

Abstract
Inhibition of NF-κB activation increases susceptibility to tumor necrosis factor (TNF)α-induced cell death, concurrent with caspases and prolonged c-Jun N-terminal kinase (JNK) activation, and reactive oxygen species (ROS) accumulation. However, the detailed mechanisms are unclear. Here we show that cellular FLICE-inhibitory protein (c-FLIP) is rapidly lost in NF-κB activation-deficient, but not wild-type fibroblasts upon TNFα stimulation, indicating that NF-κB normally maintains the cellular levels of c-FLIP. The ectopic expression of the long form of c-FLIP (c-FLIPL) inhibits TNFα-induced prolonged JNK activation and ROS accumulation in NF-κB activation-deficient fibroblasts. Conversely, TNFα induces prolonged JNK activation and ROS accumulation in c-Flip−/− fibroblasts. Moreover, c-FLIPL directly interacts with a JNK activator, MAP kinase kinase (MKK)7, in a TNFα-dependent manner and inhibits the interactions of MKK7 with MAP/ERK kinase kinase 1, apoptosis-signal-regulating kinase 1, and TGFβ-activated kinase 1. This stimuli-dependent interaction of c-FLIPL with MKK7 might selectively suppress the prolonged phase of JNK activation. Taken that ROS promote JNK activation and activation of the JNK pathway may promote ROS accumulation, c-FLIPL might block this positive feedback loop, thereby suppressing ROS accumulation.
Keywords: c-FLIP, c-Jun N-terminal kinase, NF-κB, reactive oxygen species, tumor necrosis factor
Introduction
NF-κB is a collective term of dimeric transcriptional factors that belong to the Rel family of proteins, and regulates expression of various inflammatory cytokines, chemokines, and adhesion molecules (Ghosh and Karin, 2002). NF-κB is activated by various inflammatory cytokines and cellular stress including tumor necrosis factor (TNF)α, interleukin-1 (IL-1), UV, and γ-irradiation. Moreover, NF-κB, especially the RelA-containing complex, inhibits cell death induced by TNFα, Fas ligand, TRAIL, and genotoxic stress. Currently, the antiapoptotic functions of NF-κB are supported to be mainly mediated by the upregulation of antiapoptotic genes such as cellular FLICE-inhibitory protein (c-Flip), the members of the Bcl-2 family, and X-chromosome-linked inhibitor of apoptosis (Xiap) (Karin and Lin, 2002). However, the detailed molecular mechanisms are not completely understood.
The regulation of cell death and survival is also controlled in part by another signaling cascade activated by the mitogen-activated protein kinase (MAPK) following cellular stress or cytokine signaling (Davis, 2000; Kyriakis and Avruch, 2001). In mammals, the MAPK cascades are composed of three distinct signaling modules, the c-Jun N-terminal kinase (JNK), the p38MAPK, and the extracellular signal-regulated kinase (ERK) cascades. Each MAPK is activated by sequential protein phosphorylation through a MAPK module. In the case of the JNK cascade, the MAP kinase kinase kinases (MAPKKKs) include apoptosis-signal-regulating kinase (ASK)1, MAP/ERK kinase kinases (MEKKs), MTK1 (also known as MEKK4), and TGFβ-activated kinase (TAK)1. These MAPKKKs activate MAP kinase kinase (MKK)4 and/or MKK7, which in turn activate JNKs (Davis, 2000; Kyriakis and Avruch, 2001). Cytokines and growth factors including TNFα and IL-1 induce rapid (within 10 min) yet transient MAPK activation, cellular stress, such as UV- or γ-irradiation, induces prolonged MAPK activation. Several lines of evidence suggest that transient MAPK activation is associated with gene expression, proliferation, and differentiation, whereas prolonged MAPK activation promotes cell death in a cell-type- and stimuli-dependent manner (Chen et al, 1996; Guo et al, 1998).
In the past, the contributions of the NF-κB and JNK pathways have been discussed independently. However, two recent studies have revealed that inhibition of NF-κB activation induces prolonged JNK activation that promotes TNFα-induced cell death (De Smaele et al, 2001; Tang et al, 2001). Later studies have shown that TNFα-induced accumulation of reactive oxygen species (ROS) is enhanced in NF-κB activation-deficient cells including RelA−/−, IκB kinase β−/−, and TNF receptor-associated factor (traf)2−/−traf5−/− murine embryonic fibroblasts (MEFs) (Sakon et al, 2003; Pham et al, 2004; Ventura et al, 2004; Kamata et al, 2005). Notably, this TNFα-induced ROS accumulation and prolonged JNK activation are completely suppressed by antioxidants, but not caspase inhibitors, indicating that TNFα-induced ROS accumulation is caspase-independent in NF-κB activation-deficient MEFs. Furthermore, expression of antioxidant enzyme genes including manganese-dependent superoxide dismutase (Mnsod) and ferritin heavy chain (fhc) is upregulated by TNFα in an NF-κB-dependent manner, and the ectopic expression of these genes inhibits TNFα-induced ROS accumulation in NF-κB-deficient MEFs (Pham et al, 2004; Kamata et al, 2005). Although these studies have revealed that the caspase-independent pathway plays a crucial role in ROS accumulation, a recent study has shown that activated caspase-3 cleaves the p75 subunit of complex I of the mitochondrial electron transport chain, which would impair the function of complex I, resulting in ROS accumulation (Ricci et al, 2004). In addition, Giorgio et al (2005) have shown that proapoptotic signals induce the release of p66Shc from a putative inhibitory complex and the released p66Shc then oxidizes reduced cytochrome c, thereby generating ROS. Therefore, the mechanisms whereby TNFα induces ROS accumulation are still controversial (Papa et al, 2004b; Luo et al, 2005; Nakano et al, 2006).
c-FLIP, also designated as CASH or Casper, was first identified as a molecule that interacts with FADD or is structurally related to procaspase-8 (Goltsev et al, 1997; Irmler et al, 1997; Shu et al, 1997). c-Flip encodes two splicing variants, long form of c-FLIP (c-FLIPL) and short form of c-FLIP (c-FLIPs). While c-FLIPS only contains two N-terminal death effector domains (DEDs), c-FLIPL consists of the N-terminal DEDs and a C-terminal caspase-like domain that does not possess enzymatic activity. Many studies have shown that both c-FLIPL and c-FLIPS inhibit death receptor-induced apoptosis by binding to and inhibiting caspase-8 activation via DED–DED interaction (Thome and Tschopp, 2001). Consistently, c-Flip−/− cells showed an increased susceptibility to TNFα- and Fas ligand-induced cell death (Yeh et al, 2000). Moreover, recent studies have shown that c-FLIP and viral FLIP are also involved in the Wnt signaling pathways (Naito et al, 2004; Nakagiri et al, 2005).
In the present study, we have shown that c-FLIPL is rapidly degraded after TNFα stimulation, and the ectopic expression of c-FLIPL inhibits the TNFα-induced ROS accumulation and prolonged JNK activation in NF-κB activation-deficient MEFs. Conversely, TNFα induces prolonged JNK and ERK activation, and ROS accumulation in c-Flip−/− cells. We have also shown that c-FLIPL binds to MKK7 and MEK1 in a TNFα-dependent manner, resulting in suppression of the MKK7–JNK and MEK1–ERK pathways. Taken that ROS promote JNK activation and activation of the JNK pathway may induce ROS accumulation, c-FLIPL might block this positive feedback loop, thereby suppressing ROS accumulation. Collectively, c-FLIPL is essential for suppression of TNFα-induced prolonged JNK activation and ROS accumulation.
Results
NF-κB protects signal-dependent degradation of c-FLIPL
To explore signaling intermediate(s) that link the TNF receptor to ROS accumulation, we first compared protein expression levels of downstream signaling molecules of the TNF receptor in wild-type (WT), RelA−/−, and traf2−/−traf5−/− MEFs before and after TNFα stimulation. Among various signaling molecules investigated, we found that TNFα stimulation induced a rapid and progressive loss of c-FLIPL in both RelA−/− and traf2−/−traf5−/− MEFs, concurrent with the appearance of a degradation fragment of 43 kDa (Figure 1A). Notably, MEFs appeared to express 55 kDa c-FLIPL, but not 26 kDa c-FLIPs. A similar degradation fragment was also observed in WT MEFs after TNFα stimulation, while the levels of c-FLIPL were not apparently reduced. When pretreated with a protein synthesis inhibitor, cycloheximide (CHX), c-FLIPL was progressively lost also in WT MEFs (Figure 1B). These results indicate that TNFα induces rapid degradation of c-FLIP in both WT and NF-κB activation-deficient MEFs, but the expression levels of c-FLIPL are maintained in WT MEFs by NF-κB-dependent de novo synthesis. It has been shown that the processing of c-FLIPL to the p43 fragment is mediated by caspase-8 (Shu et al, 1997; Kataoka and Tschopp, 2004), and c-FLIP levels are also controlled in part by the ubiquitin/proteasome pathway (Kreuz et al, 2001; Micheau et al, 2001). Consistently, the TNFα-induced degradation of c-FLIPL at early time points was partially inhibited by z-VAD-fmk or a proteasome inhibitor, MG132, in RelA−/− and traf2−/−traf5−/− MEFs (Supplementary Figure S1). Therefore, the TNFα-induced rapid degradation of c-FLIPL may be at least partly mediated by caspases and the ubiquitin/proteasome pathway.
Figure 1.
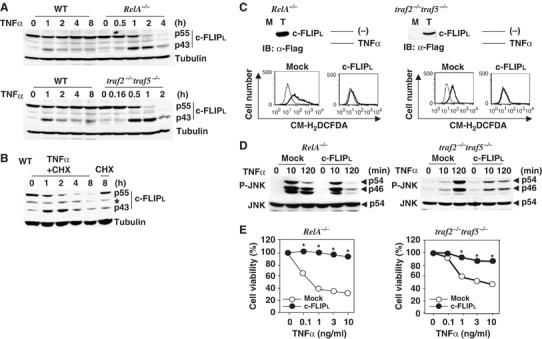
TNFα induces rapid degradation of c-FLIPL and ectopic expression of c-FLIPL inhibits TNFα-induced accumulation of ROS, prolonged JNK activation, and cell death in RelA−/− and traf2−/−traf5−/− MEFs. (A) WT, RelA−/−, and traf2−/−traf5−/− MEFs were stimulated with TNFα (10 ng/ml) for the indicated times, then the lysates were analyzed by immunoblotting with anti-c-FLIP (upper panel) and anti-tubulin (lower panel) antibodies. p55 and p43 indicate full and degraded form of c-FLIPL, respectively. (B) WT MEFs were stimulated with TNFα (10 ng/ml) in the presence of CHX (10 μg/ml) for the indicated times. Immunoblotting were performed as in (A). The asterisks indicate nonspecific bands. (C) Transfection of c-FLIPL inhibits TNFα-induced ROS accumulation in RelA−/− and traf2−/−traf5−/− MEFs. Total cell lysates from mock (M) or respective transfectants (T) were immunoblotted with anti-Flag antibody. The transfectants were unstimulated (thin line) or stimulated (bold line) with TNFα (10 ng/ml) for 4 h, and then the cells were labeled with CM-H2DCFDA (1 μM) and analyzed by flow cytometry. (D) c-FLIPL inhibits prolonged, but not early JNK activation in RelA−/− and traf2−/−traf5−/− MEFs. Mock- or c-FLIPL-transfected RelA−/− and traf2−/−traf5−/− MEFs were stimulated with TNFα (10 ng/ml) for the indicated times, and then the lysates were immunoblotted with anti-phospho-JNK (upper panel) and anti-total JNK (lower panel) antibodies. (E) c-FLIPL inhibits TNFα-induced cell death in RelA−/− and traf2−/−traf5−/− MEFs. Mock- or c-FLIPL-transfected RelA−/− and traf2−/−traf5−/− MEFs were stimulated with the indicated amounts of TNFα for 16 h. Cell viability was determined by WST assay. Results are presented as the mean of triplicate samples and represent three independent experiments with similar results. Standard errors are within 5%. *P<0.05 compared to mock transfectant.
c-FLIPL inhibits TNFα-induced ROS accumulation and prolonged JNK activation
Consistent with our previous study (Sakon et al, 2003), TNFα-induced ROS accumulation and prolonged JNK activation are suppressed by an antioxidant, butylated hydroxyanisole (BHA), but not a broad caspase inhibitor, z-VAD-fmk in RelA−/− and traf2−/−traf5−/− MEFs (Supplementary Figure S2A). This indicates that ROS accumulation and prolonged JNK activation are induced in a caspase-independent manner in these cells. To test directly whether the degradation of c-FLIPL is responsible for the TNFα-induced ROS accumulation and prolonged JNK activation, we ectopically expressed c-FLIPL in RelA−/− and traf2−/−traf5−/− MEFs. Expression of c-FLIPL almost completely inhibited TNFα-induced ROS accumulation in RelA−/− and traf2−/−traf5−/− MEFs (Figure 1C), suggesting that c-FLIPL might also suppress the caspase-independent pathway leading to ROS accumulation. TNFα-induced JNK activation at 10 min was not different between WT and RelA−/− MEFs, but this early phase JNK activation was severely impaired in traf2−/−traf5−/− MEFs (Supplementary Figure S2B). Intriguingly, c-FLIPL specifically inhibited the sustained (at 120 min), but not early (at 10 min), phase of JNK activation in RelA−/− and traf2−/−traf5−/− MEFs (Figure 1D). Moreover, expression of c-FLIPL substantially increased cell viability after TNFα stimulation in RelA−/− and traf2−/−traf5−/− MEFs (Figure 1E). Taken that TNFα induces caspase-dependent and -independent cell death in NF-κB activation-deficient cells (Sakon et al, 2003), these results indicate that ectopic expression of c-FLIPL is sufficient for suppression of TNFα-induced ROS accumulation, prolonged JNK activation, and cell death through suppression of the caspase-dependent and -independent pathways in NF-κB activation-deficient cells.
TNFα induces ROS accumulation and prolonged JNK activation in c-Flip−/− MEFs
To test directly whether c-FLIPL is essential for suppression of JNK activation and ROS accumulation, we first investigated the time course of caspase-3 activities after TNFα stimulation in c-Flip−/− MEFs. TNFα did not induce activation of caspase-3 in WT MEFs, unless the cells were stimulated with TNFα in the presence of CHX (Figure 2A). In sharp contrast, TNFα stimulation induced a robust activation of caspase-3 at 1 h and then continued at 2 h, which was completely suppressed in the presence of z-VAD-fmk or z-VAD-fmk plus BHA, but not BHA in c-Flip−/− MEFs (Figure 2A). We next tested whether JNK activation is prolonged in c-Flip−/− MEFs. While TNFα stimulation only induced transient JNK activation in WT MEFs, the sustained phase of JNK activation gradually appeared at 1 h after stimulation in c-Flip−/− MEFs, which was completely suppressed by z-VAD-fmk (Figure 2B). However, JNK activation continued at 2 h even in the presence of z-VAD-fmk, and this activation was substantially inhibited by the further addition of BHA. These results suggest that the caspase-dependent pathway plays a dominant role in JNK activation at 1 h. This is then followed by the caspase-independent ROS-inducing pathway leading to JNK activation at 2 h. Similarly, TNFα-induced ERK activation was prolonged in c-Flip−/− MEFs. Phosphorylation of JNK and ERK in TNFα plus BHA-treated cells at 2 h appeared to decrease compared to TNFα plus zVAD-fmk-treated cells. Taken that BHA alone inhibited neither caspase activation nor ROS accumulation at 2 h (Figure 2A and C), these decreases might not be due to direct inhibitory effect of BHA on JNK and ERK activation, but rather due to massive cell death of TNFα plus BHA-treated c-Flip−/− MEFs (data not shown).
Figure 2.
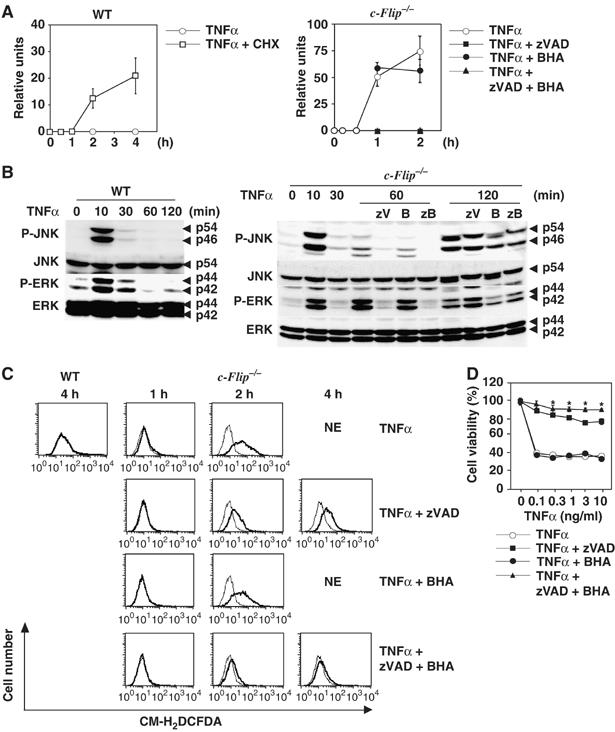
TNFα induces ROS accumulation and prolonged JNK activation in c-Flip−/− MEFs. (A) WT MEFs and c-Flip−/− MEFs were stimulated with TNFα (10 ng/ml) in the absence or presence of CHX (10 μg/ml), z-VAD-fmk (zVAD) (50 μM), BHA (100 μM), or z-VAD-fmk (50 μM) plus BHA (100 μM) for the indicated times. Caspase-3 activities were measured by using fluorogenic substrates. Results are presented as mean±s.e.'s of triplicate samples. Caspase activities were not detected in z-VAD-fmk- or z-VAD-fmk plus BHA-treated cells. (B) WT and c-Flip−/− MEFs were stimulated as in (A), and then the lysates were analyzed as in Figure 1D. zB indicates z-VAD-fmk (50 μM) plus BHA (100 μM) treatment. (C) WT and c-Flip−/− MEFs were unstimulated (thin line) or stimulated (bold line) with TNFα (10 ng/ml) as in (A) for the indicated times, accumulated ROS were analyzed as in Figure 1C. NE (not examined) indicates that ROS accumulation was not analyzed due to massive cell death. (D) c-Flip−/− MEFs were stimulated with the indicated amounts of TNFα in the absence or presence of z-VAD-fmk (zVAD) (50 μM), BHA (100 μM), or z-VAD-fmk (50 μM) plus BHA (100 μM) for 14 h. Cell viability was determined by WST assay as in Figure 1E. *P<0.05 compared to TNFα plus z-VAD-fmk-treated cells.
We tested whether TNFα induces ROS accumulation in c-Flip−/− MEFs. ROS slightly accumulated at 1 h, and then progressively accumulated at 2 h after stimulation in c-Flip−/− MEFs (Figure 2C). In contrast to NF-κB activation-deficient cells (Supplementary Figure S2A), TNFα-induced ROS accumulation was significantly suppressed by z-VAD-fmk at 2 h after simulation. However, a substantial amount of ROS still accumulated in c-Flip−/− MEFs at 2 and 4 h even in the presence of z-VAD-fmk (Figure 2C). We could not evaluate ROS accumulation in TNFα- or TNFα plus BHA-stimulated c-Flip−/− MEFs at 4 h due to cell death. Consistent with an inhibitory effect on TNFα-induced JNK activation, BHA plus z-VAD-fmk almost completely suppressed TNFα-induced ROS accumulation at 2 and 4 h after stimulation. Together, these results suggest that both caspase-dependent and -independent pathways may play crucial roles in TNFα-induced prolonged JNK activation and ROS accumulation in c-Flip−/− MEFs. The reason why BHA could not inhibit caspase-dependent ROS accumulation is currently unknown. Similarly, another antioxidant, N-acetyl cystein, could not inhibit TNFα-induced ROS accumulation in c-Flip−/− MEFs (data not shown). Consistent with these results, z-VAD-fmk plus BHA treatment more efficiently increased cell viability of c-Flip−/− MEFs than z-VAD-fmk alone (Figure 2D). Collectively, c-FLIP is essential for suppression of TNFα-induced ROS accumulation, prolonged JNK activation, and cell death.
We also tested whether deficiency of c-FLIP could affect the duration of the JNK pathway induced by other stimuli. Consistent with a previous study (Chen et al, 1996), anisomycin and UV induced prolonged JNK activation even in WT MEFs (Supplementary Figure S3A and B). Similarly, these stimuli induced prolonged JNK activation in c-Flip−/− MEFs. In contrast, IL-1 induced transient JNK activation in both WT and c-Flip−/− MEFs (Supplementary Figure S3C), suggesting that c-FLIP-dependent suppression of the JNK pathway is specific to TNFα-induced JNK activation.
TNFα-induced NF-κB activation and expression of fhc and Mnsod genes are not different between WT and c-Flip−/− MEFs
Previous studies have shown that NF-κB-dependent upregulation of antioxidant enzyme genes including fhc and Mnsod are responsible for elimination of ROS (Pham et al, 2004; Kamata et al, 2005). Therefore, TNFα induces ROS accumulation in NF-κB activation-deficient cells. On the other hand, several studies have shown that c-FLIPL activates NF-κB under particular conditions (Kataoka et al, 2000; Golks et al, 2006). To test the possibility that accumulation of ROS is due to impaired upregulation of fhc and/or Mnsod genes in c-Flip−/− MEFs, we first investigated whether TNFα-induced IκBα degradation is impaired in c-Flip−/− MEFs. Consistent with a previous study (Yeh et al, 2000), TNFα-induced degradation of IκBα was not impaired in c-Flip−/− MEFs (Figure 3A). Moreover, TNFα-induced upregulation of fhc and Mnsod mRNAs were not significantly different up to 120 min after TNFα stimulation in WT and c-Flip−/− MEFs (Figure 3B). We could not investigate the induction of mRNAs of these genes at later time points after 120 min, as almost all c-Flip−/− MEFs died.
Figure 3.
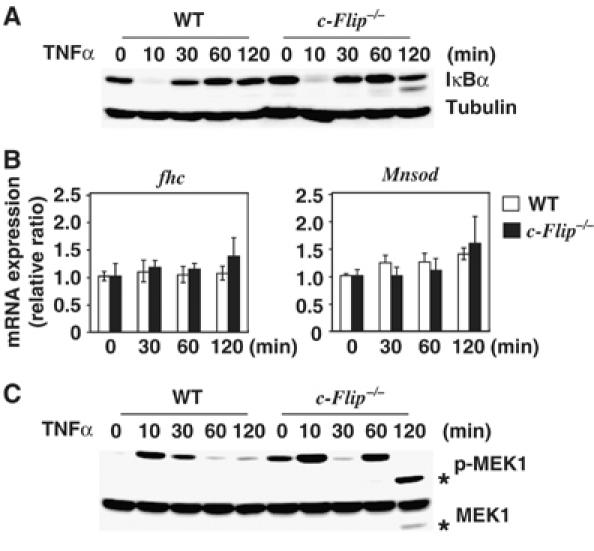
TNFα-induced NF-κB activation and expression of fhc and Mnsod mRNAs are not different between WT and c-Flip−/− MEFs. (A) TNFα induces degradation of IκBα in WT and c-Flip−/− MEFs. WT and c-Flip−/− MEFs were stimulated with TNFα (10 ng/ml) for the indicated times, then the lysates were analyzed by immunoblotting with anti-IκBα (upper panel) and anti-tubulin (lower panel) antibodies. (B) Induction of fhc and Mnsod mRNAs upon TNFα stimulation. WT or c-Flip−/− MEFs were stimulated with TNFα as in (A), and then total RNAs were extracted. Expression of the indicated genes was analyzed by real-time PCR. mRNA levels of the indicated genes were compared as a ratio to the unstimulated time points. The data are shown as mean±s.e.'s of triplicates samples. (C) TNFα induces prolonged MEK1 activation in c-Flip−/− MEFs. MEFs were stimulated as in (A), and the lysates were analyzed by immunoblotting with anti-phospho MEK1 (upper panel) and anti-total MEK1 (lower panel) antibodies. The asterisks indicate the degraded form of MEK1.
To further explore the mechanism underlying c-FLIP-dependent suppression of the ERK and JNK pathways, we examined whether TNFα-induced MEK1 phosphorylation is prolonged in c-Flip−/− MEFs. Phosphorylation of endogenous MEK1 was detected at 10 min upon TNFα stimulation in both WT and c-Flip−/− MEFs. Notably, phosphorylation of MEK1 reappeared at 60 min and persisted up to 120 min in c-Flip−/− MEFs (Figure 3C), whereas phosphorylation of MEK1 disappeared at 60 min in WT MEFs. The decrease in the size of phosphorylated MEK1 at 120 min in c-Flip−/− MEFs might be due to a degradation of MEK1. However, at least under our experimental conditions, we could not detect phosphorylation of endogenous MKK7 upon TNFαstimulation possibly due to a poor sensitivity of the antibody to recognize phosphorylated MKK7 (data not shown). Collectively, these results indicate that TNFα induces prolonged ERK and JNK activation through activation of MEK1 and possibly MKK7 in c-Flip−/− MEFs, which are not suppressed by NF-κB-inducible genes other than c-Flip.
c-FLIPL inhibits JNK and ERK activation
Taken that TNFα-induced ROS accumulation is abolished in Jnk1−/−Jnk2−/− (Jnk−/−) MEFs (Ventura et al, 2004) and the above results together, we surmised that c-FLIPL might directly inhibit the JNK pathway. Thus, we first tested whether c-FLIPL inhibits phosphorylation of MAPKs triggered by MAPKKKs including MEKK1, ASK1, and TAK1, which have been implicated in TNFα-induced MAPK activation (Nishitoh et al, 1998; Yuasa et al, 1998; Sato et al, 2005). We transfected HEK293 cells with HA-tagged MAPKs along with a constitutively active form of MEKK1 (MEKK1ΔN) or ASK1 in the presence or absence of c-FLIPL. Expression of c-FLIPL substantially inhibited the MEKK1ΔN-induced JNK and ERK phosphorylation (Figure 4A and B). c-FLIPL also inhibited the ASK1-induced JNK, but not p38 phosphorylation (Figure 4C and D). Collectively, c-FLIPL preferentially inhibits the JNK and ERK, but not p38 pathways. As we could not detect the interaction of c-FLIPL with JNK, ERK, or p38 in co-immunoprecipitation experiments (data not shown), we speculated that c-FLIPL inhibits the JNK and ERK pathways by suppressing their activators such as MAPKKs or MAPKKKs. Therefore, we next tested whether c-FLIPL inhibits MEKK1ΔN- and ASK1-triggered phosphorylation of MAPKKs in their activation loop motifs. c-FLIPL substantially inhibited the MEKK1ΔN- and ASK1-induced phosphorylation of MKK7 (Figure 4E and F). Similarly, c-FLIPL moderately inhibited the MEKK1ΔN-induced MEK1 and MKK4 phosphorylation (Figure 4G and H). In contrast, c-FLIPL did not inhibit the ASK1-induced MKK6 phosphorylation (Figure 4I). Moreover, c-FLIPL inhibited the TAK1-induced MKK7, MKK4, and JNK, but inhibitory effect of c-FLIPL on p38 phosphorylation was marginal. Similarly, c-FLIPL inhibited Tpl-2-induced JNK and ERK activation (Supplementary Figure S4). These results indicate that c-FLIPL selectively inhibits the MKK7–JNK, MKK4–JNK, and MEK1–ERK, but not MKK6–p38 pathways.
Figure 4.
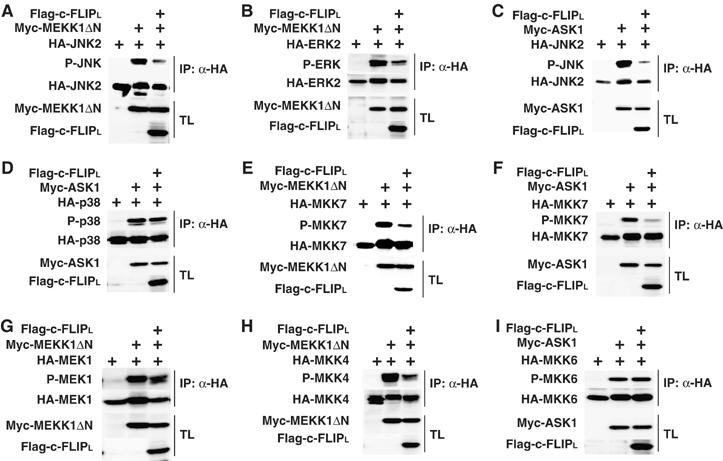
c-FLIPL inhibits the MKK7–JNK, MKK4–JNK, and MEK1–ERK pathways. (A–I) HEK293 cells were transfected with the indicated expression vectors. After immunoprecipitation (IP) with anti-HA antibody, the phosphorylation of MAPKs or MAPKKs was analyzed by immunoblotting with the indicated phospho-specific antibodies (top panels). Expression levels of the transfected proteins in the immunoprecipitates (IP) (second panels) and the total lysates (TL) (third and bottom panels) were analyzed by immunoblotting with anti-HA, anti-Myc, and anti-Flag antibodies.
c-FLIPL binds to MKK7 and MEK1 in a TNFα-dependent manner, and disrupts their interactions with MEKK1, ASK1, and TAK1
The inhibition of the MEKK1-induced MEK1, MKK4, and MKK7 phosphorylation by c-FLIPL prompted us to investigate whether c-FLIPL interacts with MAPKKs. Intriguingly, c-FLIPL interacted with MEK1 and MKK7, and their interactions were significantly enhanced in the presence of MEKK1ΔN (Figure 5A and D). These results suggest that the MEKK1-induced phosphorylation and/or conformational changes of MEK1 and MKK7 might enhance their binding to c-FLIPL. Similarly, MKK4 weakly interacted with c-FLIPL, but MKK6 did not (Figure 5B and C). We tested whether c-FLIPL could interact with MKK7 and MEK1 under more physiological conditions. We first used RelA−/− MEFs stably expressing Flag-c-FLIPL. Interactions of c-FLIPL with endogenous MKK7 and MEK1 were undetectable in unstimulated cells, but their interactions were weakly induced at 5 min and increased up to 2 h after TNFα stimulation (Figure 5E). Notably, the interactions of MKK7 or MEK1 with Flag-c-FLIPL were specific, as anti-Flag, but not control antibody, specifically co-immunoprecipitated MKK7 and MEK1. More importantly, the interaction of endogenous c-FLIPL with endogenous MKK7 was induced upon TNFα stimulation in HEK293 cells (Figure 5F). Together, c-FLIPL physically interacts with MKK7 and MEK1 in a TNFα-dependent manner.
Figure 5.
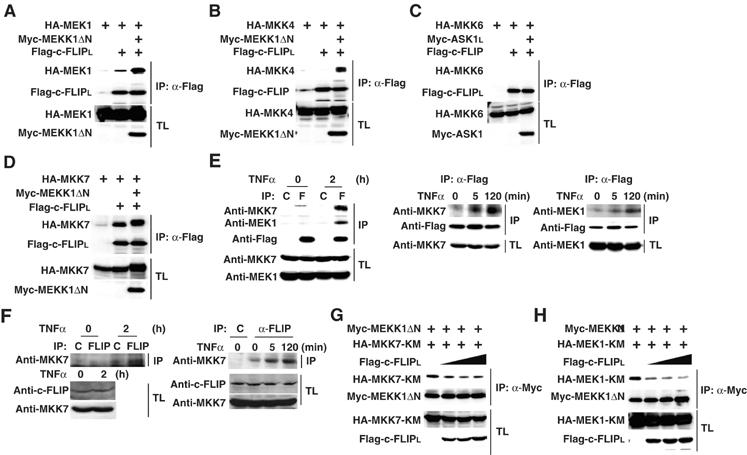
Interactions of c-FLIPL with MEK1 and MKK7 are induced in a TNFα-dependent, and c-FLIPL disrupts their interactions with MEKK1. (A–D) HEK293 cells were transfected with the indicated expression vectors. After immunoprecipitation (IP) with anti-Flag antibody, co-immunoprecipitated MAPKKs were detected by immunoblotting with anti-HA antibody (top panels). Expression levels of the transfected proteins in the immunoprecipitates (IP) (second panels) and the total lysates (TL) (third and bottom panels) were analyzed by immunoblotting with anti-Flag, anti-HA, and anti-Myc antibodies. (E) RelA−/− MEFs stably expressing Flag-c-FLIPL were stimulated with TNFα (10 ng/ml) for the indicated times. After immunoprecipitation with control (C) or anti-Flag (F) antibody, co-immunoprecipitated MKK7 and MEK1 were detected by immunoblotting with anti-MKK7 and anti-MEK1 antibodies, respectively. Expression levels of Flag-c-FLIPL in the immunoprecipitates (IP), MKK7 and MEK,1 in the total lysates (TL) were analyzed by immunoblotting with the indicated antibodies. (F) HEK293 cells were stimulated with TNFα (10 ng/ml) for the indicated times. After immunoprecipitation with control (C) or anti-c-FLIP antibodies, co-immunoprecipitated MKK7 was detected by immunoblotting with anti-MKK7 antibody (top panel). Expression levels of c-FLIPL and MKK7 in the total lysates (TL) (second and bottom panels) were analyzed by immunoblotting with the indicated antibodies. (G, H) HEK293 cells were transfected with the indicated expression vectors. After immunoprecipitation (IP) with anti-Myc antibody, co-immunoprecipitated MKK7-KM or MEK1-KM were detected by immunoblotting with anti-HA antibody (top panels). Expression levels of the transfected proteins were analyzed as in (A).
We next investigated the mechanism whereby c-FLIPL inhibits the MKK7–JNK and MEK1–ERK pathways. One possible scenario is that the binding of c-FLIPL to MKK7 or MEK1 might disrupt the interaction of MKK7 or MEK1 with MEKK1, thereby suppressing the MEKK1-induced MKK7 or MEK1 activation. A previous study has shown that once MEKK1 binds to and phosphorylates MKK4, MEKK1 rapidly dissociates with the phosphorylated MKK4 (Xia et al, 1998). Consistently, we could not detect the interaction of the WT MKK7 with MEKK1 in co-immunoprecipitation experiments (data not shown). As we previously observed the stable interaction of a kinase-inactive mutant MKK7 (MKK7-KM) with MEKK1 in co-transfection experiments (Takekawa et al, 2005), we tested whether c-FLIPL inhibits the interaction of MKK7-KM with MEKK1. Transfection of c-FLIPL inhibited the interaction of MKK7-KM with MEKK1ΔN in a dose-dependent manner (Figure 5G). Similarly, c-FLIPL inhibited the interaction of MEK1-KM with MEKK1ΔN (Figure 5H). Collectively, c-FLIPL inhibits the MKK7–JNK and MEK1–ERK pathways by disrupting the interactions of MKK7 or MEK1 with MEKK1. We also found that c-FLIPL inhibited the interaction of MKK7-KM with ASK1 or TAK1 (Supplementary Figure S4). We could not detect the interaction of c-FLIPL with ASK1 or MEKK1 (data not shown).
The C-terminal caspase-like domain of c-FLIPL is responsible for the binding to MKK7
We determined the functional domain of c-FLIPL that binds to and suppresses the MKK7–JNK pathway. We constructed expression vectors for c-FLIPs and c-FLIPΔN lacking the N-terminal DEDs. We investigated whether these mutants interact with MKK7. While both c-FLIPL and c-FLIPΔN efficiently interacted with MKK7, c-FLIPs did not (Figure 6B). Consistently, expression of c-FLIPΔN, but not c-FLIPs inhibited the MEKK1ΔN-induced MKK7 and JNK phosphorylation (Figure 6C and D), indicating that the binding of c-FLIPL to MKK7 is tightly correlated with the inhibitory effect of c-FLIPL on the MKK7–JNK pathway. To further delineate the binding region of c-FLIPL to MKK7, we constructed a series of deletion mutants of c-FLIPL and tested their ability to interact with MKK7 (Figure 6A). Notably c-FLIPLΔC(−220) interacted with MKK7, but could not inhibit the MKK7–JNK pathway (Figure 6E–G). Taken that c-FLIPLΔC(−153) bound to and inhibited MKK7 activation, the residues 203–260 are sufficient for the binding of c-FLIPL to MKK7, but additional residues 261–327 are required for inhibition of the MKK7–JNK pathway. Consistent with this notion, the residues 261–327 was required to inhibit the interaction of MEKK1 with MKK7, as c-FLIPLΔC(−220) lost the ability to inhibit the binding of MEKK1 to MKK7 (Figure 6H).
Figure 6.
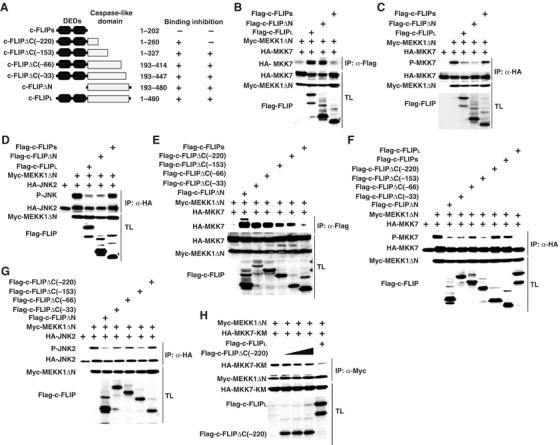
An intimate correlation between binding of c-FLIPL to MKK7 and inhibition of the MKK7–JNK pathway. (A) Schematic diagrams of deletion mutants of c-FLIPL, and the summary of their binding and inhibition experiments. (B, E) HEK293 cells were transfected with the indicated expression vectors. After immunoprecipitation (IP) with anti-Flag antibody, co-immunoprecipitated MKK7 was detected by immunoblotting with anti-HA antibody (top panels). Expression levels of the transfected proteins were analyzed as in Figure 5A. (C, D, F, G) HEK293 cells were transfected with the indicated expression vectors. After immunoprecipitation (IP) with anti-HA antibody, the phosphorylation of MKK7 (C, F) or JNK (D, G) was analyzed by immunoblotting with anti-phospho-JNK or anti-phospho-MKK7 antibodies (top panels). Expression levels of the transfected proteins were analyzed as in Figure 4A. (H) HEK293 cells were transfected with the indicated expression vectors. After immunoprecipitation (IP) with anti-Myc antibody, co-immunoprecipitated MKK7-KM was detected by immunoblotting with anti-HA antibody (top panels). Expression levels of the transfected proteins were analyzed as in Figure 5A.
Reconstitution of c-FLIPL, but not c-FLIPs, completely inhibits TNFα-induced ROS accumulation, JNK activation, and cell death in c-Flip−/− MEFs
To show directly a role for the C-terminal caspase-like domain of c-FLIPL in suppression of the JNK pathway under more physiological conditions, we reconstituted c-Flip−/− MEFs with c-FLIPL or c-FLIPs. While TNFα could not induce prolonged JNK activation in c-FLIPL transfectant, JNK activation gradually increased at 2 h and continued at 4 h after TNFα stimulation in c-FLIPs transfectant (Figure 7A). Similarly, TNFα-induced ROS accumulation was induced in c-FLIPs, but not c-FLIPL transfectants (Figure 7B). Reconstitution of c-FLIPL or c-FLIPs substantially increased cell viability of these transfectants compared to mock transfectant; however, c-FLIPL more efficiently protected cells from TNFα-induced cell death (Figure 7C). Together, the C-terminal caspase-like domain is required for c-FLIP-dependent survival signals through suppression of the JNK pathway and ROS accumulation.
Figure 7.
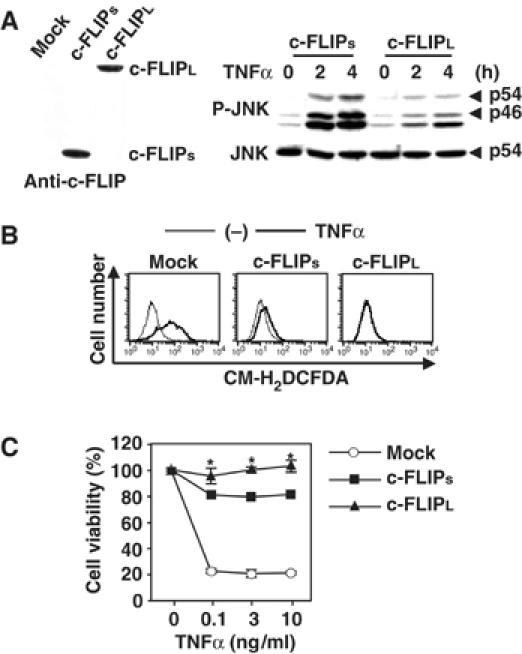
c-FLIPL more efficiently suppresses TNFα-induced ROS accumulation, prolonged JNK activation, and cell death than c-FLIPs. (A) c-FLIPL, but not c-FLIPs, completely inhibits TNFα-induced prolonged JNK activation in c-Flip−/− MEFs. Total cell lysates from mock, c-FLIPs, or c-FLIPL transfectants were immunoblotted with anti-c-FLIP antibody. c-FLIPs and c-FLIPL transfectants were stimulated with TNFα (10 ng/ml) for the indicated times, and then the lysates were analyzed as in Figure 1D. (B) c-FLIPL, but not c-FLIPs, completely inhibits TNFα-induced ROS accumulation in c-Flip−/− MEFs. The transfectants were unstimulated (thin line) or stimulated (bold line) with TNFα (10 ng/ml) for 2 h (for mock transfectant) or 8 h (for c-FLIP transfectants), then accumulated ROS were analyzed as in Figure 1C. (C) c-FLIPL, but not c-FLIPs, completely inhibits TNFα-induced cell death in c-Flip−/− MEFs. Mock or c-FLIP transfectants were stimulated with the indicated amounts of TNFα for 16 h. Cell viability was determined by WST assay as in Figure 1E. *P<0.05 compared to c-FLIPs transfectant.
Discussion
In the present study, we have demonstrated an essential role for c-FLIPL in suppression of TNFα-induced ROS accumulation and prolonged JNK activation using NF-κB activation-deficient and c-Flip−/− MEFs. c-FLIPL and c-FLIPs have been shown to bind directly to FADD and caspase-8 via DEDs, thereby inhibiting death receptor-mediated apoptosis (Goltsev et al, 1997; Irmler et al, 1997; Shu et al, 1997; Yeh et al, 2000). In addition to this well-known antiapoptotic function of c-FLIP, our present study has revealed that c-FLIPL, but not c-FLIPs, directly interacts with MKK7 and downregulates JNK activation. c-FLIPL might inhibit the JNK pathway through suppression of the interactions of MKK7 with MAPKKKs, including MEKK1, ASK1, and TAK1 (Figure 5 and Supplementary Figure S4). Together, c-FLIPL has two distinct functions, one is inhibition of the caspase pathway via the N-terminal DED, and the other is suppression of the JNK pathway via the C-terminal caspase-like domain.
Although our present study has revealed that c-FLIPL-dependent suppression of the JNK pathway is to disrupt the interaction of MKK7 with MAPKKKs, it is also likely that c-FLIPL might suppress the MKK7-induced JNK activation. Consistent with this possibility, we observed that c-FLIPL inhibited JNK activation induced by a constitutive active form MKK7, MKK7-3E, in which two serines and one threonine at the activation loop motif were mutated to glutamic acids (Holtmann et al, 1999) (Supplementary Figure S5). Moreover, our preliminary study showed that the binding domain of MKK7 to c-FLIPL was mapped to the N-terminal region of the MKK7 (data not shown), which has been shown to be responsible for the interaction with JNK (Tanoue et al, 2000). This suggests that c-FLIPL disrupts the binding of MKK7 to JNK. Further study will be required to address this possibility. Taken that the interaction of c-FLIPL with MKK7 is induced in a TNFα-dependent manner (Figure 5E and F), c-FLIPL might selectively downregulate the prolonged phase, but not the early phase JNK activation (Figure 1D). Taken that the interactions of c-FLIPL with MKK7 or MEK1 were weakly detected at 5 min after stimulation (Figure 5E and F), these early and weak interactions might not be sufficient for inhibiting MKK7 and MEK1 activation. Consistently, TNFα-induced JNK and ERK activation at 10 min were not different between WT and c-Flip−/− MEFs, whereas JNK and ERK activation were enhanced at 1 and 2 h after TNFα stimulation in c-Flip−/− MEFs compared to WT MEFs (Figure 2B).
Previous studies have shown that impaired upregulation of antioxidant enzyme genes including fhc and Mnsod is responsible for TNFα-induced ROS accumulation in NF-κB activation-deficient cells (Pham et al, 2004; Kamata et al, 2005). Then, accumulated ROS inactivate MKPs, resulting in prolonged JNK activation (Kamata et al, 2005). However, this scenario does not appear to be the case of c-Flip−/− MEFs, as expression of fhc and Mnsod mRNAs were not significantly different between WT and c-Flip−/− MEFs up to 2 h after TNFα stimulation (Figure 3B). Taken that MKPs dephosphorylate and inactivate MAPKs, but not MAPKKs, these results suggest that, in addition to ROS-dependent inactivation of MKPs, another mechanism might operate to induce prolonged MEK1 and possibly MKK7 activation in c-Flip−/− MEFs. Consistent with this notion, c-FLIPL inhibited activation of MEK1 and MKK7 (Figure 4 and Supplementary Figure S4).
A previous study has shown that GADD45β, which is induced in an NF-κB-dependent manner, binds to MKK7 and inhibits MKK7's kinase activity as competitive inhibition of ATP (Papa et al, 2004a). Taken that both c-FLIPL and GADD45β bind to MKK7, it is interesting to test whether c-FLIPL could affect the interaction of GADD45β with MKK7. However, under our experimental conditions, we could not detect a substantial interaction of GADD45β with MKK7 in HEK293 cells (data not shown). One possible explanation to explain this discrepancy is that the interaction of GADD45β with MKK7 might take place in a cell-type-specific manner. Consistently, GADD45β-dependent JNK inhibition is predominant in T cells, but not MEFs (Amanullah et al, 2003; Papa et al, 2004a). Therefore, the relation between the interactions of MKK7 with GADD45β and c-FLIPL is currently unknown and will be investigated in the future study. Collectively, c-FLIPL and GADD45β might cooperatively or independently inhibit the JNK pathway in a context- and cell type-dependent manner (Figure 8A).
Figure 8.
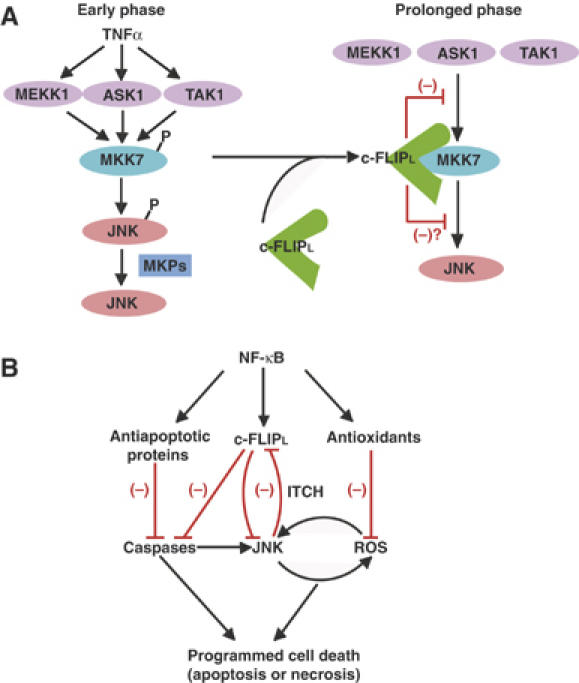
NF-κB-dependent survival signals. (A) A model for c-FLIPL-dependent suppression of the prolonged activation of the MKK7–JNK pathway. Upon TNFα stimulation, MEKK1, ASK1, and TAK1 phosphorylate and activate MKK7, which in turn activates JNK. MKPs subsequently dephosphorylate JNK, resulting in termination of the activation of JNK. In addition to this mechanism, the interaction of c-FLIPL with MKK7 is induced in a TNFα-dependent manner. This interaction inhibits the prolonged activation of the MKK7–JNK pathway by disrupting the interactions of MKK7 with MAPKKKs. Thus, c-FLIPL selectively inhibits the prolonged, but not early phase JNK activation. (B) NF-κB upregulates antioxidants and antiapoptotic proteins including MnSOD, FHC, GADD45β, XIAP, and c-FLIPL. Antiapoptotic proteins and antioxidants inhibit the caspase pathway and ROS accumulation, respectively. c-FLIPL plays a crucial role for suppression of the caspase-dependent and -independent pathways leading to JNK activation and ROS accumulation. In contrast, the prolonged JNK activation promotes degradation of c-FLIPL through activation of a ubiquitin ligase, ITCH. The delicate balance of these pathways could determine cell survival or cell death, including apoptosis and necrosis. The black and red lines indicate the positive and negative signals, respectively.
Chang et al (2006) have recently reported that JNK phosphorylates and activates a ubiquitin ligase, ITCH, that subsequently interacts with the caspase-like domain of c-FLIPL and promotes degradation of c-FLIPL. In this respect, the fact that c-FLIPL, but not c-FLIPs, binds to and inhibits JNK activation is particularly interesting (Figure 6). Under normal conditions, expression levels of c-FLIPL are sufficient for inhibition of the caspase cascade and the JNK pathway. However, once the expression levels of c-FLIPL are downregulated due to various pathological conditions, JNK activation is gradually induced. This prolonged JNK activation further promotes degradation of c-FLIPL in an ITCH-dependent manner (Chang et al, 2006). This vicious cycle along with downregulation of antioxidants and other antiapoptotic proteins ultimately induces caspase activation and/or ROS accumulation, resulting in cell death through apoptosis or necrosis (Figure 8B).
Given that c-FLIPL does not appear to have an antioxidant function, but directly binds to MKK7 and inhibits JNK activation (Figures 4 and 5), c-FLIPL might inhibit ROS accumulation through suppression of the JNK pathway. Consistently, a previous study has shown that TNFα-induced ROS accumulation is abolished in Jnk−/− MEFs (Ventura et al, 2004). Taken that ROS promote JNK activation, activation of the JNK pathway may induce ROS accumulation, which in turn amplifies JNK activation. c-FLIPL might block this positive feedback loop, thereby suppressing ROS accumulation in NF-κB activation-deficient cells. To address the possibility, it is essential to generate c-Flip−/−Jnk−/− mice. However, we cannot formally exclude the possibility that c-FLIPL directly suppresses ROS accumulation in an undetermined mechanism. In addition to this mechanism, c-FLIP also suppresses caspase-dependent ROS accumulation (Figure 2C). The caspase-dependent ROS accumulation was observed in c-Flip−/− MEFs and c-FLIP-knockdown tumor cell lines, such as HeLa, A549, and HCT116 cells, but not NF-κB activation-deficient cells (Figure 2C, Supplementary Figure S2A, and data not shown). One possible explanation is that under these conditions, in which c-FLIP is lost before stimulation, caspase activation is rapidly induced, resulting in caspase-dependent ROS accumulation along with caspase-independent ROS accumulation (Figure 2A and C). In contrast, in NF-κB activation-deficient cells, expression levels of c-FLIPL decrease upon TNFα stimulation (Figure 1). Under these conditions, caspase activation is gradually and weakly induced (Supplementary Figure S2C), and this gradually and weakly increased caspase activation is not sufficient for promoting ROS accumulation. Therefore, synergistic interplay between the JNK pathway and ROS accumulation may play a dominant role in ROS accumulation (Ventura et al, 2004). Taken that BHA, but not z-VAD-fmk, almost completely inhibited TNFα-induced ROS accumulation in NF-κB activation-deficient cells (Sakon et al, 2003) (Supplementary Figure S2A), BHA may specifically inhibit caspase-independent ROS accumulation. Further study will be required to elucidate the interplay between caspase-dependent and -independent ROS accumulation.
Upregulation of c-FLIPL has been shown to be correlated with resistance to Fas-induced apoptosis in vitro in certain tumor cell lines and also associated with autoimmune diseases (Thome and Tschopp, 2001). One of the strategies to treat these pathological conditions is to block the c-FLIPL-mediated suppression of caspase activation, thereby promoting apoptosis of tumor cells and autoreactive T cells. In addition, blocking the suppressive function of c-FLIPL on JNK activation might be an alternative strategy to treat these diseases, resulting in prolonged JNK activation-induced cell death. In this respect, the interaction of c-FLIPL with MKK7 might be an attractive target to develop new drugs to treat various diseases such as cancers and autoimmune diseases.
Materials and methods
Plasmids
pcDNA3-HA-MEK1, pcDNA3-HA-MEK1-KM, and pcDNA3-HA-MKK6 were described previously (Takekawa et al, 2005). pCR-HA-MKK4, pCR-HA-MKK7, and pCR-HA-JNK2 were generated by cloning PCR-amplified products from pCR-MKK4, pCR-MKK7, and pET-SAPKα (JNK2) (provided by E Nishida) as templates into pCR-HA vector, respectively. pCR-HA-MKK7-KM (K165M) was generated by using an in vitro mutagenesis kit (Stratagene). pCR-HA-p38 and pCR-HA-ERK2 were generated by cloning RT–PCR products of p38 and ERK2 into pCR-HA vector, respectively. pcDNA3-Myc-TAK1, pcDNA3-Myc-TAB1, pcDNA3-Myc-ASK1, and pcDNA3-Myc-Tlp-2 were provided by K Matsumoto, H Ichijo, and S Ley. An expression vector for a constitutively active MEKK1 (pcDNA3-Myc-MEKK1ΔN) was constructed by deleting N-terminal regulatory region of MEKK1 (1–1119) by PCR from pSRHis-MEKK1 (provided by S Ohno). pCR-Flag-c-FLIPL and pCR-Flag-c-FLIPs were generated by cloning PCR-amplified products from pcDNA3-CASHα and pcDNA3-CASHβ (provided by D Wallach) into pCR-Flag vector, respectively. pCR-Flag-c-FLIPΔN was constructed by PCR to delete the N-terminal DEDs. A series of deletion mutants of c-FLIPL was generated by PCR to delete the indicated amino acids from the C-terminus.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Data
Acknowledgments
We thank D Wallach, E Nishida, H Ichijo, K Matsumoto, S Ohno, S Ley, Y Gotoh, H Nishina, T Kitamura, M Kracht, P Holland, GP Nolan, and T Doi for providing reagents, and RelA−/− MEFs. This work was supported in part by Grants-in-Aid for 21st Century COE Research and Scientific Research (B) from Japan Society for the Promotion of Science, Japan, Takeda Science Foundation, and by a Grant from Human Frontier Science Program (HFSP). AN is a Research Fellow of the Japan Society for the Promotion of Science.
Competing financial interests The authors declare that they have no competing financial interests.
References
- Amanullah A, Azam N, Balliet A, Hollander C, Hoffman B, Fornace A, Liebermann D (2003) Cell signalling: cell survival and a Gadd45-factor deficiency. Nature 424: 741; discussion 742 [DOI] [PubMed] [Google Scholar]
- Chang L, Kamata H, Solinas G, Luo JL, Maeda S, Venuprasad K, Liu YC, Karin M (2006) The E3 ubiquitin ligase itch couples JNK activation to TNFα-induced cell death by inducing c-FLIP(L) Turnover. Cell 124: 601–613 [DOI] [PubMed] [Google Scholar]
- Chen YR, Wang X, Templeton D, Davis RJ, Tan TH (1996) The role of c-Jun N-terminal kinase (JNK) in apoptosis induced by ultraviolet C and γ radiation. Duration of JNK activation may determine cell death and proliferation. J Biol Chem 271: 31929–31936 [DOI] [PubMed] [Google Scholar]
- Davis RJ (2000) Signal transduction by the JNK group of MAP kinases. Cell 103: 239–252 [DOI] [PubMed] [Google Scholar]
- De Smaele E, Zazzeroni F, Papa S, Nguyen DU, Jin R, Jones J, Cong R, Franzoso G (2001) Induction of gadd45β by NF-κB downregulates pro-apoptotic JNK signalling. Nature 414: 308–313 [DOI] [PubMed] [Google Scholar]
- Ghosh S, Karin M (2002) Missing pieces in the NF-κB puzzle. Cell 109 (Suppl): S81–S96 [DOI] [PubMed] [Google Scholar]
- Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S, Marcaccio M, Pinton P, Rizzuto R, Bernardi P, Paolucci F, Pelicci PG (2005) Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 122: 221–233 [DOI] [PubMed] [Google Scholar]
- Golks A, Brenner D, Krammer PH, Lavrik IN (2006) The c-FLIP-NH2 terminus (p22-FLIP) induces NF-κB activation. J Exp Med 203: 1295–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goltsev YV, Kovalenko AV, Arnold E, Varfolomeev EE, Brodianskii VM, Wallach D (1997) CASH, a novel caspase homologue with death effector domains. J Biol Chem 272: 19641–19644 [DOI] [PubMed] [Google Scholar]
- Guo YL, Baysal K, Kang B, Yang LJ, Williamson JR (1998) Correlation between sustained c-Jun N-terminal protein kinase activation and apoptosis induced by tumor necrosis factor—α in rat mesangial cells. J Biol Chem 273: 4027–4034 [DOI] [PubMed] [Google Scholar]
- Holtmann H, Winzen R, Holland P, Eickemeier S, Hoffmann E, Wallach D, Malinin NL, Cooper JA, Resch K, Kracht M (1999) Induction of interleukin-8 synthesis integrates effects on transcription and mRNA degradation from at least three different cytokine- or stress-activated signal transduction pathways. Mol Cell Biol 19: 6742–6753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J (1997) Inhibition of death receptor signals by cellular FLIP. Nature 388: 190–195 [DOI] [PubMed] [Google Scholar]
- Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M (2005) Reactive oxygen species promote TNFα-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 120: 649–661 [DOI] [PubMed] [Google Scholar]
- Karin M, Lin A (2002) NF-κB at the crossroads of life and death. Nat Immunol 3: 221–227 [DOI] [PubMed] [Google Scholar]
- Kataoka T, Budd RC, Holler N, Thome M, Martinon F, Irmler M, Burns K, Hahne M, Kennedy N, Kovacsovics M, Tschopp J (2000) The caspase-8 inhibitor FLIP promotes activation of NF-κB and Erk signaling pathways. Curr Biol 10: 640–648 [DOI] [PubMed] [Google Scholar]
- Kataoka T, Tschopp J (2004) N-terminal fragment of c-FLIP(L) processed by caspase 8 specifically interacts with TRAF2 and induces activation of the NF-κB signaling pathway. Mol Cell Biol 24: 2627–2636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuz S, Siegmund D, Scheurich P, Wajant H (2001) NF-κB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol Cell Biol 21: 3964–3973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J (2001) Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev 81: 807–869 [DOI] [PubMed] [Google Scholar]
- Luo JL, Kamata H, Karin M (2005) IKK/NF-κB signaling: balancing life and death—a new approach to cancer therapy. J Clin Invest 115: 2625–2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J (2001) NF-κB signals induce the expression of c-FLIP. Mol Cell Biol 21: 5299–5305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito M, Katayama R, Ishioka T, Suga A, Takubo K, Nanjo M, Hashimoto C, Taira M, Takada S, Takada R, Kitagawa M, Matsuzawa S, Reed JC, Tsuruo T (2004) Cellular FLIP inhibits β-catenin ubiquitylation and enhances Wnt signaling. Mol Cell Biol 24: 8418–8427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagiri S, Murakami A, Takada S, Akiyama T, Yonehara S (2005) Viral FLIP enhances Wnt signaling downstream of stabilized β-catenin, leading to control of cell growth. Mol Cell Biol 25: 9249–9258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano H, Nakajima A, Sakon-Komazawa S, Piao JH, Xue X, Okumura K (2006) Reactive oxygen species mediate crosstalk between NF-κB and JNK. Cell Death Differ 13: 730–737 [DOI] [PubMed] [Google Scholar]
- Nishitoh H, Saitoh M, Mochida Y, Takeda K, Nakano H, Rothe M, Miyazono K, Ichijo H (1998) ASK1 is essential for JNK/SAPK activation by TRAF2. Mol Cell 2: 389–395 [DOI] [PubMed] [Google Scholar]
- Papa S, Zazzeroni F, Bubici C, Jayawardena S, Alvarez K, Matsuda S, Nguyen DU, Pham CG, Nelsbach AH, Melis T, Smaele ED, Tang WJ, D'Adamio L, Franzoso G (2004a) Gadd45β mediates the NF-κB suppression of JNK signalling by targeting MKK7/JNKK2. Nat Cell Biol 6: 146–153 [DOI] [PubMed] [Google Scholar]
- Papa S, Zazzeroni F, Pham CG, Bubici C, Franzoso G (2004b) Linking JNK signaling to NF-κB: a key to survival. J Cell Sci 117: 5197–5208 [DOI] [PubMed] [Google Scholar]
- Pham CG, Bubici C, Zazzeroni F, Papa S, Jones J, Alvarez K, Jayawardena S, De Smaele E, Cong R, Beaumont C, Torti FM, Torti SV, Franzoso G (2004) Ferritin heavy chain upregulation by NF-κB inhibits TNFα-induced apoptosis by suppressing reactive oxygen species. Cell 119: 529–542 [DOI] [PubMed] [Google Scholar]
- Ricci JE, Munoz-Pinedo C, Fitzgerald P, Bailly-Maitre B, Perkins GA, Yadava N, Scheffler IE, Ellisman MH, Green DR (2004) Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell 117: 773–786 [DOI] [PubMed] [Google Scholar]
- Sakon S, Xue X, Takekawa M, Sasazuki T, Okazaki T, Kojima Y, Piao JH, Yagita H, Okumura K, Doi T, Nakano H (2003) NF-κB inhibits TNF-induced accumulation of ROS that mediate prolonged MAPK activation and necrotic cell death. EMBO J 22: 3898–3909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, Matsumoto K, Takeuchi O, Akira S (2005) Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol 6: 1087–1095 [DOI] [PubMed] [Google Scholar]
- Shu HB, Halpin DR, Goeddel DV (1997) Casper is a FADD- and caspase-related inducer of apoptosis. Immunity 6: 751–763 [DOI] [PubMed] [Google Scholar]
- Takekawa M, Tatebayashi K, Saito H (2005) Conserved docking site is essential for activation of mammalian MAP kinase kinases by specific MAP kinase kinase kinases. Mol Cell 18: 295–306 [DOI] [PubMed] [Google Scholar]
- Tang G, Minemoto Y, Dibling B, Purcell NH, Li Z, Karin M, Lin A (2001) Inhibition of JNK activation through NF-κB target genes. Nature 414: 313–317 [DOI] [PubMed] [Google Scholar]
- Tanoue T, Adachi M, Moriguchi T, Nishida E (2000) A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat Cell Biol 2: 110–116 [DOI] [PubMed] [Google Scholar]
- Thome M, Tschopp J (2001) Regulation of lymphocyte proliferation and death by FLIP. Nat Rev Immunol 1: 50–58 [DOI] [PubMed] [Google Scholar]
- Ventura JJ, Cogswell P, Flavell RA, Baldwin AS Jr, Davis RJ (2004) JNK potentiates TNF-stimulated necrosis by increasing the production of cytotoxic reactive oxygen species. Genes Dev 18: 2905–2915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Wu Z, Su B, Murray B, Karin M (1998) JNKK1 organizes a MAP kinase module through specific and sequential interactions with upstream and downstream components mediated by its amino-terminal extension. Genes Dev 12: 3369–3381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh WC, Itie A, Elia AJ, Ng M, Shu HB, Wakeham A, Mirtsos C, Suzuki N, Bonnard M, Goeddel DV, Mak TW (2000) Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity 12: 633–642 [DOI] [PubMed] [Google Scholar]
- Yuasa T, Ohno S, Kehrl JH, Kyriakis JM (1998) Tumor necrosis factor signaling to stress-activated protein kinase (SAPK)/Jun NH2-terminal kinase (JNK) and p38. Germinal center kinase couples TRAF2 to mitogen-activated protein kinase/ERK kinase kinase 1 and SAPK while receptor interacting protein associates with a mitogen-activated protein kinase kinase kinase upstream of MKK6 and p38. J Biol Chem 273: 22681–22692 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Data