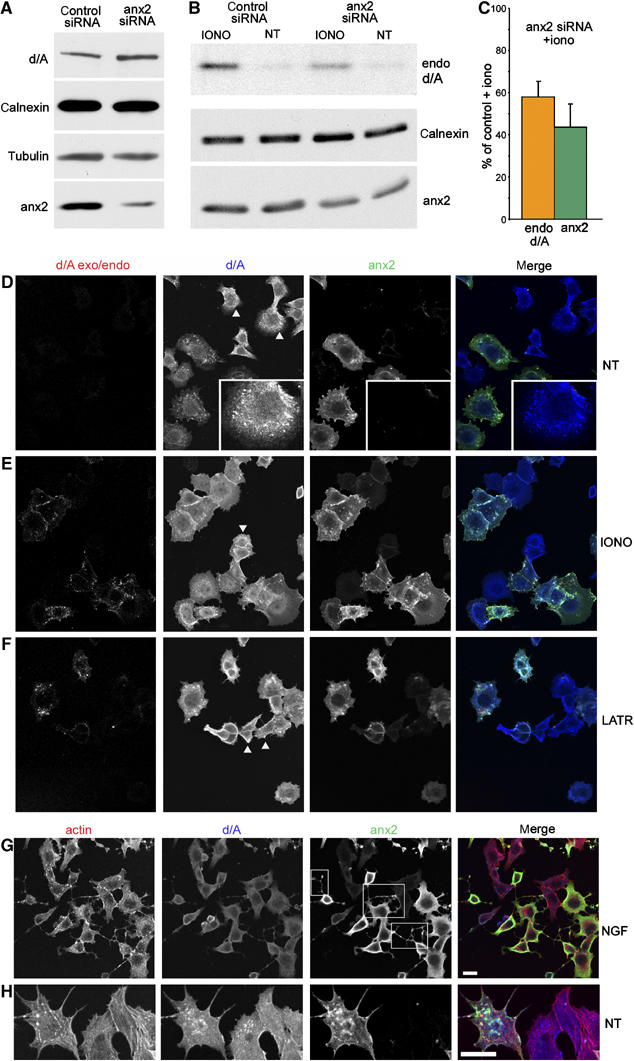
Downregulation of anx2 blocks enlargeosome exocytosis. (A) bottom row: compared to scrambled siRNA (control), anx2 siRNA induces in PC12-27 a considerable decrease of the protein without any parallel decrease of d/A, calnexin and tubulin. (B) Compares the amount of d/A mAb endocytosed by cells treated with either the scrambled or the anx2 siRNA, exposed (IONO) or not (NT) to ionomycin. The cells treated with anx2 siRNA, which express decreased levels of anx2 (bottom row) and normal levels of calnexin (middle row), show greatly reduced exo/endocytic responses. Quantification (averages of three experiments) of the endocytosed anti-d/A Ab and of anx2 in siRNA-treated cell populations exposed to ionomycin is shown in (C). The immunofluorescence of the d/A exo/endocytosis (exo/endo), and of anx2 and d/A expression in PC12-27 cells treated with anx2 siRNA, non-treated (NT) or stimulated with ionomycin (IONO), is shown in (D, E). Of the cells expressing d/A, only some are positive also for anx2 (merge image in the right frame), and only the latter show enlargeosome exo/endocytosis upon ionomycin, whereas the others show no appreciable responses (d/A exo/endo, left frame). (F) Analogous results obtained using latrunculinA (LATR), instead of ionomycin, to stimulate exocytosis. Actin (revealed by phalloidin), d/A and anx2 triple immunolabelling of a group of anx2 siRNA-treated PC12-27 cells exposed to NGF (2 days) is illustrated in (G) at higher magnification, and in non-differentiated cells, in (H). Only the cells maintaining normal expression of anx2 (squares in the anx2 frame) exhibit long dendrites, whereas those showing downregulation of the protein do not. Actin cytoskeleton maintains the usual morphology also in cells depleted of anx2. The bar in the merged frame of (G), valid also for (D–F), and that in the merged frame (H) correspond to 10 μm.
Immunofluorescence of siRNA-treated PC12-27 revealed coexistence, in the cell population, of a majority almost completely free of anx2, showing however an unchanged distribution of d/A (insets in Figure 7D and arrowheads in d/A panels of Figure 7D–F), with a minority showing normal levels and distribution of both anx2 and d/A (Figure 7D). In control cells, treated with scrambled siRNA, expression of anx2 was largely homogeneous (not shown). Downregulated and control cells were stimulated for 5 min with 5 μM ionomycin in the presence of the anti-d/A mAb (Figure 7E). After washing and permeabilization, the cells were exposed first to the anti-mouse TRITC-Ab, to label in red the surface and endocytosed d/A, then to anti-d/A together with anti-anx2 Abs followed by their secondary Abs coupled to Alexa 647 and FITC, respectively. In control cells and in those still expressing anx2, the ionophore induced the usual redistribution of both d/A and anx2 from the cytoplasm to the plasma membrane and then of d/A to an endocytic compartment (Figure 7E). In contrast, in cells lacking anx2, the intracellular d/A was apparently unchanged; however, its surface appearance upon stimulation was largely or even completely blocked (Figure 7E). A similar inhibition was observed when the downregulated cells, instead of ionomycin, were exposed to latrunculinA (Figure 7F).
Previous evidence (Borgonovo et al, 2002) had suggested the participation of enlargeosomes in a few cellular functions, including growth of dendrites during NGF-induced differentiation. Interestingly, in the population of PC12-27 cells exposed to anx2 siRNA, only those maintaining the protein were able to grow dendritic fibers (Figure 7G). Dendrite growth appears therefore controlled by one or more anx2-dependent mechanism(s), possibly including enlargeosome exocytosis. In the absence of anx2, both with and without NGF (Figure 7G and H), the actin cytoskeleton appeared unchanged. Together with the latrunculinA data of Figure 7F, this result excludes alterations of the cytoskeleton to have a mechanistic role in the block of exocytosis induced by anx2 downregulation.
The anx2 N-terminal domain is critical for enlargeosome membrane binding
In PC12-27 cells transfected with anx2 cDNA coupled, at its C-terminus, to that of GFP (anx2-GFP), the construct distribution was mostly punctate and co-localized with both d/A (Figure 8A and B) and S100A10 (Figure 8F), that is, it exhibited the same distribution of wt anx2 (compare Figure 8A and B to Figures 3E and 4F). Moreover, anx2-GFP did show significant co-localization neither with early and recycling endosomes nor with lysosomes (Figure 8C–E). Based on these data, anx2-GFP appears fully adequate to investigate the properties and mechanisms of the anx2 binding to the enlargeosome membrane.
Figure 8.
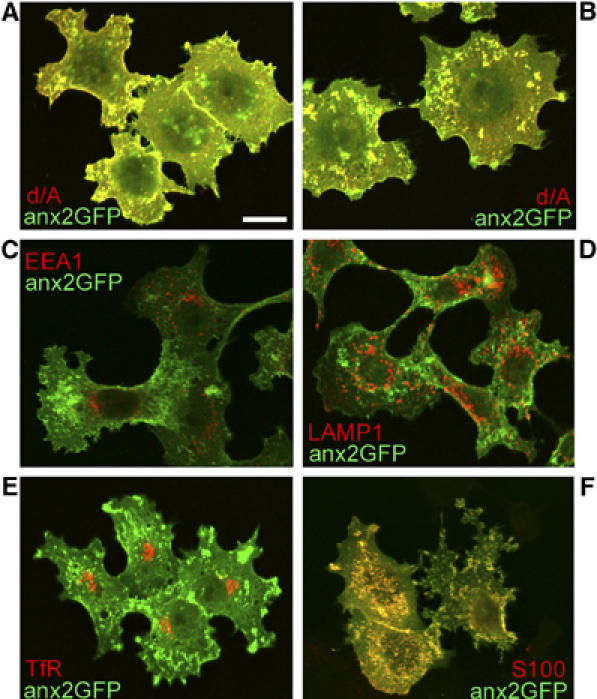
Distribution of anx2-GFP: comparison with d/A, EEA1, LAMP1, transferrin receptor and S100A10. Notice the large co-localization of anx2-GFP with d/A (A, B) and S100A10 (S100, F), whereas with the markers of early (EEA1, C) and recycling (TfR, E) endosomes, and of lysosomes (LAMP1, D) co-localization is below the level of specificity. The bar in (A), valid also for (B–F), corresponds to 10 μm.
For these studies, we used a number of anx2 mutated constructs (Figure 9A) exhibiting: (i) truncation of the 26-aminoacid N-terminal domain (ΔN26-GFP), which includes the site of interaction with S100A10; (ii) replacement of the isoleucine 6 and leucine 7 with two glutamic acids (ΔIL-GFP) (Thiel et al, 1991), which also precludes the binding of S100A10; (iii) replacement of the five acidic amino acids present in the anx2 core domain, with elimination of the Ca2+-binding sites (ΔCa-GFP); and (iv) both the first and the third changes (ΔN26/ΔCa-GFP). Western blot and immunofluorescence of PC12-27 transiently transfected with these constructs are illustrated in Figure 9B and C. The recovery of anx2-GFP in the total particulate and final supernatant fractions (Figure 9B) was similar to that of wt anx2 (Figure 4A). In contrast, the ΔN26-GFP and the ΔIL-GFP mutants were recovered almost completely in the final supernatant, strongly suggesting a key role of the N terminal domain in the anx2-membrane binding. On the other hand, the ΔCa-GFP mutant was recovered almost completely in the particulate fraction (Figure 9B). Thus, Ca2+-binding sites may not favor, but rather prevent binding unless enough Ca2+ is present (see also Filipenko et al, 2000). The distribution of the ΔN26/ΔCa-GFP was intermediate, with predominance however of the particulate fraction (Figure 9B). Therefore, the increase of membrane binding due to the loss of the Ca2+ binding sites largely compensates for the loss of membrane binding by the truncation. By d/A immunofluorescence (Figure 9C), the cells transfected with ΔN26-GFP and ΔIL-GFP constructs showed no trace of dually labelled puncta but a diffuse GFP labelling in the cytoplasm and in the nucleus (Figure 9C). The nuclear export sequence of anx2 is in fact located in the N-terminal domain (Eberhard et al, 2001). Also the ΔN26/ΔCa-GFP cells showed diffuse labelling, however, with a significant punctate co-localization with d/A (Figure 9C). When the cells expressing the N-terminal-mutated anx2 were exposed to ionomycin, fluorescence was largely redistributed to the surface, establishing an apparent co-localization with d/A (Figure 9E). The Ca2+-dependent membrane relocation of these constructs was confirmed also by subcellular fractionation of ΔN26-GFP-expressing cells homogenized and centrifuged in buffered sucrose containing either low (0.1 μM) or high (1 mM) [Ca2+] (Figure 9H).
Figure 9.
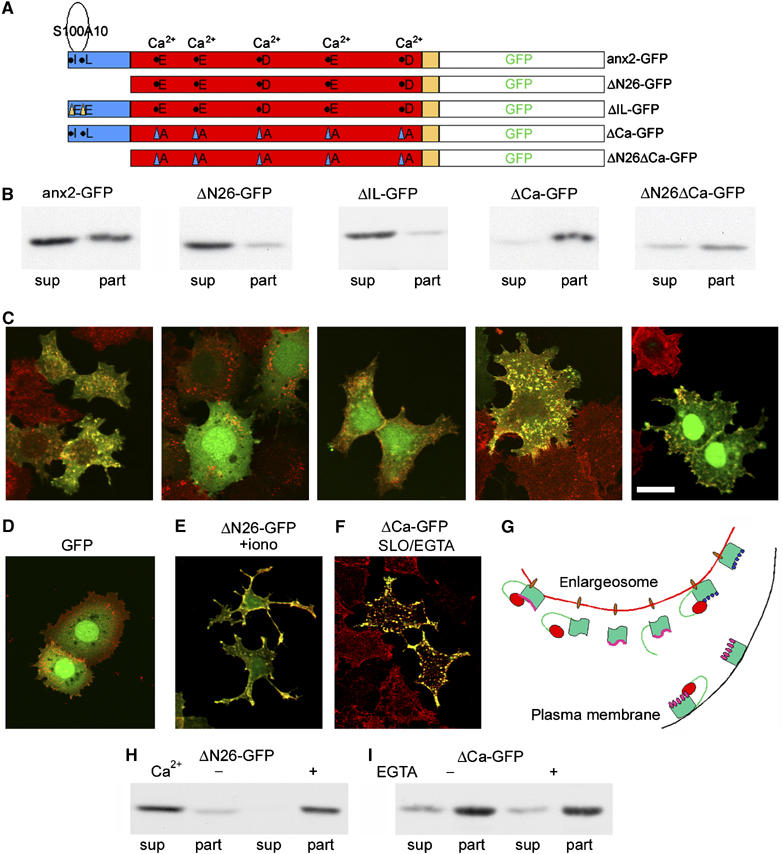
Expression and intracellular distribution of anx2 mutants coupled to GFP. (A) The mutations induced in anx2 C-terminally coupled to GFP. In anx2-GFP the sequence is unchanged; Δ26-GFP is truncated of its N-terminal domain (blue); ΔIL is mutated at two amino acids (isoleucine 6 and leucine 7) of the N-terminal domain, needed for the tetrameric assembly with S100A10; ΔCa-GFP is point-mutated at the five acidic amino acids of the core (red), the Ca2+ binding sites of anx2; ΔN26ΔCa-GFP is both N-terminal truncated and point mutated at the Ca2+-binding sites. (B) The expression and distribution, in the final supernatant (sup) and particulate (part) subcellular fractions, of anx2-GFP and its mutants, indicated as in (A). (C) The corresponding immunofluorescence of anx2-GFP and its mutants (green) and of d/A (red). (D) The distribution of GFP, mostly nuclear and cytosolic, and of d/A. (E) The redistribution, from the cytosol to the plasma membrane, of ΔN26-GFP after 5 min stimulation with 5 μM ionomycin. (F) The punctate retention of ΔCa-GFP in transiently transfected PC12-27 cells permeabilized with SLO and incubated for 30 min at 0.01 μM [Ca2+], pH 7.4 (SLO/EGTA). Under these conditions anx2-GFP is completely released (see Figure 5). The bar in (C, right panel), valid also for (D–F), corresponds to 10 μm. (H) The supernatant/particle distribution of ΔN26-GFP in buffered sucrose containing 0.1 μM (−) and 1 mM (+) Ca2+; (I) the supernatant/particle distribution of ΔCa-GFP in buffered sucrose (0.1 μM Ca2+), without (−) or with (+) 1 mM EGTA to further decrease [Ca2+] (below 0.01 μM). (G) A model summarizing the membrane binding of the various anx2 forms, depending on their domains and also on the [Ca2+]i. The enlargeosome membrane (red) bears putative anx2 receptors (brown oval) bound, at basal [Ca2+] (pink line), to wt anx2 (left) associated to S100A10 (red dot); and, independently on [Ca2+]i, to ΔCa-anx2 (with mutations in the Ca-binding sites, blue dots), and to the dually mutated ΔN26ΔCa-anx2 (right). Various forms bind to the enlargeosome membrane only when [Ca2+]i increases, and remain in the cytosol when [Ca2+]i is low (wt anx2, second from left) or even basal (the forms that cannot bind S100A10 (central): truncated of the N-terminal domain (ΔN26) and mutated in the ile6-leu7 residues (ΔIL). Binding of wt anx2 to the raft-lipids of the plasma membrane (black) occurs after increase of [Ca2+]i, when its Ca2+-binding sites are occupied by the cation (pink dots). Under the same conditions, also ΔN26-anx2 appears to bind the plasma membrane. Alternatively, it could bind the enlargeosome membrane and be redistributed to the plasma membrane following exocytosis.
In the cells expressing ΔCa-GFP, the punctate co-localization with d/A was much stronger than that of anx2-GFP, and the diffuse background less evident (Figure 9C). In fact, ΔCa-GFP remained mostly associated with the d/A-positive puncta even when the cells, permeabilized with SLO, were incubated at very low [Ca2+] (compare Figures 5D and 9F). Membrane association of ΔCa-GFP at low [Ca2+] was confirmed by its recovery in the particulate fraction even when the samples were analyzed in the presence of EGTA, buffering [Ca2+] to 0.01 μM (Figure 9I). A model summarizing the role of the two domains in the anx2 binding to the enlargeosome and the plasma membranes is shown in Figure 9G.
The anx2-GFP construct: a tool to investigate the traffic of enlargeosomes in living cells
PC12-27 cells stably transfected with anx2-GFP were imaged by time-lapse video microscopy, either at rest or after ionomycin (5 μM). In resting cells, fluorescent puncta were seen to move randomly at a slow rate (Figure 10B; Supplementary Movie 1). Upon ionomycin addition, the puncta switched to faster waves oriented toward the periphery of the cell, with surface accumulation of fluorescence. Concomitantly, the cytosolic fluorescence vanished (Figure 10C; Supplementary Movie 2). Average anx2 movements, near the nucleus and near the plasma membrane, were recorded in 15 spots each from three different cells, both control and ionomycin-treated (Figure 10A). While in resting cells fluorescence was constant, upon ionomycin it decreased in the cytoplasm and increased in the plasma membrane area, reaching a plateau in ∼2 min. A cell preloaded with BAPTA, in which movement of the fluorescent spots remained slow and random, was used as specificity reference (Supplementary Movie 3).
Figure 10.
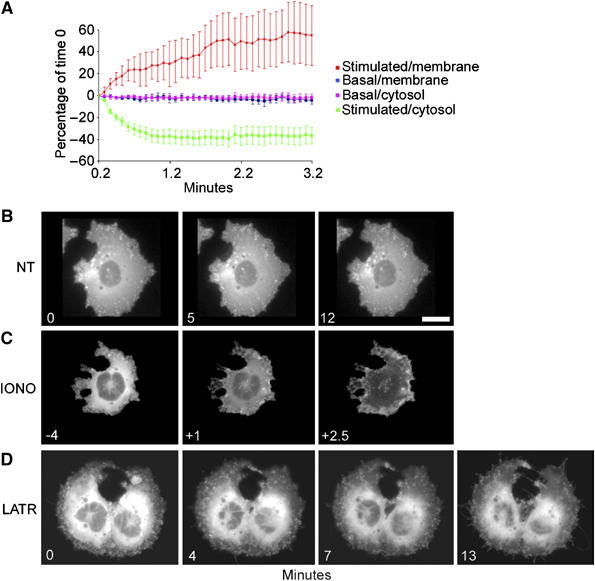
Time course of anx2 re-localization and enlargeosome dynamics in resting cells, and after application of ionomycin or latrunculinA. (A) The average distribution±s.d. of anx2-GFP fluorescence in three different PC12-27 cells, 10 fields/cell, 5 close to the plasma membrane (membrane), 5 to the nucleus (cytosol), both at rest (basal) and after 5 μM ionomycin (stimulated). At rest the overall distribution of anx2 is stable; upon stimulation, both the soluble and the particulate pools decline in the cytoplasm, concomitantly with a considerable increase in the plasma membrane. The images of (B–D) were extracted from the videos of Supplementary data 3, 4 and 6. (B) The distribution of anx2-GFP in resting PC12-27. The puncta move apparently at random, and their overall pattern does not change significantly during recording. The rapid and profound changes induced by ionomycin, with redistribution to the plasma membrane of both puncta and diffuse fluorescence, are illustrated in (C). (D) The changes induced by latrunculinA (5 μM). The diffuse fluorescence appears little affected whereas the puncta decrease progressively, with transfer of their fluorescence to the plasma membrane. The bar in (B, right frame), valid also for (C,D), corresponds to 10 μm.
Videos were recorded also after administration of latrunculinA (10 μM). During the first 5 min, the cells emitted long filamentous protrusion, establishing contacts and even continuities with adjacent cells. Fluorescent puncta speeded up their movements, penetrating the protrusions and even moving from one cell to the next (Figure 10D, Supplementary Movie 4). Surface accumulation of fluorescence was also seen, developing however at rates much slower than after ionomycin. In contrast, background fluorescence was unchanged. The movement of GFP in GFP-vector transfected cells was monitored as a control. Neither upon ionomycin nor latrunculinA treatments the cytosolic and nuclear GFP re-localized to the cell membrane (Supplementary Movies 5 and 6).
Discussion
The role of the cytoskeleton in regulated exocytosis can be quite different. In some cells, the secretory organelles bind to microtubules or microfilaments only during their intracellular travel, before docking to their fusion sites; in others they keep their binding and remain at some distance from the plasma membrane until stimulation occurs. These differences may explain why in some cells treatments that affect the cytoskeleton have little effect on exocytic responses, while in others they are strongly stimulatory or inhibitory (Bader et al, 2004; Dillon and Goda, 2005). Therefore, the role of the cytoskeleton in an exocytic system cannot be predicted a priori but requires a specific investigation, which in the case of enlargeosomes had never been made.
Enlargeosomes are small organelles rapidly exocytosed upon stimulation and then recycled by a form of endocytosis distinct from the clathrin-dependent process (Borgonovo et al, 2002; Cocucci et al, 2004). Here, we show that exo/endocytosis of enlargeosomes, similar to those of dense-core granules of chromaffin and PC12 cells (Bader et al, 2004), is only slightly inhibited by disruption of microtubules. In contrast, disruption of microfilaments induces considerable increase of both non-stimulated and stimulated exocytosis, and inhibition of endocytosis. In other exocytic cells, a few small G proteins and their effectors (Frantz et al, 2002; Bader et al, 2004), as well as the anxs (Gerke et al, 2005), were shown to be involved in the control of exo/endocytosis. In particular, anx2 was investigated for its binding to membrane rafts and phosphoinositides (Zobiack et al, 2003; Rescher et al, 2004), and for its role in both exocytosis (Knop et al, 2004; Chasserot-Golaz et al, 2005) and clathrin-dependent endocytosis (Engqvist-Goldstein and Drubin, 2003; Zobiack et al, 2003). Our present finding that anx2 is highly concentrated in the subcellular fraction mostly enriched in enlargeosomes first suggested the involvement of this protein in the generation and/or exocytosis of these organelles.
Two properties of the anx2/enlargeosome interaction, revealed in PC12-27, HeLa and BSC1, appear unique. First, it is much more extensive than those with other structures (Jost et al, 1997; Konig and Gerke, 2000; Zobiack et al, 2003), accounting for over 50% of the cellular anx2; second, it is extensive also at rest, and not only after stimulation. Previous electrophysiological data in PC12-27 had shown the total enlargeosome membrane area to be <20% of the resting plasma membrane (Kasai et al, 1999). The latter accounts for ∼15% of the total membrane of the cell (unpublished). The particulate pool of anx2 appears therefore concentrated on the membrane of a small compartment, only ∼3% of the total cell membranes in PC12-27.
What could be the mechanisms of this binding/concentration of anx2 on the enlargeosome membrane? Previous studies had revealed two mechanisms for anx2 membrane-binding. The first, Ca2+-dependent, involves the five Ca2+-binding sites in the protein and phosphoinositides and lipids raft in the membranes (Filipenko et al, 2000; Konig and Gerke, 2000; Gerke et al, 2005). The second, Ca2+-independent, involves the N-terminal domain in the protein (Jost et al, 1997; Zeuschner et al, 2001; Zobiack et al, 2003). The membrane composition of enlargeosomes is still unknown. However, they resist Triton X-100 solubilization (Cocucci et al, 2004), a property typical of raft-rich membranes. Therefore, the Ca2+-dependent/lipid raft mechanism could have a role. However, the anx2 construct lacking the Ca2+-binding sites did not loose the enlargeosome binding competence, even at the very low [Ca2+]. In the last condition, therefore, the unique concentration of anx2 construct at the enlargeosome surface might depend on the N-terminal domain of the protein, including the two residues necessary for its tetramer assembly with S100A10. Taken together these results suggest that one (or more) specific conformation(s), induced in anx2 by the inactivation of its negative charges at the Ca2+-binding sites and/or by the S100A10 assembly to its N-terminal domain, are needed for the binding to the enlargeosome membrane to take place. In addition to appropriate lipids, also a membrane receptor might be involved. In other membranes, a receptor-mediated anx2 binding had already been hypothesized (Jost et al, 1997; Konig and Gerke, 2000).
The identification of anx2 as a second marker, after d/A, gave us the opportunity to investigate new properties of enlargeosomes. Our evidence in PC12-27, HeLa, BSC1 and differentiated wt PC12 cells clearly shows that the co-localization of anx2 and d/A is not due to their direct interaction, but to their independent binding to either face of the enlargeosome membrane. In previous studies, however, both anx2 and d/A had been reported to be located in the cytosol (Sussman et al, 2001; Gerke et al, 2005). Moreover, Benaud et al (2004) reported the two proteins to become associated to the cytosolic surface of the plasma membrane during MDCK cell polarization. We show here that in cells devoid of enlargeosomes, such as U373, d/A may indeed be located in the cytosol. In the cells that express the enlargeosome, however, d/A appears to be transported across the membrane by an ABC transporter (Herget and Tampe, 2006), and accumulated inside the organelle. ABC transporters were believed to transport peptides and relatively small proteins (interleukin-1β, basic FGF, galectin, anx1) only at the plasma and ER membranes. Recently, however, their membrane distribution was shown to be wider (Kubo et al, 2005; Herget and Tampe, 2006) and their transport to include macromolecules (Prehm and Schumacher, 2004). Translocation of d/A across the enlargeosome membrane is inhibited by two well-known blockers of ABC transporters, verapamil and glybenclamide, not by probenecid, MK571 and DIDS. These results suggest d/A transport to involve a member of the ABCB family, possibly ABCB1 expressed in PC12-27 and not in wt PC12. Further studies will be needed to identify the transporter.
Video analysis of living PC12-27 cells expressing anx2-GFP provided the first information about motility of enlargeosomes. When [Ca2+]i was raised by ionomycin not only the enlargeosome but also the cytosolic pool of anx2 was rapidly redistributed to the plasma membrane. In contrast, when exocytosis was stimulated by latrunculinA, only the enlargeosome pool was redistributed. These results demonstrate that the plasma membrane redistribution of the cytosolic pool, which occurs also with the construct truncated of the N-terminal domain, is a consequence of the [Ca2+]i rise and not of enlargeosome exocytosis.
Finally, anx2 is needed for the regulated exocytosis of enlargeosomes. With other exocytic organelles, conflicting results had been reported. In endothelia anx2 is necessary for the exocytosis of Weibel–Palade bodies but not for that of the plasminogen activator organelles (Knop et al, 2004); in catecholamine-secreting cells, for the exocytosis of dense-core granules of chromaffin cells (Chasserot-Golaz et al, 2005), and not of PC12 cells. Accordingly, wt PC12 exocytize their dense-core granules efficiently (Kasai et al, 1999) in spite of their low levels of anx2. In chromaffin cells, the effect of anx2 on exocytosis was attributed to the induction of raft domains at the fusion sites of the plasma membrane (Chasserot-Golaz et al, 2005), a mechanism unlikely for enlargeosome exocytosis, which is little affected by cell extraction of cholesterol, the main component of rafts (Cocucci et al, 2004). Also, the interaction of anx2 with actin seems to play no direct role. When actin is disassembled by latrunculinA, anx2 remains associated with the enlargeosome surface, and exocytosis is not inhibited, but stimulated. Moreover, in anx2 downregulated cells the cytoskeleton appears unaffected, yet exocytosis induced by ionomycin, and also by latrunculinA, is inhibited. The latter result excludes the inhibition to be mediated by a tightening of the cytoskeleton consequent to anx2 depletion (Hayes et al, 2006). The hypothesys that remain is that the anx2 surface coating of enlargeosomes is needed for enlargeosomes to acquire their exocytic competence. Further studies will be needed to identify partners and mechanisms involved in these processes.
Materials and methods
Materials
Antibodies used were: monoclonal anti-anx2 and monoclonal anti-paxillin (BD Transduction Labs); polyclonal anti-anx1 and monoclonal anti-TfR (Zymed); monoclonal anti-d/A (Borgonovo et al, 2002); monoclonals anti-S100A10 (BD and R&D Systems); monoclonal anti-β-tubulin (Sigma-Aldrich); polyclonal anti-calnexin (Stressgen); monoclonal anti-Na+/K+ ATPase (Upstate Biotechnology); monoclonal and polyclonal anti-EEA1 (BD and ABR); monoclonal anti-LAMP1 (Calbiochem). FITC- and TRITC-conjugated secondary antibodies were from SBA. Alexa 647-conjugated anti-mouse antibody, Texas red- and fluorescein-coupled phalloidin were from Molecular Probes.
Cell cultures
PC12-27 and wt PC12 clones (Borgonovo et al, 2002) were grown in DMEM (BioWhittaker), 5% FCIII and 10% donor horse serum (EuroClone), 2 mM ultraglutamine and 100 U/ml penicillin/streptomycin (BioWhittaker); U373 and HeLa cells in DMEM, 10% FCIII, 1 mM Na pyruvate 2 mM ultraglutamine and 100 U/ml penicillin/streptomycin; BSC1 cells in DMEM, 10% FBS, 2 mM ultraglutamine and 100 U/ml penicillin/streptomycin.
Immunofluorescence
Cells grown on coverslips were fixed for 10 min in 4% paraformaldehyde and quenched with 0.1 M glycine. For whole-cell immunolabelling they were permeabilized in 0.1%Triton X-100, incubated for 1 h with primary antibodies in PBS +1% BSA, washed and processed with the secondary antibodies. For labelling the antigens exposed to the surface, cells were incubated for 5 min with the primary antibodies together with ionomycin (Calbiochem) 1–5 μM, then fixed, permeabilized and processed with secondary antibodies. To remove d/A exposed on the surface and reveal only the endocytosed d/A, the cells exposed for 5 min to the anti-d/A antibody and ionomycin were washed with medium supplemented with 300 mM NaCl, then fixed and processed as above. Images of blue autofluorescence were collected at excitation wavelength of 405 nm to reveal the distribution of cells in the fields. Average fluorescence values±s.d. were calculated based on intensity/cell values established by the ImageJ program (ImageJ, NIH, rsb.info.nih.gov/ij/) on five fields, of about 10 cells each, per experimental condition. Fluorescent images were collected using a confocal laser scanning microscope TCS SP2 from Leica Microsystem with 63 × /1.4 plan-APOCHROMAT oil immersion objective.
Western Blots, 2D PAGE and peptide sequencing
SDS–PAGE and Western blots of cell preparations were made and quantified as described by Borgonovo et al (2002). 2D PAGE analysis of the fractions collected from the gradients was performed by the IPGphor method (Amersham Biosciences). After separation, the spots of interest were excised, processed for mass-spectrometric analysis and sequenced as described (Wilm et al, 1996).
FM 1-43 assay of membrane area
PC12-27 cells were resuspended in KRH and transferred to a fluorometer cuvette. FM 1-43 dye (4 μM) was added under continuous stirring, and the changes of fluorescence intensity were recorded at excitation and emission wavelengths of 479 and 598 nm. After 5-min incubation, 5 μM latrunculinA or the same volume of 10% DMSO was added to the experimental and control samples. After further 3.5 min, 1 μM ionomycin was added to both samples, and the recording continued for 10 more min.
Subcellular fractionation
Monolayers were resuspended and the cells homogenized in HB (0.32 M sucrose, 5 mM HEPES pH 7.4 and protease inhibitor mixture from Sigma) with various [Ca2+], passed through a 27G1/2 needle and centrifuged for 5 min at 100 g. The supernatant was centrifuged for 30 min at 100 000 r.p.m. in the ultraTL100 centrifuge to obtain a pellet (particulate fraction) and the final supernatant. Postnuclear supernatants in HB, prepared from PC12-27 and wt PC12 homogenized by a cell cracker (EMBL; clearance 14 μm), were loaded on top of 0.32–1.8 M sucrose gradients and centrifuged for 24 h at 82 000 g. Of the 12 fractions, one was diluted to 0.3 M sucrose with 5 mM HEPES and sedimented at 110 000 g for 1 h, resuspended in HB, and loaded on a second, 0.32–1.8 M sucrose gradient centrifuged for 15 min at 90 000 g. Of the six fractions, two were diluted and centrifuged at 110 000 g as above, and the pellets were analyzed by 2D gel electrophoresis.
Cytoskeleton depolymerization/stabilization and drug inhibition of ABC transporters
Cells were incubated at 37°C in complete medium containing 5 μM latrunculinA (Molecular Probes, 15 min) 30 μM nocodazole (SIGMA-Aldrich, 1 h) or 100 nM jasplakinolide (Molecular Probes, 1 h). For recovery, after the 15 min with latrunculinA, the cells were washed and re-incubated with fresh medium for 24 h.
To inhibit ABC transporters, PC12-27 cells were incubated without serum for 16 h at 37°C in DMEM with either 50 μM verapamil, 100 μM glybenclamide, 100 μM probenecid, 100 μM DIDS (Sigma) or 100 μM MK571 (Alexis Biochemicals). After washing, the cells were fixed and processed for immunofluorescence, either immediately or after re-incubation in complete medium for 4 or 24 h.
Plasma membrane permeabilization with streptolysin-O
Cell monolayers, incubated at 4°C for 30 min with 1 μg/ml streptolysin-O (S Bhakdi, Mainz, Germany) in Kglu buffer (20 mM HEPES, 120 mM potassium glutamate, 20 mM potassium acetate, 0.1% BSA, 200 μM ATP, 200 μM MgCl2 and adjusted to 0.01 and 10 μM [Ca2+]), as such or supplemented with 0.1 M Na2CO3, pH 11, were washed and incubated at 37°C for 30 min. Cells were washed and processed for immunofluorescence and Western blot; media, precipitated overnight at −20°C with acetone, for Western blot.
ABC transporter mRNAs. Anx2 constructs and siRNAs
Expression of 11 ABC transporter mRNAs was determined by a Multi-gene-12 RT–PCR Profiling kit (Super Array Bioscience Corporation), according to the instructions.
Human anx2 and S100A10 cDNAs were the kind gifts of T Hunter and V Gerke, respectively. Anx2-GFP was constructed by PCR using the following primers: forward 5′-CCGGTCGACGCCACCATGTCTACTGTTCACGAAATC and the reverse 5′-CCGGGATCCGCGTCATCTCCACCACACAGG. The primers for ΔN26-GFP were: forward 5′-CCGGTCGACGCCACCATGGTCAAAGCCTATACTAACTTT and the reverse 5′ as above. Anx2-GFP and ΔN26-GFP were mutated for inactivation of the five Ca2+-binding sites (E52A, E95A, D161A, E246A and D321A). Anx2-GFP was also mutated on the S100A10 binding site I6L7E (Thiel et al, 1991). The PCR products, processed with the SalI and BamHI restriction enzymes, were inserted into the pEGFP-N1 vector (Clonetech).
RNA oligonucleotides (sense sequences GUGCCUAUGGGUCGGUCAAtt and CUUCGACGCUGAGAGGGAUtt, from Ambion), chosen by homology to human sequences (Zobiack et al, 2003; Benaud et al, 2004) were used for anx2 gene silencing. Control was a 19-bp scrambled sequence with 3′ dT overhangs from Ambion, showing no significant homology to any known gene sequence.
PC12-27 monolayers were transfected twice at 48 h distance with the lipofectamine reagent (Invitrogen) and a mix of either the two siRNA or the control siRNA (100 nM). Seventy-two hours after the first transfection, the cells were incubated for 5 min without or with 5 μM ionomycin or latrunculinA together with the anti-d/A antibody. Cells were fixed and processed as described in Immunofluorescence. Intensity of quantified Western blot bands (averages of three different experiments) was expressed as % of control cells.
Time-lapse videomicroscopy and image processing
Fluorescent images were collected using a confocal laser scanning microscope TCS SP2 from Leica Microsystem with 63 × /1.4 plan-APOCHROMAT oil immersion objective.
Videomicroscopy of a PC12-27 stable cell line expressing anx2-GFP and of cells transiently transfected with pEGFPN1 vector as a control was performed by using a DELTAVISION apparatus (Widefield Microscopy System from Applied Precision) equipped with an IL-70 microscope (Olympus), a plan-apo × 60 oil immersion objective (1.4 numerical aperture), a CoolSnap HQ digital camera (Roper Scientific) and GFP optical filter sets (Ex. 490/20, Em. 528/38, from Omega Optical).
Cells were plated on a glass bottom dish 35 mm diameter (MaTek Corporation). Five micrmolar ionomycin or latrunculinA were added after 4 min to 1 ml KRH medium containing 2 mM Ca2+, and images were acquired in epifluorescence time series at 37°C. After addition of ionomycin, images were collected for 3 min at 5 s intervals; after addition of latrunculinA, for 13 min at 10 s intervals. As a control, cells were preloaded with 50 μM BAPTA for 30 min prior to addition of ionomycin. For quantification of the data, 30 fields, 2 μm2 each, half near the nucleus and half near the plasma membrane, were chosen at the various time points from three different cells, and corrected for bleaching with the ImageJ program (bleach constant calculated from the video of the non-treated cell). Values were expressed as % of the fluorescence intensity at time zero. Series of images were assembled in avi files using a free image analysis software (ImageJ, NIH). Images were processed using Photoshop 8.0 (Adobe).
Supplementary Material
Supplementary Movie 1
Supplementary Movie 2
Supplementary Movie 3
Supplementary Movie 4
Supplementary Movie 5
Supplementary Movie 6
Supplementary Figure 1
Supplementary Figure 2
Acknowledgments
We thank Tony Hunter for the anx2 construct, Volker Gerke for the S100A10 construct and Marco Bianchi for suggestions in the ABC studies; Rosalba D'Alessandro, Vitor L Sousa and Gabriella Racchetti for assistance in some of the experiments. This work was founded in part by grants from the Telethon Fondazione ONLUS (GGGP030234), the European Community (APOPIS-LSHM-CT-2003-503330) and the FIRB 2004 Program of the Italian Minister of Research (MIUR).
References
- Bader MF, Doussau F, Chasserot-Golaz S, Vitale N, Gasman S (2004) Coupling actin and membrane dynamics during calcium-regulated exocytosis: a role for Rho and ARF GTPases. Biochim Biophys Acta 1742: 37–49 [DOI] [PubMed] [Google Scholar]
- Benaud C, Gentil BJ, Assard N, Court M, Garin J, Delphin C, Baudier J (2004) AHNAK interaction with the annexin 2/S100A10 complex regulates cell membrane cytoarchitecture. J Cell Biol 164: 133–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgonovo B, Cocucci E, Racchetti G, Podini P, Bachi A, Meldolesi J (2002) Regulated exocytosis: a novel, widely expressed system. Nat Cell Biol 4: 955–962 [DOI] [PubMed] [Google Scholar]
- Cerny J, Feng Y, Yu A, Miyake K, Borgonovo B, Klumperman J, Meldolesi J, McNeil PL, Kirchhausen T (2004) The small chemical vacuolin-1 inhibits Ca(2+)-dependent lysosomal exocytosis but not cell resealing. EMBO Rep 5: 883–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chasserot-Golaz S, Vitale N, Umbrecht-Jenck E, Knight D, Gerke V, Bader MF (2005) Annexin 2 promotes the formation of lipid microdomains required for calcium-regulated exocytosis of dense-core vesicles. Mol Biol Cell 16: 1108–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocucci E, Racchetti G, Podini P, Rupnik M, Meldolesi J (2004) Enlargeosome, an exocytic vesicle resistant to nonionic detergents, undergoes endocytosis via a nonacidic route. Mol Biol Cell 15: 5356–5368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon C, Goda Y (2005) The actin cytoskeleton: integrating form and function at the synapse. Annu Rev Neurosci 28: 25–55 [DOI] [PubMed] [Google Scholar]
- Eberhard DA, Karns LR, VandenBerg SR, Creutz CE (2001) Control of the nuclear–cytoplasmic partitioning of annexin II by a nuclear export signal and by p11 binding. J Cell Sci 114: 3155–3166 [DOI] [PubMed] [Google Scholar]
- Engqvist-Goldstein AE, Drubin DG (2003) Actin assembly and endocytosis: from yeast to mammals. Annu Rev Cell Dev Biol 19: 287–332 [DOI] [PubMed] [Google Scholar]
- Filipenko NR, Kang HM, Waisman DM (2000) Characterization of the Ca2+-binding sites of annexin II tetramer. J Biol Chem 275: 38877–38884 [DOI] [PubMed] [Google Scholar]
- Fox MT, Prentice DA, Hughes JP (1991) Increases in p11 and annexin II proteins correlate with differentiation in the PC12 pheochromocytoma. Biochem Biophys Res Commun 177: 1188–1193 [DOI] [PubMed] [Google Scholar]
- Frantz C, Coppola T, Regazzi R (2002) Involvement of Rho GTPases and their effectors in the secretory process of PC12 cells. Exp Cell Res 273: 119–126 [DOI] [PubMed] [Google Scholar]
- Gerke V, Creutz CE, Moss SE (2005) Annexins: linking Ca(2+) signalling to membrane dynamics. Nat Rev Mol Cell Biol 6: 449–461 [DOI] [PubMed] [Google Scholar]
- Hayes MJ, Shao D, Bailly M, Moss SE (2006) Regulation of actin dynamics by annexin 2. EMBO J 25: 1816–1826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herget M, Tampe R (2006) Intracellular peptide transporters in human—compartmentalization of the ‘peptidome'. Pflugers Arch 18 May [E-pub ahead of print] [DOI] [PubMed] [Google Scholar]
- Jacovina AT, Zhong F, Khazanova E, Lev E, Deora AB, Hajjar KA (2001) Neuritogenesis and the nerve growth factor-induced differentiation of PC-12 cells requires annexin II-mediated plasmin generation. J Biol Chem 276: 49350–49358 [DOI] [PubMed] [Google Scholar]
- Jost M, Zeuschner D, Seemann J, Weber K, Gerke V (1997) Identification and characterization of a novel type of annexin–membrane interaction: Ca2+ is not required for the association of annexin II with early endosomes. J Cell Sci 110 (Part 2): 221–228 [DOI] [PubMed] [Google Scholar]
- Kasai H, Kishimoto T, Liu TT, Miyashita Y, Podini P, Grohovaz F, Meldolesi J (1999) Multiple and diverse forms of regulated exocytosis in wild-type and defective PC12 cells. Proc Natl Acad Sci USA 96: 945–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M, Aareskjold E, Bode G, Gerke V (2004) Rab3D and annexin A2 play a role in regulated secretion of vWF, but not tPA, from endothelial cells. EMBO J 23: 2982–2992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig J, Gerke V (2000) Modes of annexin–membrane interactions analyzed by employing chimeric annexin proteins. Biochim Biophys Acta 1498: 174–180 [DOI] [PubMed] [Google Scholar]
- Kubo Y, Sekiya S, Ohigashi M, Takenaka C, Tamura K, Nada S, Nishi T, Yamamoto A, Yamaguchi A (2005) ABCA5 resides in lysosomes, and ABCA5 knockout mice develop lysosomal disease-like symptoms. Mol Cell Biol 25: 4138–4149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prehm P, Schumacher U (2004) Inhibition of hyaluronan export from human fibroblasts by inhibitors of multidrug resistance transporters. Biochem Pharmacol 68: 1401–1410 [DOI] [PubMed] [Google Scholar]
- Rescher U, Ruhe D, Ludwig C, Zobiack N, Gerke V (2004) Annexin 2 is a phosphatidylinositol (4,5)-bisphosphate binding protein recruited to actin assembly sites at cellular membranes. J Cell Sci 117: 3473–3480 [DOI] [PubMed] [Google Scholar]
- Shtivelman E, Bishop JM (1993) The human gene AHNAK encodes a large phosphoprotein located primarily in the nucleus. J Cell Biol 120: 625–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussman J, Stokoe D, Ossina N, Shtivelman E (2001) Protein kinase B phosphorylates AHNAK and regulates its subcellular localization. J Cell Biol 154: 1019–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel C, Weber K, Gerke V (1991) Characterization of a Ca(2+)-binding site in human annexin II by site-directed mutagenesis. J Biol Chem 266: 14732–14739 [PubMed] [Google Scholar]
- Vaudry D, Chen Y, Ravni A, Hamelink C, Elkahloun AG, Eiden LE (2002) Analysis of the PC12 cell transcriptome after differentiation with pituitary adenylate cyclase-activating polypeptide (PACAP). J Neurochem 83: 1272–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RB, Kuo CL, Lien LL, Lien EJ (2003) Structure–activity relationship: analyses of p-glycoprotein substrates and inhibitors. J Clin Pharm Ther 28: 203–228 [DOI] [PubMed] [Google Scholar]
- Wilm M, Shevchenko A, Houthaeve T, Breit S, Schweigerer L, Fotsis T, Mann M (1996) Femtomole sequencing of proteins from polyacrylamide gels by nano-electrospray mass spectrometry. Nature 379: 466–469 [DOI] [PubMed] [Google Scholar]
- Zeuschner D, Stoorvogel W, Gerke V (2001) Association of annexin 2 with recycling endosomes requires either calcium- or cholesterol-stabilized membrane domains. Eur J Cell Biol 80: 499–507 [DOI] [PubMed] [Google Scholar]
- Zobiack N, Rescher U, Ludwig C, Zeuschner D, Gerke V (2003) The annexin 2/S100A10 complex controls the distribution of transferrin receptor-containing recycling endosomes. Mol Biol Cell 14: 4896–4908 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Movie 1
Supplementary Movie 2
Supplementary Movie 3
Supplementary Movie 4
Supplementary Movie 5
Supplementary Movie 6
Supplementary Figure 1
Supplementary Figure 2