

Abstract
Organisms rely heavily on protein phosphorylation to transduce intracellular signals. The phosphorylation of a protein often induces conformational changes, which are responsible for triggering downstream cellular events. Protein kinases are themselves frequently regulated by phosphorylation. Recently, we and others proposed the molecular mechanism by which phosphorylation at a hydrophobic motif (HM) regulates the conformation and activity of many members of the AGC group of protein kinases. Here we have developed specific, low molecular weight compounds, which target the HM/PIF-pocket and have the ability to allosterically activate phosphoinositide-dependent protein kinase 1 (PDK1) by modulating the phosphorylation-dependent conformational transition. The mechanism of action of these compounds was characterized by mutagenesis of PDK1, synthesis of compound analogs, interaction-displacement studies and isothermal titration calorimetry experiments. Our results raise the possibility of developing drugs that target the AGC kinases via a novel mode of action and may inspire future rational development of compounds with the ability to modulate phosphorylation-dependent conformational transitions in other proteins.
Keywords: AGC kinase, PDK1, PIF-pocket, protein conformation, protein phosphorylation
Introduction
Fifty years after its discovery, protein phosphorylation is the most widely studied intracellular regulatory mechanism (Pawson and Scott, 2005). Phosphorylation of proteins often induces conformational changes with physiological outcomes, such as increased or decreased activity of enzymes. Phosphorylation-mediated conformational transitions are likely to be of general relevance, as it is estimated that up to one-third of cellular proteins are phosphorylated. Protein phosphorylation is catalyzed by protein kinases, which transfer the terminal phosphate from an nucleotide triphosphate (NTP) (generally ATP) to substrate proteins. In fact, protein kinases are often regulated by phosphorylation, which triggers conformational changes in their catalytic domains (Huse and Kuriyan, 2002). As deregulation of protein kinases can lead to disorders such as cancer (Blume-Jensen and Hunter, 2001), they have emerged as one of the major groups of drug targets in the pharmaceutical industry (Cohen, 2002).
We and others have previously gained insight into the biochemical, molecular and structural aspects of the mechanism by which a family of protein kinases termed AGC kinases are regulated via phosphorylation within a hydrophobic motif (HM, Phe-Xaa-Xaa-Phe-Ser/Thr(P)-Tyr), which is usually located 45–60 residues C-terminal to the protein kinase catalytic core (Etchebehere et al, 1997; Parker and Parkinson, 2001; Pearl and Barford, 2002; Biondi and Nebreda, 2003; Newton, 2003). In AGC kinases, the HM phosphorylation site acts in concert with the ‘activation loop' phosphorylation site to stabilize the active conformation. The mechanism by which HM phosphorylation triggers activation relies on the docking of the phosphorylated HM to a particular HM-binding pocket in the protein kinase catalytic domain. The HM-binding pocket was first defined in the cAMP-dependent protein kinase (PKA) structure (Knighton et al, 1991) (Figure 1A). In the phosphoinositide-dependent protein kinase 1 (PDK1) the pocket was characterized as a regulatory site and was termed the ‘PIF-pocket' (Biondi et al, 2000). In PDK1, the HM/PIF-pocket docks the HM of substrate protein kinases, for example, p90 ribosomal S6 kinase (RSK), 70 S6 kinase (S6K) and serum- and glucocorticoid-induced protein kinase (SGK) only when they are phosphorylated. This interaction not only provides docking for the substrates but it also activates PDK1 to enable phosphorylation, and hence activation of RSK, S6K and SGK (Frodin et al, 2000; Biondi et al, 2001; Collins et al, 2003). The equivalent HM/PIF-pocket was subsequently found to be a regulatory site in many AGC kinases (Frodin et al, 2002; Yang et al, 2002). Significantly, inactive structures of the AGC kinases protein kinase B/Akt (PKB) and mitogen and stress-activated protein kinase (MSK) show that the HM-pocket is disturbed; two of its lining walls, the conserved α-C helix and the α-B helix, are either disordered or replaced by an unusual β-sheet (Yang et al, 2002; Huang et al, 2003; Smith et al, 2004). Concomitant to this change in the HM/PIF-pocket, the structure of the ATP-binding site in the inactive catalytic domains of AGC kinases is significantly different from that observed in the active kinase. In order to activate the kinases, the binding of the phosphorylated HM sequence (P-HM) to the HM/PIF-pocket regulatory site must bring about a conformational change, which directly involves the HM/PIF-pocket and allosterically affects the ATP-binding site. Thus, the inactive–active transition involves a phosphorylation-dependent conformational change (Figure 1D). The model of allostery between the active site and the regulatory site suggests that the interaction of a phosphorylated HM to the HM/PIF-pocket activates PDK1 by stabilizing the α-C helix in the active form. In this process, a conserved Glu residue (Glu130 in PDK1) correctly positions a key active site Lys residue (Lys111 in PDK1) which directly interacts with the phosphate from ATP (Biondi et al, 2002; Pearl and Barford, 2002; Biondi, 2004). Biochemical and structural studies on PDK1 and AGC kinases have defined a phosphate-binding site next to the HM/PIF-pocket (Biondi et al, 2002; Frodin et al, 2002). This phosphate-binding site is responsible for triggering the binding of the HM to the HM/PIF-pocket only when it is phosphorylated. However, overexpression of PDK1 can trigger the phosphorylation of substrates, which lack the HM sequences (Frodin et al, 2002), indicating a subtle regulation of the interaction. For this reason, the requirement of the HM/PIF-pocket in PDK1 for intermolecular interactions with substrates in vivo has been studied using knock-in experiments, where the levels of PDK1 are kept at physiological levels (Collins et al, 2003, 2005). On the other hand, the regulation by HM phosphorylation in AGC kinases can be viewed as a particular example of an induced intramolecular interaction regulated by the presence of the phosphate.
Figure 1.
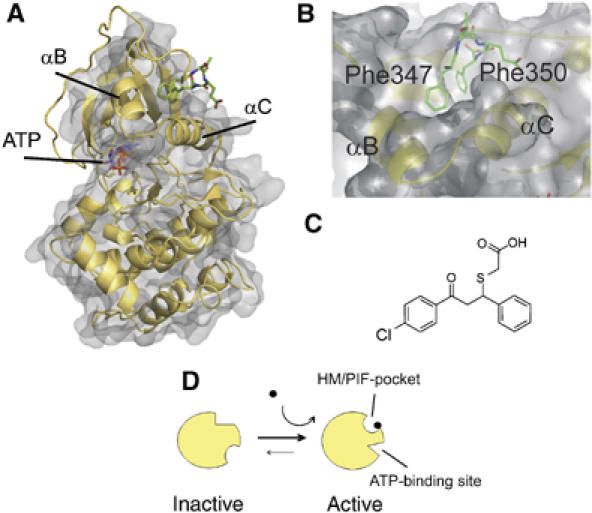
C-terminal hydrophobic motif and the PIF-pocket of PKA. Structure of compound mimicking the phosphorylated HM. (A) The PKA catalytic domain surface structure is shown with underlying ribbon representation. The C-terminal Phe-X-X-Phe-COOH residues and the ATP molecule are shown as sticks. (B) Closeup representation of the HM/PIF-pocket. α-Helices lining the pocket (αB and αC) are indicated. (C) Structure of compound 1. (D) Scheme of AGC protein kinase activation by phosphorylation. The red circle represents protein phosphorylation. The represented activation of the kinase involves conformational changes in the HM/PIF-pocket and in the ATP-binding site, as described for PKB and MSK.
Other well known molecular mechanisms of regulation by phosphorylation employ a similar intramolecular phosphorylation-dependent interaction strategy. Such is the case of tyrosine kinases (Sicheri et al, 1997; Xu et al, 1997) and the glycogen synthase protein kinase 3 (GSK3) (Dajani et al, 2001; Frame et al, 2001), where phosphorylation at a site outside the catalytic domain triggers the intra-molecular binding of the phosphorylated sequence to a phosphate-binding site in the catalytic domain and the regulation of GSK3 activity. Although the molecular events that trigger the conformational changes by phosphorylation in most proteins are not known (Johnson and Lewis, 2001), it is expected that an analogous mechanism involving intra- or intermolecular phosphorylation-dependent docking may promote conformational changes in a larger range of proteins.
In this study, we present the rational design and characterization of small molecules that, binding to the HM/PIF-pocket regulatory site in PDK1, allosterically activate the kinase by mimicking the conformational transitions physiologically triggered by phospho-peptide docking. These findings open a novel field in the development of small modulators of protein kinase activity. In addition, our work may have implications for modulating conformational transitions in other proteins.
Results
Discovery of small molecules that increase PDK1 activity
Phosphorylation can sometimes be mimicked by replacement of the phosphorylatable residue with an acidic residue. This occurs physiologically in the HM of the protein kinase C related protein kinase 2 (PRK2), from where the 24-amino-acid polypeptide PIFtide is derived. PIFtide can activate both PDK1 and PKB with high potency (Biondi et al, 2000; Yang et al, 2002). This suggested to us that other chemical groups, distinct from phosphate, may mimic the required interactions and trigger the allosteric conformational changes. In addition, PIFtide has considerably higher affinity for PDK1 than any other HM and P-HM tested (Biondi et al, 2001). However, a relatively large polypeptide comprising the complete hydrophobic motif present in PIFtide (GFRDFDY) did not activate PDK1 at concentrations up to 500 μM (data not shown). Therefore, in order to test if small compounds can trigger the phosphorylation-dependent conformational changes in AGC kinases, we next evaluated whether nonpeptide small molecules could be suitable as allosteric modulators of PDK1.
As a first step in the rational design of small compounds to mimic the phosphorylation-dependent transition, we compared the structure of the PDK1 HM/PIF-pocket with that of the closed, active conformation of PKA. The HM/PIF-pocket contains two subpockets, where the two Phe residues from the HM dock (Figure 1A and B). In the PDK1 structure, one of these sub-pockets appeared significantly diminished in depth owing to the positioning of Phe157 (PDB 1H1W) (Biondi et al, 2002). In addition, even in the presence of ATP, PDK1 crystallized in an ‘intermediate' form, with active site residues not positioned correctly for catalysis. Therefore, to enable the development of compounds to mimic the active form of AGC kinases, we decided to use the closed, active structure of PKA and, in particular, the C-terminal HM to define a pharmacophore model. We performed in silico screening of a chemical library consisting of 60 000 low-molecular-weight compounds and selected compounds predicted to possess features similar to Phe347 and Phe350 within the HM of PKA crystal structure (1ATP; Figure 1B).
Depending on the parameters imposed, between 250 and 2500 different compounds were identified in the in silico screenings. The selected compounds were visually evaluated and 220 compounds were further tested in vitro. The results revealed that two compounds significantly increased the intrinsic activity of PDK1 toward a polypeptide substrate that comprises the activation loop residues of PKB, known as T308tide (Biondi et al, 2000). As a control, these two compounds did not modify the activity of PDK1 in the presence of an excess of the HM polypeptide, PIFtide. Based on the structure and the common characteristics of the hits, we further evaluated related small compounds with different chemical scaffolds. We selected compound 1 (Figure 1C) for further characterization because, in comparison to other active compounds, it possessed a low AC50, produced high maximal activation levels and appeared relatively specific towards PDK1.
Activation of PDK1 by P-HM polypeptides and compound 1 requires the presence of a negative charge
The initial compounds that activated PDK1 possessed a carboxyl group. Therefore, we first characterized the requirement of the carboxyl on compound 1 and compared the results to the requirement of the phosphate on P-HM polypeptides. PDK1 was activated by polypeptides P-HM-PKB and P-HM-RSK, derived from the phosphorylated HMs of AGC kinases PKB and p90 RSK, respectively (Figure 2A). The corresponding nonphosphorylated polypeptides did not activate PDK1, confirming that the activation of PDK1 by the HM of substrates was dependent on the phosphorylation of the HM. Similarly to P-HM polypeptides, compound 1 activated PDK1 with an AC50 of 25 μM (Figure 2B and Table II, R4=H). In contrast, a compound bearing a methyl-ester instead of a free carboxyl group (compound 2) was inactive across a wide range of concentrations (Figure 2B and Table II, R4=CH3), suggesting that the carboxylate ion was necessary, possibly mimicking contacts physiologically achieved by the phosphate moiety, which is also negatively charged.
Figure 2.
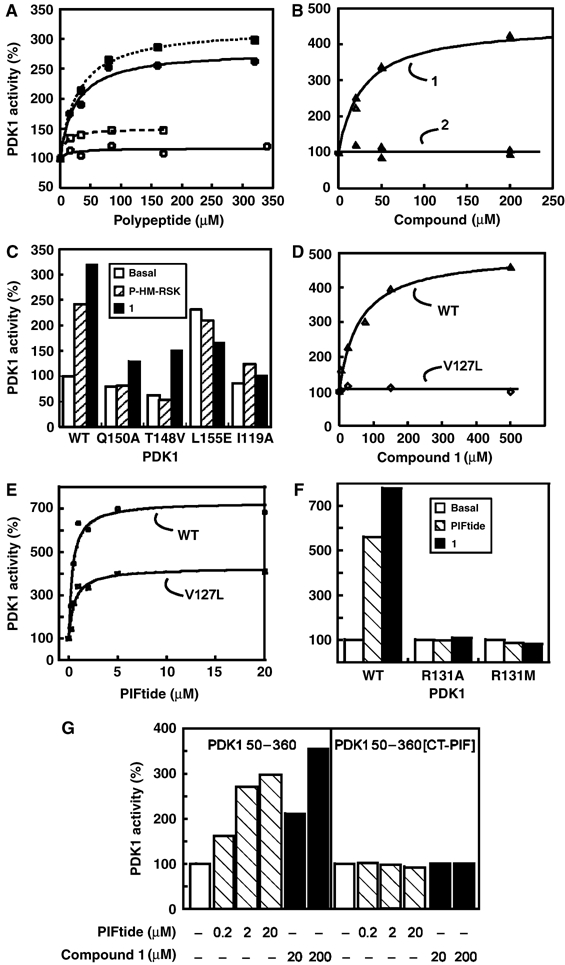
Activation of PDK1 and mutants by phosphopeptides and small compounds designed to target the HM/PIF-pocket. The intrinsic activity of purified PDK1 and the indicated mutants were measured in vitro using the polypeptide T308tide as a substrate in the presence or absence of the indicted P-HM polypeptides, PIFtide or small compounds. (A) Effect of phosphopeptides (closed symbols), dephosphopeptides (open symbols), derived from the HM of PKB (squares) or RSK (circles) on the specific activity of PDK1. (B) Effect of compound 1,comprising a carboxyl group (open triangles), and its methyl-ester form, compound 2 (closed triangles), on PDK1 activity. (C) Effect of P-HM-RSK and compound 1 (both at 20 μM) on PDK1 WT and PIF-pocket PDK1 mutants [Gln150Ala], [Thr148Val], [Leu155Glu] and [Ile119Ala]. (D) Compound 1 activated PDK1 WT (open triangles) but not PDK1[Val127Leu] (open rhombi). (E) PDK1[Val127Leu] retained the ability to be activated by PIFtide with similar AC50 as PDK1 WT. Basal activity and maximal activation levels varied in different preparations of both of these enzymes. (F) Arg131, which forms part of the PDK1 P-HM-binding site adjacent to the HM/PIF-pocket, is required for the activation by PIFtide and compound 1. (G) The activity of a PDK1 50–360[CT-PIF] chimera is not affected by PIFtide or compound 1. PDK1 50–360[CT-PIF] comprises the catalytic domain of PDK1 joined to the C-terminal residues from protein kinase PRK2, comprising the PDK1-interacting fragment (PIF).
Table 2.
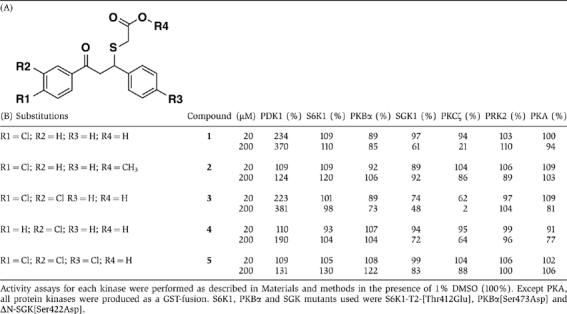
Specificity of low-molecular-weight compounds toward PDK1 and a panel of AGC kinases. (A) Basic scaffold indicating positions of R1, R2, R3 and R4; and (B) Effect of R1, R2, R3 and R4 on the activity of small compounds
 |
Activation of PDK1 by compounds is abolished by mutations in the HM/PIF-pocket
We then evaluated the mode of action of compound 1 by analyzing how its activating properties were affected by a series of mutations in the HM/PIF-pocket and surrounding residues. Table I summarizes the results obtained with PDK1 mutants. As described previously (Biondi et al, 2000), most PDK1 proteins mutated along the HM/PIF-pocket showed only partially reduced binding to PIFtide and could still be activated by PIFtide, although they required higher concentrations for maximal activation. We also tested the effect of P-HM peptides and small compounds on PDK1 proteins mutated at different positions within the HM/PIF-pocket (Figure 2C). Mutation of Gln150, Thr148 or Ile119 completely abolished the activation of PDK1 by the phosphorylated polypeptides P-HM-RSK (20 μM) and P-HM-PKB (Figure 2C and not shown). On the other hand, as expected from the interaction with a much smaller compound, most PIF-pocket mutants were still activated by compound 1 (e.g., mutants PDK1[Gln150Ala] and PDK1[Thr148Val]). Constitutively active PDK1 mutants, in which the hydrophobic residue Leu155 is replaced with Glu or Ser (Biondi et al, 2000), could not be further activated by P-HM peptides nor by compound 1 (Figure 2C and Table I).
Table 1.
Effect of mutations in PDK1 on basal activity and activation by HM polypeptide PIFtide and compound 1
| Basal activity (U/mg) | (%) | PIFtide | Compound 1 | |||
|---|---|---|---|---|---|---|
| AC50 (μM) | Max. activity (U/mg) | AC50 (μM) | Max. activity (U/mg) | |||
| GST-PDK1 WT | 0.2 | 100 | 0.11±0.03 | 0.9±0.05 | 34±9 | 1.1±0.07 |
| GST-PDK1 [Ile119Ala] | 0.65 | 331 | 0.66±0.14 | 2.58±0.17 | 90±20 | 2.1±0.23 |
| GST-PDK1 [Val124Ala] | 0.48 | 241 | 0.21±0.03 | 1.91±0.06 | >200 | 2.6±0.5 |
| GST-PDK1 [Val124Leu] | 0.4 | 200 | 0.35±0.2 | 0.59±0.04 | 180±60 | 1.1±0.13 |
| GST-PDK1 [Val127Leu] | 0.6 | 301 | 0.12±0.03 | 0.99±0.08 | No effect | — |
| GST-PDK1 [Val127Thr] | 1.2 | 597 | 0.14±0.07 | 2.36±0.15 | 104±58 | 1.69±0.2 |
| GST-PDK1 [Arg131Ala] | 0.72 | 358 | 3.7±2 | 1.24±0.8 | >200 | 1.09±0.06 |
| GST-PDK1 [Arg131Met] | 0.2 | 101 | 0.29±0.05 | 0.34±0.06 | No effect | — |
| GST-PDK1 [Arg131Lys] | 0.3 | 149 | 0.13±0.02 | 0.77±0.02 | 51±15 | 1.13±0.09 |
| GST-PDK1 [Ser135Ala] | 0.89 | 443 | 0.18±0.02 | 3.4±0.09 | 109±33 | 2.82±0.4 |
| GST-PDK1 [Thr148Val] | 0.59 | 295 | INH | NA | 102±19 | 2.44±0.26 |
| GST-PDK1 [Gln150Ala] | 0.3 | 150 | 0.5±0.06 | 1.55±0.06 | 55±9 | 1.12±0.06 |
| GST-PDK1 [Gln150Glu] | 0.57 | 285 | No effect | — | >200 | 2.5±0.7 |
| GST-PDK1 [Gln150Lys] | 0.36 | 182 | 0.29±0.05 | 2.7±0.2 | 15±4 | 1.37±0.06 |
| GST-PDK1 [Gln150Met] | 0.61 | 305 | 0.86±0.25 | 2.11±0.15 | 177±64 | 2.28±0.26 |
| GST-PDK1 [Leu155Glu] | 0.60 | 300 | No effect | — | No effect | — |
| GST-PDK1 [Leu155Ser] | 2.75 | 915 | No effect | — | No effect | — |
| GST-PDK1 [Leu155Val] | 1.46 | 732 | No effect | — | No effect | — |
| GST-PDK1 [Leu155Ala] | 1.05 | 525 | 3.7±0.66 | 3.46±0.15 | >200 | 2.02±0.61 |
| GST-PDK1 [Phe157Leu] | 0.7 | 349 | 0.32±0.19 | 1.4±0.14 | ND | ND |
| GST-PDK1 [Phe157Met] | 0.5 | 250 | 1.52±0.59 | 1.7±0.24 | 140±100 | 0.9±0.3 |
| GST-PDK1 [Lys115Met] | 0.32 | 160 | No effect | — | >200 | NA |
| PDK1 50–360 | 0.65 | 325 | 0.17±0.09 | 2.62±0.18 | 38±8 | 3.46±0.19 |
| PDK1 50–360 [CT-PIF] | 1.02 | 510 | No effect | — | No effect | — |
| PDK1 activity was measured at room temperature using a polypeptide substrate derived from the activation loop phosphorylation site of PKB, T308tide (0.1 mM). The listed data indicate the basal activity of the kinases (U/mg), their relative activity as a comparison to GST-PDK1 WT (%), AC50 (μM) as well as the maximal activity in the presence of PIFtide and compound 1 (U/mg). GST-PDK1 WT was purified in numerous occasions and the maximal activation level of different batches varied between three- and eight-fold activation with PIFtide or compound 1. Variations in the basal activity and fold activation were also observed in mutants. Therefore, comparisons in the maximal level of activation between mutants should be performed with care. The data shown are from an experiment where PIFtide concentrations were tested between 50 nM and 20 μM and compound 1 concentrations were between 5 and 200 μM, in triplicate. AC50 and max. activity were estimated by fitting the data to a hyperbola using the Kaleidagraph software. One unit of PDK1 activity was defined as the amount required to catalyze the phosphorylation of 1 nmol of the T308tide in 1 min. ND, not determined; no effect, addition of PIFtide or compound 1 did not have any effect on the basal activity (−) of the mutant kinase; INH, the addition of PIFtide to this mutant inhibited its activity; NA, the data could not be accurately estimated within the concentrations tested. | ||||||
Mutation of Val127 (at the base of the HM/PIF-binding pocket) to Leu abolished activation by small compounds (Figure 2D). On the other hand, PDK1[Val127Leu] was activated by PIFtide and P-HM-RSK (Figure 2E), with an AC50 similar to that of the wild-type (WT) protein. Val127 is a nonconserved residue forming part of the base of the HM/PIF-pocket in PDK1. In PKA, its equivalent is Thr88, which is located at the base of the Phe347 subpocket (Figure 1B). Replacement of Val127 by Thr generated a PDK1 HM/PIF-pocket mutant that had increased specific activity and could be further activated by PIFtide (260%) and compound 1 (240%), suggesting that a Thr at this position did not abolish the ability of compounds to bind and activate the kinase. Altogether, the results suggested that compound 1 targeted the intended site, the HM/PIF-pocket in PDK1, and that the identity of residues forming the hydrophobic pocket could determine the ability of compounds to activate PDK1. Thus, our results provide evidence that the residues forming part of the HM/PIF-pocket could provide specificity to the compounds.
PDK1 Arg131 is required for activation by compound 1
Structural and biochemical studies have previously defined the residues that form the phosphate-binding site responsible for docking the P-HM onto PDK1 (Biondi et al, 2002; Frodin et al, 2002). Thus, the crystal structure of PDK1 defined a sulfate-binding site next to the HM/PIF-pocket, comprising Arg131, Thr148, Lys76 and Gln150. Biochemical analyses of PDK1 mutants identified Gln150 and Arg131 as the most important residues that allowed binding and promoted activity of PDK1 by the phosphorylated HM polypeptides. As described above (Figure 2C), PDK1[Gln150Ala] and PDK1[Thr148Val] were still activated by compound 1, suggesting that the carboxylate group from compound 1 did not make any specific interactions with these residues. We next examined if the positively charged Arg131 was required for the activation by compound 1. Indeed, when Arg131 was mutated to Met or Ala, the resulting PDK1 mutants were no longer activated by compound 1 (Figure 2F). In order to evaluate if the guanidinium group from Arg131 was required for the activation by compound 1, we generated a PDK1 mutant with a Lys residue in place of Arg131. Activation of PDK1[Arg131Lys] by small compounds was unaffected (Table I). The results indicated that the activation of the enzyme by compound 1 required the positive charge at the position of Arg131.
The activity of a PDK1 50–360[CT-PIF] chimera is not affected by compound 1
We generated a PDK1 chimera consisting of the catalytic domain of PDK1 comprising residues 50–360 joined to the last 48 amino acids from PRK2, which include the sequence of PIFtide (PDK1 50–360[CT-PIF]). PIFtide binds with high affinity to PDK1. Therefore, we expected that the HM/PIF-pocket in this protein would be strongly bound to the C-terminus of PRK2. In this scenario, the specific activity of the PDK1 50–360[CT-PIF] chimera would not be affected by compounds that otherwise would bind to the PDK1 50–360 HM/PIF-pocket. The PDK1 50–360[CT-PIF] chimera had 1.5-fold higher specific activity than the PDK1 catalytic domain alone. Furthermore, the activity of PDK1 50–360[CT-PIF] chimera was not affected by PIFtide at concentrations that activated the PDK1 50–360 protein (Figure 2G). Finally, PDK1 50–360[CT-PIF] specific activity was not affected by compound 1, further supporting the notion that PIFtide and compound 1 targeted an overlapping site in the catalytic domain of PDK1.
Compound 1 blocks the interaction of PDK1 with the HM polypeptide PIFtide
We next used surface plasmon resonance to test the ability of compound 1 to compete with the interaction between glutathione-S-transferase (GST)-PDK1 1–556 to PIFtide. We first probed the binding of PDK1 to the biotin-PIFtide-coated chip at different PDK1 concentrations (Figure 3A). The interaction of PDK1 with biotin-PIFtide indicated a KD of approximately 65 nM. Preincubation of PDK1 with unlabelled PIFtide abolished this interaction (Figure 3B), verifying that the PDK1–biotin-PIFtide interaction was specific. Therefore, in order to allow a sensitive measurement of any displacement of binding by small compounds, the competition experiments were performed at a PDK1 concentration of 45 nM. Under the established conditions, compound 1 was able to disrupt the binding of PDK1 to PIFtide in a concentration-dependent manner (Figure 3C). By contrast, the ester form (compound 2) did not displace the binding significantly, suggesting that it did not interact with PDK1 at the HM/PIF-pocket site with comparable affinities. These experiments further confirmed that compound 1 interacted with the PIF-binding pocket in PDK1. Furthermore, these results suggested that the lack of activity of compound 2 may be due to its inability to bind the HM/PIF-pocket on PDK1.
Figure 3.
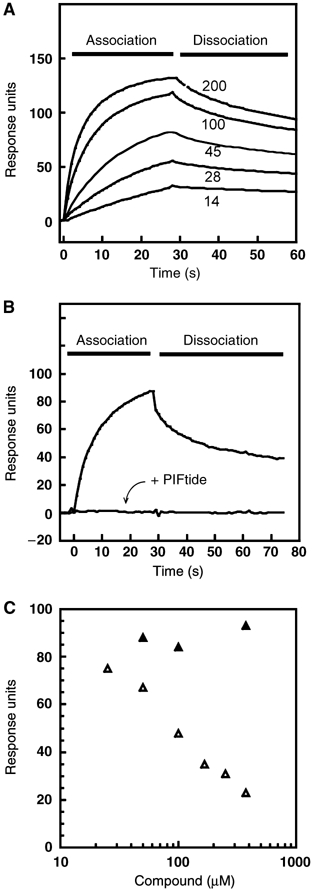
Small compounds displace the binding of PDK1 1–556 to the HM peptide derived from its substrate PRK2, PIFtide. Surface plasmon resonance measurements were carried out on a BiaCore system to measure the interaction between GST-PDK1 and biotin-PIFtide coupled to a streptavidin-coated chip. (A) Direct measurement of PDK1 interaction with biotin-PIFtide. PDK1 was injected at the indicated concentrations (nM). For the following competition experiments, PDK1 45 nM was used. (B) PDK1 interaction with biotin-PIFtide was abolished by preincubation with PIFtide. (C) Compound 1 (open triangles) displaced the interaction of PDK1 with biotin-PIFtide whereas compound 2 (closed triangles), which lacks the carboxylate group, did not affect the binding of PDK1 to the chip. The data points represent the maximal response units obtained after 30 s injection in the presence of the indicated concentration of compound. In (A) and (B), the spikes at the start and end of injection have been eliminated for clarity.
PDK1–compound 1 interaction studied by isothermal titration calorimetry (ITC)
The interaction of compound 1 with PDK1 was further studied using ITC (Figure 4). ITC experiments indicated that compound 1 bound to PDK1 CD with a 1:1 stoichiometry, with a binding affinity in the micromolar range (KD=18 μM). This value was broadly in agreement with activity assays, although it was lower than the estimated AC50. The association reaction was enthalpy and entropy favored and driven by the entropy term (ΔH/ΔG=32%), characteristic of large hydrophobic contributions. This observation is consistent with the binding of compound 1 to the HM/PIF-pocket.
Figure 4.
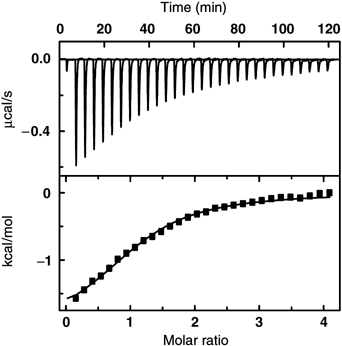
Characterization of compound 1 interaction with PDK1 50–360 by ITC. Upper panel, raw data of the titration of compound 1 into PDK1 50–360. Lower panel, integrated heats of injection, corrected for the heat of dilution, with the solid line corresponding to the best fit of the data using OriginTM software. The best fit corresponded to a model with one type of sites. The parameters defining the fitted curve are KD=18+2 μM, ΔH=−2.1+0.1 kcal/mol, TΔS=4.3+0.2 kcal/mol and N=1.0+0.1.
The identity of R1, R2 and R3 substituents in compound 1 confer specificity toward PDK1
In order to confirm that the compound action on PDK1 was specific, we then synthesized a series of related compounds containing chlorine substituents at different positions (Table IIA). Compounds with chlorine at R1, or in both R1 and R2 positions were capable to activate PDK1, whereas compounds with chlorine only at R2 or in the three positions R1, R2 and R3 had greatly reduced activity towards PDK1 (Table II, first column). This indicated that substitutions in R1, R2 and R3 conferred specificity for PDK1. Substitutions in R1 and R2 were permissive and the resulting compounds were active towards PDK1. In sharp contrast, the substitution at the second benzyl ring (R3) greatly decreased the compound's ability to activate PDK1. Thus, the specific location of the chlorine substitutions in analog compounds had markedly different effects on PDK1 and provided evidence of a specific interaction.
Compound 1 does not affect the intrinsic activity of related AGC kinases
As the initial compounds were selected in silico based on properties of the HM-pocket that are common between different members of the AGC kinase family, we evaluated the protein kinase specificity of compound 1 by testing its effect on other AGC kinases (Table IIB). At a concentration of 20 μM, compound 1 activated PDK1, but had no significant effect on the AGC kinases PKBα/AKT1, SGK1, PRK2, protein kinase Cζ (PKCζ), S6K or PKA. At higher concentrations, compound 1 did not increase the activity of any other AGC kinase tested. On the other hand, its methyl-ester form (compound 2), without the negative charge, did not affect significantly the activity of any protein kinase tested. Interestingly, a variation of component with chlorine at position R2 (compound 4) or R1 and R2 (compound 3) not only retained the ability to activate PDK1 but also significantly inhibited other protein kinases in the panel, such as SGK and PKCζ. In contrast, compound 5, with chlorine at R1, R2 and R3 did not affect the activity of any protein kinase in the panel (Table IIB). We conclude that the compounds developed to mimic the conformational transition in AGC kinases can be produced with considerable selectivity toward one AGC kinase family member.
Compound 1 blocks the phosphorylation and activation of its substrates S6K and SGK, which require the HM/PIF-pocket for docking
SGK and S6K are substrates of PDK1 that require the docking of their phosphorylated HM to the PDK1 HM/PIF-pocket to trigger their phosphorylation and activation by PDK1 (Biondi et al, 2001; Collins et al, 2003). Thus, based on those studies, it was expected that compounds that specifically target the HM/PIF-pocket in PDK1 would inhibit S6K and SGK phosphorylation and activation by PDK1. Therefore, we tested the effect of compound 1 on the phosphorylation and activation of S6K and SGK constructs containing a negatively charged residue in place of the HM phosphorylation site (Thr412Glu and Ser422Asp, respectively). These kinases are activated by PDK1 phosphorylation. Compound 1 decreased the PDK1-catalyzed phosphorylation of S6K1 (Figure 5A), in agreement with the hypothesis that compound 1 bound to the HM/PIF-pocket in PDK1. In addition, compound 1 inhibited SGK1 activation by PDK1. In contrast, SGK activation by PDK1[Val127Leu] was not inhibited by compound 1 (Figure 4B), suggesting that the inhibitory effect was not due to an interaction between compound 1 and SGK, but rather due to the binding of compound 1 to the hydrophobic HM/PIF-pocket of PDK1.
Figure 5.
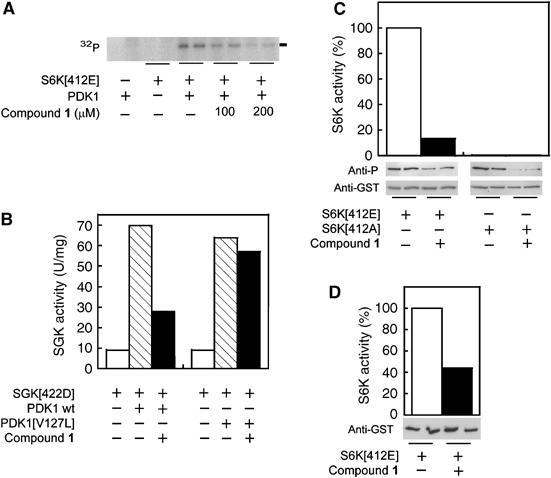
Small compounds designed to bind the HM/PIF-pocket of PDK1 can block the PDK1-dependent phosphorylation and activation of substrates that require docking to the HM/PIF-pocket of PDK1. (A) Effect of compound 1 (100 and 200 μM) on PDK1 phosphorylation of its substrate S6K1. The phosphorylation reaction was performed in vitro with [γ-32P]ATP and then separated on SDS–PAGE followed by exposure to PhosphoImager. The radioactive band corresponding to phosphorylated GST-S6K1-T2[Thr412Glu] (S6K[412E]) is indicated. (B) PDK1 or PDK1[Val127Leu] (PDK1[V127L]) were preincubated with GST-ΔN-SGK1[Ser422Asp] (SGK[422D]) in the presence or absence of compound 1 (60 μM), followed by the addition of SGK substrate to measure its activity. (C, D) Effect of compound 1 on the activation loop phosphorylation and activity of S6K mutants transfected into HEK293 cells. Cells overexpressing GST-S6K1-T2[Thr412Glu] and the inactive mutant S6K1-T2[Thr412Ala] (S6K[412E]) were treated for 90 min with 200 μM compound 1 or vehicle (DMSO), followed by cell lysis. (C) The activity of S6K was measured in vitro after glutathione sepharose purification; aliquots of purified proteins were separated on SDS–PAGE and the level of activation loop phosphorylation was visualized by immunoblotting using phospho-specific antibodies that recognize the phosphorylated activation loop, followed by stripping of the membranes and reprobing with anti-GST to verify the presence of equal amounts of protein. (D) Cells overexpressing GST-S6K1-T2[Thr412Glu] were treated as in (C), but S6K activity was measured in vitro after immunoprecipitation of the GST-fusion protein with anti-GST antibodies; the level of S6K expression was analyzed by Western blotting of crude extracts. In (C) and (D), the duplicates are from independent transfections performed in parallel.
We also attempted to verify if compound 1 could have a similar effect on S6K phosphorylation in a cell assay approach. To this end, we transfected HEK293 cells with plasmids encoding for GST-S6K-T2[Thr412Glu] and GST-S6K-T2[Thr412Ala] and purified the fusion proteins using a glutathione–sepharose resin. In agreement with the in vitro data, treatment of cells with 200 μM of compound 1 caused a decrease of S6K1-T2[Thr412Glu] activity, which correlated with a decrease in the phosphorylation at the activation loop (Figure 4C). We also tested the effect of compound 1 on S6K1-T2[Thr412Ala], an inactive mutant which also interacts with PDK1, albeit with lower affinity (Biondi et al, 2001). Again, we observed that the incubation of the cells with compound 1 significantly decreased the activation loop phosphorylation (Figure 4C). A similar decrease in the activity of GST-S6K-T2[Thr412Glu] was observed when the kinase assay was performed using anti-GST immunoprecipitates (Figure 4D). GST-S6K-T2[Thr412Glu] activity was inhibited in cells with an IC50 of 80 μM (not shown). Preliminary studies designed to measure the ability of compound 1 to permeate through an artificial lipid membrane (PAMPA assay) suggested that compound 1 would not readily enter cells. Thus, it is possible that upon treatment of cells with compound 1, the intracellular concentrations were lower than the concentrations applied; alternatively, internalization may be due to an active transport, which may influence the in vivo IC50. Altogether, the results suggested that the PDK1 HM/PIF-binding pocket is available for interaction with a small-molecular-weight compound within the environment of the cell.
Discussion
Previous work suggested a molecular model to explain the molecular mechanism of activation of AGC kinases by HM phosphorylation. We concluded that the essence of the mechanism involves a ligand binding to the regulatory site, and therefore, we here further studied the allosteric mechanism of regulation and explored if the essential aspects of the regulation could be mimicked by small molecules, which, in the future, could be developed into orally available drugs. Based on the molecular model of activation of AGC kinases and in silico analysis of the HM/PIF-pocket structure, we have now developed small-molecular-weight compounds that activate PDK1. Mutagenesis at the center of the HM/PIF-pocket (e.g., Leu155Glu and Val127Leu) blocked the effect of the compounds on PDK1, suggesting that the HM/PIF-pocket is the target site for compound 1. The ability of compounds to activate PDK1 also required the positive charge from Arg131, which forms part of the associated phosphate-binding site that physiologically interacts with the phosphate within the HM phosphorylation site from substrates (Collins et al, 2005). The activation by compound 1 required the presence of the free carboxylate group, as compound 2, possessing a methyl-ester instead, lost the ability to bind and activate PDK1. These results supported the notion that compound 1 interacted with the HM/PIF-pocket and with the positive charge from Arg131 residue which forms the adjacent phosphate-binding site. This was further supported by surface plasmon resonance experiments, which showed that the small compounds can displace the binding between PDK1 and the HM polypeptide PIFtide. In further agreement with this notion, compound 1 did not affect the activity of a PDK1 chimera, which has the sequence of PDK1 linked to the C-terminal 48 residues from PRK2 that includes the 24 amino acids present in PIFtide. In addition, the compounds were able to block PDK1 phosphorylation of its substrates S6K and SGK, which depend on the docking to the HM/PIF-pocket of PDK1 for efficient phosphorylation and activation. Therefore, we conclude that the ability of compound 1 to activate PDK1 is related to its ability to interact specifically with the HM/PIF-pocket and the associated phosphate-binding site of PDK1. On the other hand, the specificity of the effect of compounds on PDK1 was verified by synthesizing compound analogs. Some of these analogs where inactive and unable to interact with PDK1; such marked structure–activity relationship argues for selectivity of compounds and helps to define the requirements to activate PDK1. Altogether our results provide extensive evidence that small compounds can modulate the conformational inactive–active transition of PDK1 by targeting the HM/PIF-pocket regulatory site (Figure 6A and B). To our knowledge, this is the first report of a rational development of small compounds that mimic a conformational transition physiologically induced by phosphorylation.
Figure 6.
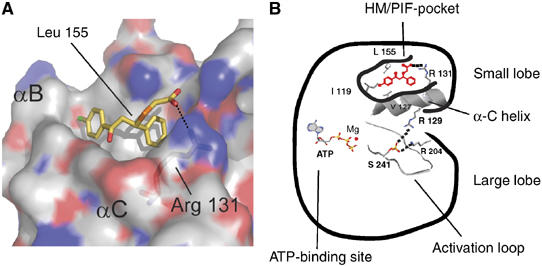
Model for compound 1 docking and activation of PDK1. (A) Model for compound 1 docking to the HM/PIF-binding pocket on PDK1. Molecular docking of compound 1 onto PDK1 crystal structure was unreliable. Therefore, the presented model was built on the basis of the mode of interaction of the C-terminal Phe347 and Phe350 to PKA, whereas the position of the carboxylate was further adapted to fit the biochemical results which suggest a specific interaction with the positive charge of Arg131. For this model, we have moved Phe157 out of the second Phe cavity by choosing another rotamer of Phe157. (B) Scheme for the activation of PDK1 by compound 1. The mechanism of activation of PDK1 by compound 1 involves the binding to the HM/PIF-pocket. We here describe Val127, Ile119, Leu155 and Arg131 as residues which are important for the activation of PDK1 by compound 1. Val127, Ile119 and Leu155 are expected to provide hydrophobic interactions to compound 1, whereas Arg131 appears to be responsible for the interaction with the carboxylate from compound 1. The activation process also requires the presence of the phosphate at the activation loop (Komander et al, 2005). As the mechanism of activation of AGC kinases appears highly conserved, the model for the activation of PDK1 by small compounds may be the starting point for the development of small compounds targeting AGC kinases by similar approaches.
We conclude that the conformational changes induced by phosphorylation of AGC kinases can be simplified to a system where a ligand binds to the HM/PIF-pocket regulatory site in a phosphorylation-dependent manner. Analogous mechanisms of intra- and intermolecular regulation by phosphorylation have been described in other systems, including glycogen phosphorylase (Johnson and Lewis, 2001), Tyr kinases (Gonfloni et al, 1997; Sicheri et al, 1997; Xu et al, 1997) and GSK3 (Dajani et al, 2001; Frame et al, 2001). Our success in the generation of small molecules that can activate PDK1 by mimicking the phosphorylation-dependent docking of substrates suggests that it may be possible to modulate phosphorylation-dependent conformational changes with small compounds in other systems.
The structure of the catalytic domain in active protein kinases is extremely conserved. In contrast, the inactive structures appear to be diverse. Thus, in the crystal structures of inactive PKB and MSK, the α-C helix is disturbed and the DFG motif is distinctly positioned as in the active structures (Yang et al, 2002; Smith et al, 2004). Thus, activation of PKB and MSK is thought to involve a large conformational transition. On the other hand, the crystal structure of the inactive PDK1 protein mutated in the activation loop phosphorylation site (Ser241Ala) has the α-C helix and the DFG motif in a similar position as in the active form (Komander et al, 2005). The authors concluded that the structure of PDK1 Ser241Ala represents the inactive structure of PDK1 and therefore the inactive–active transition may not involve similar conformational changes as thought for PKB or MSK. As a strong electron density was observed at the position equivalent of the phosphor-Ser241 in PDK1[Ser141Ala] crystal, it is also possible that this unidentified high-affinity binding ligand stabilized the active conformation of the α-C helix and the DFG motif in those crystals.
Our mutagenesis work further defines, at a molecular level, the regions of PDK1 that are required for the activation. Thus, a small molecule occupying a limited space in the HM/PIF-pocket defined by Ile119, Leu155, Val127 and Arg131 was able to activate PDK1. Mutations in these residues blocked activation by compound 1. It appears likely that the hydrophobic residues Ile119, Leu155 and Val127 form part of the hydrophobic pocket required for binding the phenyl groups of compound 1, whereas the positive charge on Arg131 may make specific electrostatic interactions with the carboxylate anion from compound 1 (Figure 6A). Mutations in Ile119, Leu155 and Arg131 also inhibited PIFtide and P-HM polypeptide binding and activation of PDK1 (Biondi et al, 2000, 2002). By contrast, other residues that were important for binding the HM polypeptide were found not to be essential for the activation process as they were not required for activation by compound 1. This was the case for Gln150, which plays a major role in PDK1 binding to PIFtide (Biondi et al, 2000; Komander et al, 2005). Thus, our work further highlights the HM/PIF-binding pocket as a minimum requirement for the modulation of the activity of PDK1.
Although the small compounds were able to mimic the conformational changes induced by phosphorylation of the HM from substrates, the small compounds were not able to mimic all the effects that the HM polypeptides exert on PDK1. Thus, while PDK1 stability to temperature is increased by 10°C in the presence of PIFtide (Biondi et al, 2000), compound 1 did not stabilize PDK1 to thermal denaturation (data not shown), suggesting that the larger set of interactions by PIFtide may be required for this increased stability. Importantly, we here demonstrate that all additional specific interactions set by physiological docking polypeptides outside the HM/PIF-pocket are not required for the activation of PDK1.
We predict that the HM/PIF-pocket regulatory site in other AGC kinases may also be targeted by small compounds, which could stabilize the inactive structures and inhibit the intrinsic activity of the enzymes. Interestingly, compounds 1 and 3 (Table II), which stimulated PDK1 intrinsic activity, inhibited PKCζ in our panel. In contrast, compounds 4 and 5, which possess single differences in the identity of substituents in R1 and R3, did not inhibit PKCζ in a comparable manner. It is thus possible that compounds 1 and 3 are allosteric inhibitors of PKCζ, which target the HM/PIF-pocket site.
Finally, compound 1 proved to be reasonably specific for PDK1 and did not affect significantly the activity of other AGC kinases. We here identified that mutation in two nonconserved residues, Ile119 and Val127, abolished activation by compound 1. In other AGC kinases, the equivalent residue to Val127 is Thr, Ile, Leu or Ala and the equivalent residue to Ile119 is Val, Leu, Asn or His. Thus, our initial characterization of the compound 1-binding site suggests that the pocket may be amenable to the development of specific compounds directed to other AGC kinases. In addition, the residue equivalent to Arg131 may not form part of a phosphate-binding site in other AGC kinases, as is the case of Lys429 in PKCθ (1XJD) (Xu et al, 2004). Future work should clarify if, in such cases, interaction of compounds with residues equivalent to Arg131 could also promote the activation of the respective AGC kinase.
In conclusion, the rational development of compound 1 proves that small-molecule compounds can provide all the necessary requirements to induce the conformational changes required for activation of AGC protein kinases, by interacting with the regulatory HM/PIF-pocket. However, a major challenge is to further develop such compounds into high-affinity, specific and active drugs at low concentrations. The activation induced by the small-molecular-weight compounds described here is physiologically achieved in AGC kinases by HM phosphorylation. The general mechanism by which the phosphorylation transduces into a conformational change in AGC kinases is a phosphorylation-dependent docking interaction of a regulatory sequence to the catalytic domain. A similar mechanism operates in other well-characterized examples of protein regulation by phosphorylation. Therefore, our work provides the first evidence that, through understanding the molecular mechanism by which a protein is regulated via phosphorylation, it may be possible to rationally design small-molecular-weight drugs to mimic the conformational states physiologically achieved by phosphorylation. Such compounds could have future applications in the treatment of disease by specifically modulating the phosphorylation-dependent conformation of a target protein.
Materials and methods
General materials and methods, the expression and purification of protein kinases, and the synthesis and characterization of compounds are described in the Supplementary data section.
Peptides
Polypeptides T308tide: KTFCGTPEYLAPEVRR; Crosstide: GRPRTSSFAEG; Kemptide: LRRASLG; HM-PIFtide: GFRDFDY and PIFtide: REPRILSEEEQEMFRDFDYIADWC, were synthesized by us and the purity verified by HPLC. Where required, the polypeptides were HPLC-purified. Polypeptides used were at least 75% pure; the identity of polypeptides was confirmed by N-terminal sequencing and mass spectrometry. P-HM-PKB: KGAGGGGFPQFS(P)YSA; HM-PKB: KGAGGGGFPQFSYSA; P-HM-RSK: KGAGGGGFRGFS(P)FVA and HM-RSK: KGAGGGGFRGFSFVA were obtained from ThermoHybaid, and identity verified by mass spectrometry. Biotinylation of PIFtide was performed by amine coupling using biotin-N-hydroxysuccinimide (Perbio) following the instructions provided by the manufacturer. Modified peptides were re-purified to over 90% purity on a Jupiter C5 HPLC column (Phenomenex) applying a linear gradient from 17% acetonitrile/0.1% trifluoroacetic acid/83% water to 70% acetonitrile/0.1% trifluoroacetic acid/30% water, lyophilized and kept at −20°C until use.
Protein kinase activity tests
Protein kinase activity tests were performed essentially as previously described (Balendran et al, 2000; Biondi et al, 2000, 2001). Substrates used were either MBP (for PKCζ) or polypeptides (Crosstide for PKB, S6K, SGK and PRK2; T308tide for PDK1, and Kemptide for PKA). PKA (Sigma) was measured in the presence of 100 μM cAMP. Alternatively, assays were performed in a 96-well format, aliquots spotted on p81 phosphocellulose papers (Whatmann), washed in 0.01% phosphoric acid, dried and then exposed and analyzed using PhosphoImager technology (Storm, Molecular Dynamics). Activity measurements were performed in duplicates with less than 10% difference between duplicate pairs. Experiments were repeated at least twice, although most of the experiments were repeated multiple times, with similar results.
PDK1 activity assay was performed in a 20 μl mix containing 50 mM Tris–HCl (pH 7.5), 0.05 mg/ml BSA, 0.1% β-mercaptoethanol, 10 mM MgCl2, 100 μM [γ-32P]ATP (5–50 c.p.m./pmol), 0.003% Brij, 150 ng PDK1 and T308tide (from 0.05 to 1 mM). We should note that the specific activity of PDK1 towards T308tide greatly depends on the peptide concentration as the Km toward T308tide is >10 mM (Biondi et al, 2000). For screening purposes, the concentration of T308tide in the assay and the specific activity of the ATP used was in the lower range, with the sole purpose of saving material. Linearity was ensured by performing the assays under conditions in which the product was less than 2% of the least concentrated substrate. All PDK1 kinetic analyses shown were performed with 0.1 mM T308tide as substrate. The effect of small compounds on PDK1 was repeated with proteins from different purification batches and with different protein constructs, comprising the full-length protein or the catalytic domain alone, with similar results. Nevertheless, some differences were observed in the maximal activation of PDK1 between different batches of the same PDK1 construct (3 × to 8 × maximal activation). This variability may be due to the PIFtide/compound-stimulated autophosphorylation of the activation loop in certain preparations having less than a 1:1 molar phosphorylation stoichiometry at this site.
Phosphorylation of S6K1-T2[T412E] and SGK1-ΔN[S422D] by PDK1 in vitro was performed as described previously (Biondi et al, 2001), with the sole difference that the reaction mix included 1% DMSO, and, where indicated, compound 1. In order to measure the effect of compound 1 on the activity and the level of activation loop phosphorylation of S6K1 in cells, pEBG2T vectors coding for GST-S6K1-T2[Thr412Glu] and GST-S6K1-T2[Thr412Ala] were transfected into HEK293 cells (3.5 cm dishes), and the cells serum starved during 16 h before treatment with compound 1 (200 μM). Cells were lysed 36 h after transfection, cleared by centrifugation and incubated with glutathione sepharose. The resin was washed two times with lysis buffer supplemented with 0.250 M NaCl, followed by two washes with a buffer containing 50 mM Tris–HCl, 0.1 mM EGTA and 0.1% β-mercaptoethanol. The GST-fusion proteins were eluted by the addition of glutathione 20 mM and the mix was cleared from resin by filtration through Spin-X tubes. The resulting proteins were used both for Western blotting and for activity measurements. Similar decrease in S6K activity was observed when the cells were treated with IGF1 for 20 min before cell lysis. Compound 1 in vivo IC50 ranged from 70 to 100 μM in three independent experiments. Activity assays of the immunoprecipitated S6K, in the presence of resin, were performed under agitation.
In silico screening of small-molecular-weight compound databases
The compound database screening was performed on the Maybridge (Tintagel) database of compounds. The commercial database contained, at the time, approx. 60 000 compounds (85% MW <400). The HM peptide of PKA bound in its pocket served as a model HM/PIF-binding pocket ligand and was used to define a minimum set of pharmacophoric features: the two phenyl rings corresponding to Phe347 and Phe350 of the PKA hydrophobic motif, and the oxygen atoms of the C-terminal carboxyl group. Distance constraints were set between the ring centers and the center of the C-terminal phenyl and the two carboxyl oxygens. In a second screening, spheres with van der Waals radii of 1 Å defined as exclusion volumes were included as an addition constraint in order to account for Leu116, which is located in the middle of the pocket. The screening was performed using the flexible search routine of Unity 4.3 (Tripos). The software adjusted conformations of the compounds in the database along the course of the search and thus treated compound flexibility to some extent. The screening yielded 2500 and 250 hits for the first and second screenings, respectively, without scoring. The screening resulted in the identification of chemical structures exhibiting at least two aromatic rings often connected by a three to nine atom membered chain and a variety of oxygen containing moieties such as carbonyl, sulfonyl, carbamoyl, amide, ester and carboxylic acid functions. This output was further refined by excluding redundant analogs of the same scaffold, compounds exhibiting bulky or polar substituents at the phenyl rings and compounds with more than four rotatable bonds connecting the rings. Two hundred and twenty compounds from the hits were tested in vitro.
Interaction of PDK1 with PIFtide and the displacement by small compounds
Binding of PDK1 to PIFtide was analyzed by surface plasmon resonance on a BiaCore 3000 system using a streptavidin-coated Sensor chip (SA) and biotin-PIFtide, as described previously (Biondi et al, 2001). Biotin-PIFtide was bound to the chip to a level of 15–25 response units in different experiments. GST-PDK1 bound to biotin-PIFtide with an affinity of 90 nM, in good agreement with previous data obtained using the BiaCore system. For interaction–displacement assays, GST-PDK1 (60 nM) was injected (30 μl/min) in a buffer containing 10 mM HEPES (pH 7.5), 150 mM NaCl, 0.005% Tween 20, 1 mM DTT and 1% DMSO, in the presence or absence of compounds. GST-PDK1 was preincubated with compounds for 1–10 min before injection into the system, with similar results. Experiments were performed at least twice using different biotin-PIFtide-coated chips, with similar results obtained on each occasion.
Isothermal titration calorimetry
The calorimetric titration was performed using the VP-ITC instrument from MicroCal Inc. (Northampton, MA, USA) as described previously (Schaeffer et al, 2002) with the following modifications. PDK1 and compound 1 were prepared in 50 mM Tris–HCl (pH 7.5), 175 mM NaCl, 1 mM DTT and 3% DMSO. Titration was performed by 30 successive injections (10 μl) of compound 1 (0.9 mM) into a 1.41 ml reaction cell containing PDK1 50–360 (50 μM). Raw calorimetric data were corrected for the heat of dilution, and analyzed using the program Origin™ provided by the manufacturer. Binding stoichiometry, enthalpy and equilibrium dissociation constants were determined by fitting corrected data to a model with a single binding site.
Supplementary Material
Supplementary data
Acknowledgments
We thank KD Zang, E Meese, C Welter, G Plotz, A Scheidig, R Zimmermann, M Freichel, V Flockerzi, R Müller and R Bernhardt (University of Saarland) for support of our research project and Nina Müller for help in some protein purifications. We are grateful to Giselle Wiggin (Samuel Lunenfeld Research Institute, Toronto) for careful reading of the manuscript, and Angel Nebreda (CNIO, Madrid), Morten Frödin and Axel Scheidig for comments on the work. In silico screenings were performed by C Kirchhof, Evotec (Hamburg). We acknowledge the support by the Europrofession Foundation (Saarbrücken, Germany) that allowed the settlement of the Research Group PhosphoSites at the University of Saarland. We also acknowledge support from HOMFOR (Uniklinik Saarland), DFG (BI 1044/2-1) and Deutsche Krebshilfe (106388) to RMB, Anschubfinanzierung (University of Saarland) to ME, and the Wilhelm Sander Foundation (2003.119.1) to AP. The Technologiebeteiligungsgesellschaft (tbg), Bonn, Germany, and the Saarland Ministry of Economics, Saarland, supported early work.
References
- Balendran A, Biondi RM, Cheung PC, Casamayor A, Deak M, Alessi DR (2000) A 3-phosphoinositide-dependent protein kinase-1 (PDK1) docking site is required for the phosphorylation of protein kinase Czeta (PKCzeta) and PKC-related kinase 2 by PDK1. J Biol Chem 275: 20806–20813 [DOI] [PubMed] [Google Scholar]
- Biondi RM (2004) Phosphoinositide-dependent protein kinase 1, a sensor of protein conformation. Trends Biochem Sci 29: 136–142 [DOI] [PubMed] [Google Scholar]
- Biondi RM, Cheung PC, Casamayor A, Deak M, Currie RA, Alessi DR (2000) Identification of a pocket in the PDK1 kinase domain that interacts with PIF and the C-terminal residues of PKA. EMBO J 19: 979–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biondi RM, Kieloch A, Currie RA, Deak M, Alessi DR (2001) The PIF-binding pocket in PDK1 is essential for activation of S6K and SGK, but not PKB. EMBO J 20: 4380–4390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biondi RM, Komander D, Thomas CC, Lizcano JM, Deak M, Alessi DR, van Aalten DM (2002) High resolution crystal structure of the human PDK1 catalytic domain defines the regulatory phosphopeptide docking site. EMBO J 21: 4219–4228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biondi RM, Nebreda AR (2003) Signalling specificity of Ser/Thr protein kinases through docking-site-mediated interactions. Biochem J 372: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume-Jensen P, Hunter T (2001) Oncogenic kinase signalling. Nature 411: 355–365 [DOI] [PubMed] [Google Scholar]
- Cohen P (2002) Protein kinases—the major drug targets of the twenty-first century? Nat Rev Drug Discov 1: 309–315 [DOI] [PubMed] [Google Scholar]
- Collins BJ, Deak M, Arthur JS, Armit LJ, Alessi DR (2003) In vivo role of the PIF-binding docking site of PDK1 defined by knock-in mutation. EMBO J 22: 4202–4211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins BJ, Deak M, Murray-Tait V, Storey KG, Alessi DR (2005) In vivo role of the phosphate groove of PDK1 defined by knockin mutation. J Cell Sci 118: 5023–5034 [DOI] [PubMed] [Google Scholar]
- Dajani R, Fraser E, Roe SM, Young N, Good V, Dale TC, Pearl LH (2001) Crystal structure of glycogen synthase kinase 3 beta: structural basis for phosphate-primed substrate specificity and autoinhibition. Cell 105: 721–732 [DOI] [PubMed] [Google Scholar]
- Etchebehere LC, Van Bemmelen MX, Anjard C, Traincard F, Assemat K, Reymond C, Veron M (1997) The catalytic subunit of Dictyostelium cAMP-dependent protein kinase – role of the N-terminal domain and of the C-terminal residues in catalytic activity and stability. Eur J Biochem 248: 820–826 [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P, Biondi RM (2001) A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol Cell 7: 1321–1327 [DOI] [PubMed] [Google Scholar]
- Frodin M, Antal TL, Dummler BA, Jensen CJ, Deak M, Gammeltoft S, Biondi RM (2002) A phosphoserine/threonine-binding pocket in AGC kinases and PDK1 mediates activation by hydrophobic motif phosphorylation. EMBO J 21: 5396–5407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frodin M, Jensen CJ, Merienne K, Gammeltoft S (2000) A phosphoserine-regulated docking site in the protein kinase RSK2 that recruits and activates PDK1. EMBO J 19: 2924–2934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonfloni S, Williams JC, Hattula K, Weijland A, Wierenga RK, Superti-Furga G (1997) The role of the linker between the SH2 domain and catalytic domain in the regulation and function of Src. EMBO J 16: 7261–7271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Begley M, Morgenstern KA, Gu Y, Rose P, Zhao H, Zhu X (2003) Crystal structure of an inactive akt2 kinase domain. Structure (Cambridge) 11: 21–30 [DOI] [PubMed] [Google Scholar]
- Huse M, Kuriyan J (2002) The conformational plasticity of protein kinases. Cell 109: 275–282 [DOI] [PubMed] [Google Scholar]
- Johnson LN, Lewis RJ (2001) Structural basis for control by phosphorylation. Chem Rev 101: 2209–2242 [DOI] [PubMed] [Google Scholar]
- Knighton DR, Xuong NH, Taylor SS, Sowadski JM (1991) Crystallization studies of cAMP-dependent protein kinase. cocrystals of the catalytic subunit with a 20 amino acid residue peptide inhibitor and MgATP diffract to 3.0 A resolution. J Mol Biol 220: 217–220 [DOI] [PubMed] [Google Scholar]
- Komander D, Kular G, Deak M, Alessi DR, van Aalten DM (2005) Role of T-loop phosphorylation in PDK1 activation, stability, and substrate binding. J Biol Chem 280: 18797–18802 [DOI] [PubMed] [Google Scholar]
- Newton AC (2003) Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem J 370: 361–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker PJ, Parkinson SJ (2001) AGC protein kinase phosphorylation and protein kinase C. Biochem Soc Trans 29: 860–863 [DOI] [PubMed] [Google Scholar]
- Pawson T, Scott JD (2005) Protein phosphorylation in signaling—50 years and counting. Trends Biochem Sci 30: 286–290 [DOI] [PubMed] [Google Scholar]
- Pearl LH, Barford D (2002) Regulation of protein kinases in insulin, growth factor and Wnt signalling. Curr Opin Struct Biol 12: 761–767 [DOI] [PubMed] [Google Scholar]
- Schaeffer F, Matuschek M, Guglielmi G, Miras I, Alzari PM, Beguin P (2002) Duplicated dockerin subdomains of Clostridium thermocellum endoglucanase CelD bind to a cohesin domain of the scaffolding protein CipA with distinct thermodynamic parameters and a negative cooperativity. Biochemistry 41: 2106–2114 [DOI] [PubMed] [Google Scholar]
- Sicheri F, Moarefi I, Kuriyan J (1997) Crystal structure of the Src family tyrosine kinase Hck. Nature 385: 602–609 [DOI] [PubMed] [Google Scholar]
- Smith KJ, Carter PS, Bridges A, Horrocks P, Lewis C, Pettman G, Clarke A, Brown M, Hughes J, Wilkinson M, Bax B, Reith A (2004) The structure of MSK1 reveals a novel autoinhibitory conformation for a dual kinase protein. Structure (Cambridge) 12: 1067–1077 [DOI] [PubMed] [Google Scholar]
- Xu W, Harrison SC, Eck MJ (1997) Three-dimensional structure of the tyrosine kinase c-Src. Nature 385: 595–602 [DOI] [PubMed] [Google Scholar]
- Xu ZB, Chaudhary D, Olland S, Wolfrom S, Czerwinski R, Malakian K, Lin L, Stahl ML, Joseph-McCarthy D, Benander C, Fitz L, Greco R, Somers WS, Mosyak L (2004) Catalytic domain crystal structure of protein kinase C-theta (PKCtheta). J Biol Chem 279: 50401–50409 [DOI] [PubMed] [Google Scholar]
- Yang J, Cron P, Thompson V, Good VM, Hess D, Hemmings BA, Barford D (2002) Molecular mechanism for the regulation of protein kinase B/Akt by hydrophobic motif phosphorylation. Mol Cell 9: 1227–1240 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data