

Abstract
Only a few methods permit researchers to study selected regions of the central and peripheral nervous systems with a spatial and time resolution sufficient to image the function of neural structures. Usually, these methods cannot analyse deep-brain regions and a high-resolution method, which could repeatedly probe dynamic processes in any region of the central and peripheral nervous systems, is much needed. Here, we show that fibred fluorescence microscopy—which uses a small-diameter fibre-optic probe to provide real-time images—has the spatial resolution to image various neural structures in the living animal, the consistency needed for a sequential, quantitative evaluation of axonal degeneration/regeneration of a peripheral nerve, and the sensitivity to detect calcium transients on a sub-second timescale. These unique features should prove useful in many physiological studies requiring the in situ functional imaging of tissues in a living animal.
Keywords: imaging, optic fibre, fluorescence, GFP, axonal regeneration, calcium transients
Introduction
The development of dynamic and functional imaging approaches is important to the study of physiology in the living animal, particularly given the growing number of genetically modified organisms. Multiphoton microscopy has been an advantage to dynamic studies in vivo; however, it can only access the adult rodent brain down to about 500 μm (Helmchen & Denk, 2002). Thus, one cannot image structures deeper than layer II of the cortex, as well as all sub-cortical areas. So far, the in vivo access of such deep structures has been limited to non-invasive imaging techniques, such as magnetic resonance imaging and positron emission tomography, which have significantly lower resolution and acquisition times compared with those of microscopy (Rudin & Weissleder, 2003). Recently, gradient refractive index lenses have been introduced into the live brain (Jung et al, 2004; Levene et al, 2004; Mehta et al, 2004), which allow one- and two-photon fluorescence imaging of individual neurons. Here, we describe fibred fluorescence microscopy (FFM), an approach that uses a flexible fibre-optic probe to provide real-time, high-resolution images of fluorescent brain areas, down to 6 mm below the surface of the rodent skull. We report the use of FFM as a non-invasive method for the analysis of peripheral neuro-epithelial organs, and a minimally invasive method for imaging peripheral nerves. The method allows a direct, rapid, yet accurate measurement of neuronal degeneration and regeneration in vivo, which can be used repeatedly to monitor the same nerve fibre in individual animals. FFM also permits the in situ recording of calcium transients within deep central nervous system nuclei in the anaesthetized animal. These unique features represent an important improvement for monitoring the structure and function of neural structures and other tissues in vivo.
Results
FFM images small nerve structures in vivo
To validate FFM, we imaged the peripheral nervous system using a 650 μm-diameter optic probe (Table 1; Fig 1A). In Thy-1 eYFP (enhanced yellow fluorescent protein) mice, which express eYFP in all sensory and motor neurons (Feng et al, 2000), FFM allowed us to observe cell bodies in the dorsal root ganglia (Fig 1B) and to follow individual fibres from there to their termination sites. We then imaged the saphenous nerve to monitor single axons (Fig 1C), nerve endings at skin receptors (Fig 1D) and neuromuscular junctions (Fig 1E). All these recordings were obtained in real time and without detectable tissue damage or loss of fluorescence. Supplementary Movie 1 online presents a typical recording, showing the pattern of nerve fibres in the saphenous nerve.
Table 1.
Characteristics of the microprobes used in this study
| Probe | S-0650 | S-0300 |
| Diameter (mm) | 0.65 | 0.30 |
| Maximum field of view* (μm × μm) | 600 × 500 | 300 circular |
| Lateral resolution (μm) | 5<lr<15 | 5<lr<15 |
| Axial resolution (μm) | 8 | 8 |
| Number of fibres* | 30,000 | 6,000 |
| Numerical aperture* | 0.32 | 0.32 |
Lateral resolution (lr) was evaluated with fluorescent beads (supplementary information and supplementary Fig 2 online).
Axial resolution: the value indicates the distance where the fluorescence intensity of a sub-resolution bead is 50% of the maximum value (supplementary information and supplementary Fig 2 online).
*Values provided by Mauna Kea Technologies.
Figure 1.
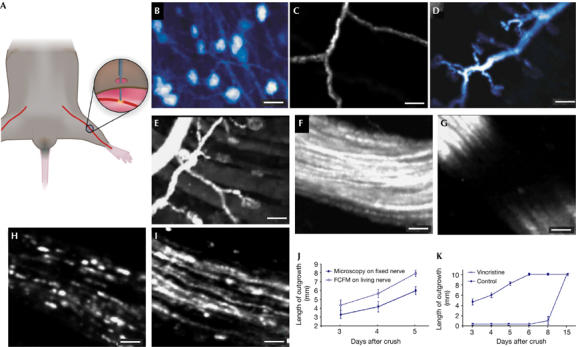
Fibred fluorescence microscopy visualizes neural structures and evaluates their regeneration. (A) View of the probe (blue) taken in contact with the saphenous nerve (red) through a small skin cut. (B–E) Fibred fluorescence microscopy (FFM) shows neuron bodies and processes in the dorsal root ganglion (B), single axons seen across the perineurium (C), and their terminations at skin receptors (D) or neuromuscular plates (E). Scale bars, 20 μm (B) and 30 μm (C–E). (F–I) FFM shows the arrangement of neurites in the saphenous nerve under control conditions (F) and 1 min after crush of the proximal part of the nerve (G). At the site of the crush, the nerve is no longer fluorescent owing to loss of yellow fluorescent protein. Fluorescent ovoids, typical of axon degeneration, are seen 1 day later (H). Four days after crush, part of the axons had already regenerated, regaining a close to normal appearance (I). Scale bars, 20 μm (F,G) and 50 μm (H,I). (J) The length of axonal outgrowth, as evaluated by FFM, had a similar time course as that evaluated by histology of the fixed nerve, but was about 30% higher, at all time points. (K) After treatment with vincristine, FFM detected the expected delay in the regeneration of crushed saphenous fibres. Values in (J,K) are mean±s.e.m. measurements performed on three (J) and five mice (K) per time point, each mouse value being the mean of three measurements per nerve.
Repeated FFM imaging of a peripheral nerve
We then used Thy-1 eYFP transgenic mice to monitor the axonal changes that occur after crush injury of the saphenous nerve (Pan et al, 2003). The crushed saphenous nerve and the contralateral nerve, which served as a control, were monitored daily for 15 days after the crush using the 650 μm-diameter probe, which was introduced through a skin incision and gently moved along the surface of the nerve (Fig 1A). FFM imaged in vivo the fibre bundles forming the main nerve trunk (Fig 1F), showing the same arrangement as observed in the fixed nerve, which was visualized by standard fluorescence microscopy (supplementary Fig 3 online). Immediately after the crush, no fluorescence was observed at the site of injury, owing to the loss of the YFP protein through the damaged cell membranes (Fig 1G). One day after the crush, the axons of the distal stump showed fragmented, YFP-rich areas (Fig 1H), corresponding to the ovoid structures that are typical of neural degeneration. These areas became smaller and less numerous over the next several days, whereas axon endings adjacent to the site of injury began to grow (Fig 1I). Each stage of this degeneration–regeneration sequence (Stoll et al, 1989; Fawcett & Keynes, 1990) was repeatedly recorded, in intervals of less than 2 min, from the same anaesthetized mice. The real-time visualization showed the main morphological features that are typically obtained by using the more time-consuming confocal microscopy analysis of fixed nerves (Frykman et al, 1988; supplementary Fig 3 online). Using FFM, the front of progression from regenerating axons was also easily identified, thus allowing fast and reliable measurements of the outgrowth length from the site of injury, under various experimental conditions.
FFM allows in vivo measurements of nerve regeneration
FFM was compared with histology in the quantification of axonal regeneration after crush injury of the saphenous nerve. When evaluated according to the protocol described in the supplementary information online, both approaches indicated a similar time-dependent rate of axonal outgrowth (Fig 1J). However, FFM consistently resulted in 30% higher values, as it was not affected by the artefactual volume shrinkage caused by the histological processing of fixed tissue.
Thy-1 eYFP transgenic mice were also treated with vincristine— a cytotoxic plant alkaloid that depolarizes microtubules and delays axonal regeneration (Shiraishi et al, 1985a, 1985b; Pan et al, 2003)—1 day after the crush of the saphenous nerve. Compared with controls, nerves exposed to vincristine and analysed by FFM showed no regenerative process up to 6 days after crushing (Fig 1K). Regenerating fibres were observed only from days 8 to 15, which was in agreement with previous reports that used standard analysis methods (Nakamura et al, 2001; Pan et al, 2003).
Non-invasive imaging of the olfactory neuroepithelium
Owing to its small diameter and flexible properties, the fibre-optic probe could also be introduced into the nasal cavity (Fig 2A) to visualize the cell bodies of olfactory receptor neurons (Fig 2B) and their axons (Fig 2C) in CaMK–eGFP mice (CaMK, Ca2+/calmodulin-dependent kinase II; eGFP, enhanced green fluorescent protein; Krestel et al, 2001). With both the 650 μm and the 300 μm probes (Table 1), we could image the olfactory nerve (arrows in Fig 2C) up to its termination in the olfactory bulb. Supplementary Movie 2 online shows a real-time sequence of this experiment. The visible movement is due to the manipulator moving the probe in and out of the nostril.
Figure 2.
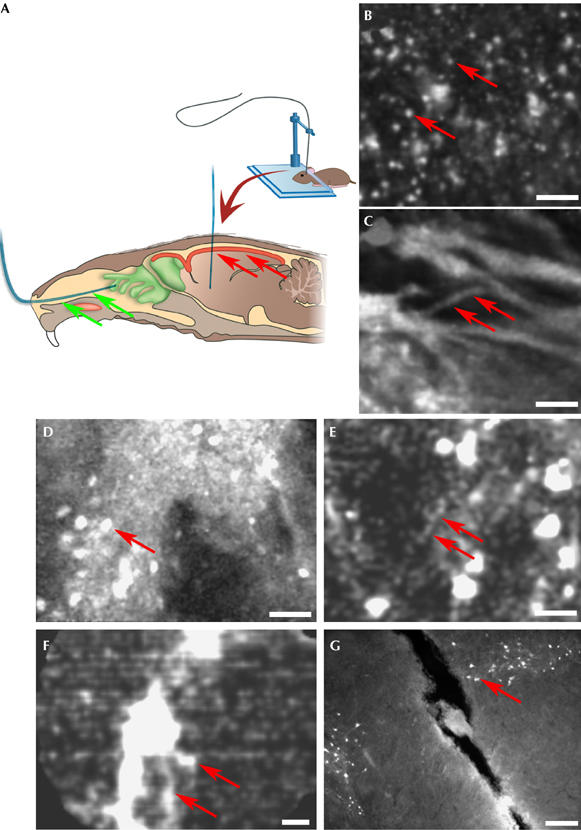
Fibred fluorescence microscopy allows non-invasive access to the olfactory epithelium and a minimally invasive approach to deep-brain nuclei. (A) Stereotactic approach for the introduction of the fibre-optic probe (blue) into the brain and for its non-invasive introduction through a nostril (green arrows). The red-coloured area on the surface of the brain (red arrows) shows the penetration depth achievable with a two-photon microscope, which also cannot non-invasively access the olfactory epithelium. (B,C) Images from supplementary Movie 2 online showing individual olfactory neurons (arrows in B) and bundles of the olfactory nerve (arrows in C) of CaMK–eGFP transgenic mice (CaMK, Ca2+/calmodulin-dependent kinase II; eGFP, enhanced green fluorescent protein). Scale bars, 25 μm (B) and 30 μm (C). (D) In the striatum of CaMK–eGFP transgenic mice, the fibred fluorescence microscopy probe shows the somata of medium-spiny neurons (arrow) and their surrounding neuropil. Penetration depth, 4 mm. Scale bar, 50 μm. (E) Lentivirus-transduced neurons in the ventral tegmental area (VTA) and individual dendrites (arrows). Penetration depth, 4.5 mm. Scale bar, 25 μm. (F) Single image from supplementary Movie 3 online showing dopaminergic neurons (arrows) in the VTA. Penetration depth, 4.5 mm. Scale bar, 25 μm. (G) Confocal image of a slice of VTA, sampled immediately after the imaging experiment, showing the trace left by the 300 μm probe used to image the TH–eGFP transgenic mouse used in (D). The arrow points to one GFP-labelled dopaminergic neuron. Scale bar, 200 μm.
In vivo deep-brain imaging
Many fluorescent gene expression markers provided animal models for the imaging of deep-brain structures, and we used the bevel-shaped 300 μm-diameter probe, positioned by stereotaxy (Fig 2A), to image these structures. Transgenic mice expressing eGFP in parts of the rostral brain (Krestel et al, 2001), in the dopaminergic neurons of the ventral tegmental area (VTA) and substantia nigra (Sawamoto et al, 2001), and mice injected in the VTA with lentiviral vectors (Maskos et al, 2005), allowed the region-specific targeting of brain cells. Using these models, we imaged deep-brain structures in situ at single-cell resolution, including the striatum (Fig 2D) and VTA (Fig 2E,F), which is 4.5 mm below the surface of the mouse skull. TH (tyrosin hydroxylase)-eGFP mice were used to specifically image dopaminergic neurons, identified by their dendrites and surrounding neuropil in the VTA (Fig 2F). Supplementary Movie 3 online shows multiple penetrations of the probe into this region, repeatedly imaging the same neuron without obvious damage. Fig 2G shows a trace of the probe penetration in the VTA. Supplementary Fig 3 online describes the detailed analysis of the consequences of deep-brain penetration on tissue viability.
Calcium imaging
To monitor neural activity in deep-brain regions, we stereotactically injected the fluorescent dye Oregon Green BAPTA-1 AM (OGB1) into the brain of anaesthetized adult rats (Stosiek et al, 2003). Using a probe with a tip polished to a conical shape (supplementary Fig 1B online), which allowed both easy penetration and adequate contact with the tissue, we obtained an image in which space is represented in polar coordinates: the centre of the field corresponds to the tip of the fibre that images the deepest structures of the tissue, whereas the periphery corresponds to regions closer to the brain surface (supplementary Movie 4 online). This fluorescent signal is the typical labelling obtained after injection of the chemical calcium indicator dye. As can be seen in supplementary Movies 4 and 6 online, the optic fibre crosses layers of different fluorescence intensities during its descent into the brain, which is due to the uneven loading of different brain regions.
We used bicuculline, an antagonist of γ-aminobutyric acid A receptors, as an inducer of hippocampal epileptiform activities (Ben-Ari et al, 1981). The probe was placed in the dentate gyrus and 50 s after intravenous injection of the drug the FFM showed a 50–100% increase in fluorescence, reflecting an increase in cytosolic free calcium (Fig 3; supplementary Movie 5 online). A parallel electroencephalogram (EEG) reported highly synchronous neuronal activities, best seen in the expanded traces showing peaks of an important synchronous activity (Fig 3D), owing to the epileptic discharges provoked by the injection of bicuculline. Fluorescence responses were measured on regions corresponding to individual cell bodies or small groups of cells (red, blue and green traces in Fig 3), as well as in the neuropil (yellow trace in Fig 3), consistent with a global increase in intracellular calcium during the seizure.
Figure 3.
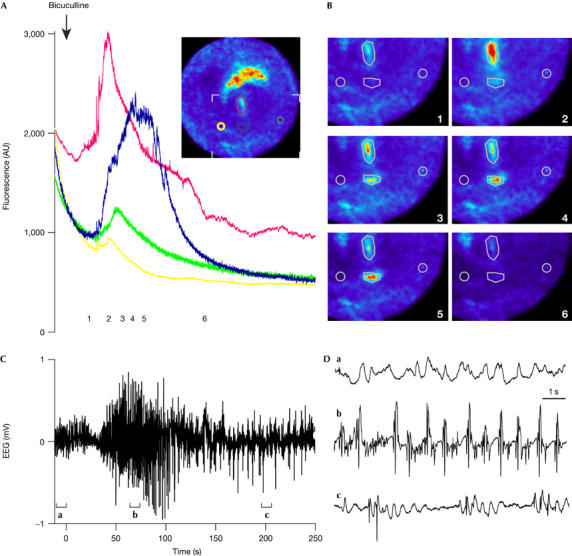
Fibred fluorescence microscopy monitors changes in intracellular calcium in the hippocampus in response to bicuculline-induced epileptiform activity. (A) Time course of the fluorescence intensity of Oregon Green BAPTA-1 measured in the four regions indicated by the coloured outlines in the inset. The radius of the inset image is 490 μm. (B) Enlarged views of the recorded region outlined by the white angles in the inset of (A) and corresponding to the time points indicated in (A). (C) An electroencephalogram (EEG) was recorded simultaneously and bicuculline was injected intravenously at the time indicated by the arrow in (A). After bicuculline injection, epileptiform activity recorded on the EEG developed synchronously with the increase in the fluorescence measured by fibred fluorescence microscopy at points 1–6 (A). (D) Magnified portions of the EEG as indicated by the brackets in (C).
In another set of experiments, we elicited neuronal activity by electrical stimulation of deep thalamic nuclei by using four trains of ten stimulations at 10 Hz, which increased rhythmic activity in the 1–2 Hz range for about 20 s. As shown by the EEG, synchronization of the network activity of the thalamus, again more evident in the expanded traces, was obtained (Fig 4B). During the electrical stimulation, an FFM probe located in the ventro-basal thalamus showed a large increase in the OGB1 fluorescence, reflecting increased neuronal activity (Fig 4; supplementary Movie 7 online). The fluorescence changes showed no correlation with heartbeat or ventilation and occurred on a much slower time frame, ruling out potential motion artefacts. Moreover, motion induced by the experimenter during positioning of the probe resulted in distinctive changes in the image (see supplementary Movies 4, 6 online), which were different from those observed during the neural response (supplementary Movie 5 online).
Figure 4.
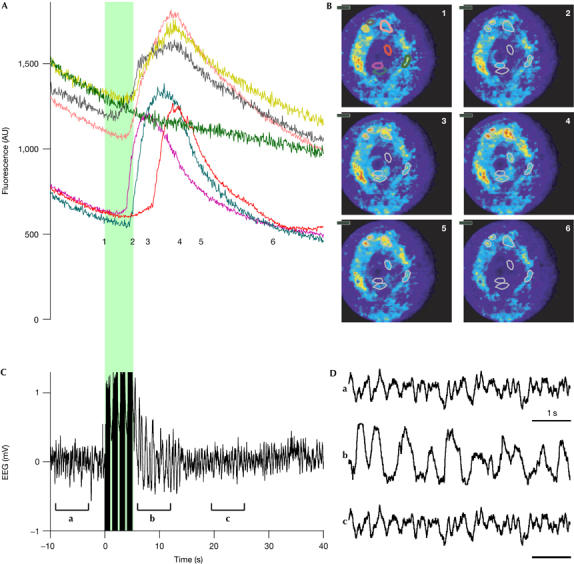
Fibred fluorescence microscopy monitors changes in intracellular calcium in the thalamus in response to electrical stimulation. (A) Time course of the fluorescence intensity measured in the seven regions indicated by the coloured outlines shown in (B). (B) Views of the recorded region at the time points indicated by the corresponding numbers in (A). (C) Electroencephalogram (EEG) recorded simultaneously. Electrical stimulation of deep thalamic nuclei, indicated by the green block, elicited a synchronous activity visible on the EEG and an increase in the fluorescence measured by fibred fluorescence microscopy in the ventro-basal thalamus (A). (D) Magnified portions of the EEG as indicated by the brackets in (C).
Discussion
We have shown that FFM is a fibre-optic microscopy approach that allows the in vivo imaging of fluorescently labelled neural structures in the anaesthetized rodent, including in regions that are inaccessible by other technologies. At present, FFM is the only method for non-invasive access to the olfactory neuro-epithelium and the olfactory nerve (Mehta et al, 2004); this relates specifically to the characteristics of the flexible probes it uses. The probe comprises many optical micro-fibres arranged in a bundle, and has a lateral resolution well below 15 μm (supplementary Figs 1,2 online). Although this resolution is less than that provided by gradient refractive index lenses (Mehta et al, 2004; Flusberg et al, 2005), it is sufficient to analyse the in situ morphology of most neurons. In addition, the optic probe can be given a shape that facilitates brain penetration: the bevelled and cone-shaped probes allowed repeated access to brain loci located to a depth of 6 mm below the surface of the skull, and provided real-time imaging of such regions at cellular resolution.
This approach allowed us to monitor for the first time calcium signals in deep-brain structures in response to either pharmacological or electrical stimulation. Large fluorescence signals originated from cell bodies as well as from the surrounding neuropil and glial cells. Although the relative contribution of neurons and glial cells to the fluorescence change could not be determined, the recorded signals were clearly related to the large synchronous neuronal activities reported by the EEG. The use of novel GFP variants and fusion proteins—which can serve as indicators of the physiological state of a neuron (Hasan et al, 2004; Miesenbock, 2004) or neural circuit (Maskos et al, 2002)—makes such an in situ imaging of the living rodent most timely. Although the OGB1 used in the study is prone to bleaching under constant laser illumination, such as the one used here, this problem should be solved by an intermittent laser activation that would reduce the overall illumination. Furthermore, viral vectors and an increasing number of animal models, locally expressing a variety of indicator molecules, will undoubtedly become amenable to monitoring by FFM.
We have also documented that FFM allows highly reliable measurements of peripheral nerve outgrowth, which parallel those obtained by standard microscopic analysis of fixed nerves, but which can be obtained in a much shorter time frame, repeatedly in the same mice, and without interference of artefacts, notably owing to fixation-induced shrinkage of cells and tissues that are unavoidably caused during tissue preparation (Frykman et al, 1988). These unique features markedly decrease the number of animals required for a study. Even though the current implementation of the system requires the probe to be in contact with or relatively close to the specimen, our study has shown no deleterious effects of such a contact on the kinetics of axonal damage and recovery. Rather, the approach has shown the feasibility to image individual nerve fibres and terminals, which should facilitate the analysis of conditions targeted to specific neuronal sub-populations (Pan et al, 2003). As an example, FFM easily documented the transient effect of a single dose of vincristine, which has been previously reported to delay neuronal regeneration both by preventing the outgrowth of some axons and by slowing down that of others (Shiraishi et al, 1985a; Pan et al, 2003).
At present, the FFM approach requires the animal to be deeply anaesthetized during the recordings. However, the approach is potentially amenable to the in vivo imaging of the central nervous system in an awake, unrestricted animal, similar to experiments reported recently by Adelsberger et al (2005). A long-term implantation of the fibre-optic bundle is feasible in the mouse. In addition, the probes have a diameter comparable with that of microdialysis probes, and are flexible so that they can be precisely positioned stereotactically without major brain damage (supplementary Fig 4 online). Such developments are urgently needed for imaging physiological neural functions in the freely moving animal.
In summary, by providing a minimally invasive, easy and online acquisition of data, FFM should facilitate the in vivo study of neurons and their responses to experimental treatments. The approach can also be immediately extended to other tissues, organs and conditions, therefore providing an important tool for the in vivo investigation of many biological questions under a variety of physiological and pathological conditions.
Methods
Animals. For imaging peripheral nerves, transgenic Thy1–YFP mice of line 16 were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). One group of ten mice was anaesthetized by intraperitoneal injection of ketamine. The posterior leg was mechanically shaved and a skin incision was made to expose the saphenous nerve. The nerve was crushed near the groin by ligaturing it for 2 min. The tips of a pair of fine forceps dipped in China ink were used to mark the crush site. The contralateral nerve was similarly processed, except for the crush, and used as an internal control. In another series of experiments, mice were injected intraperitoneally 1 day after the crush with 0.5 mg/kg vincristine sulphate (100 mg/ml solution in 0.9% NaCl; Sigma-Aldrich Chimie, Lyon, France). Control mice received saline alone. The quantitative evaluation of nerve degeneration and regeneration is described in supplementary information online.
For mouse brain imaging, adult CaMK–eGFP mice (Krestel et al, 2001) were anaesthetized using ketamine/xylazine in PBS, and the GFP-expressing regions were imaged by introducing a bevelled fibre-optic probe through a hole in the skull (Fig 2A); the probe was positioned by using the electrode holder of a stereotaxic device (Stoelting Co., Wood Dale, IL, USA).
Calcium imaging experiments were carried out on adult male Sprague–Dawley rats (250–300 g) anaesthetized with urethane (1.5 g/kg, intraperitoneally), placed on a stereotaxic frame and maintained at 37°C. The heart rate and EEG were monitored to ensure adequate anaesthesia. A catheter was placed in the jugular vein for injection of bicuculline (0.5 mg/kg, 0.5 mg/ml in 1 M HCl; Tocris Bioscience, Bristol, UK; Berman et al, 2000). One microlitre of 1 mM OGB1 (Molecular Probes Invitrogen Ltd, Paisley, UK) was injected using air pressure through a glass pipette of 60 μm diameter, set to eject 0.5 μl over about 5 min. Injection coordinates were (in mm): for the dentate gyrus, 4 caudal to bregma, 7 lateral and 3.4 deep; for the ventro-postero median nucleus of the thalamus, 2.9 caudal to bregma, 2.7 lateral and 5.6 deep. Optical recording started 1 h later. For electrical stimulation, a monopolar tungsten electrode of 80 μm diameter with 300 μm exposed tip was placed 800 μm deeper than the tip of the optic probe.
Imaging set-up. The imaging device is described in the supplementary information online. Briefly, tissues were excited with a 488 nm laser light, and emission was measured between 500 and 650 nm. Images were continuously acquired at 12 Hz, using microprobes made of bundles of optic fibres, 300 or 650 μm in diameter (Table 1).
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
Supplementary Movie 1
Supplementary Movie 2
Supplementary Movie 3
Supplementary Movie 4
Supplementary Movie 5
Supplementary Movie 6
Supplementary Movie 7
Supplementary Information
Acknowledgments
We thank J.P. Bourgeois and S. Pons for comments on the manuscript, C. Galliot for the drawings, C. Custody for helpful discussion and Mauna Kea Technologies for making the Cell-vizio instrument available. This work was supported by grants from the Swiss National Foundation (310000-109402), the Juvenile Diabetes Research Foundation International (1-2005-46) and the National Institutes of Health (DK-63443-01) to P.M., the Association de Recherche sur le Cancer ARC and the ANR programme ‘Neuroscience et Psychiatrie' to U.M.
Competing Interest Statement P.V. and R.L. are consultants for Mauna Kea Technologies.
References
- Adelsberger H, Garaschuk O, Konnerth A (2005) Cortical calcium waves in resting newborn mice. Nat Neurosci 8: 988–990 [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Tremblay E, Riche D, Ghilini G, Naquet R (1981) Electrographic, clinical and pathological alterations following systemic administration of kainic acid, bicuculline or pentetrazole: metabolic mapping using the deoxyglucose method with special reference to the pathology of epilepsy. Neuroscience 6: 1361–1391 [DOI] [PubMed] [Google Scholar]
- Berman RF, Fredholm BB, Aden U, O'Connor WT (2000) Evidence for increased dorsal hippocampal adenosine release and metabolism during pharmacoligically induced seizures in rats. Brain Res 872: 44–53 [DOI] [PubMed] [Google Scholar]
- Fawcett JW, Keynes RJ (1990) Peripheral nerve regeneration. Annu Rev Neurosci 13: 43–60 [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR (2000) Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28: 41–51 [DOI] [PubMed] [Google Scholar]
- Flusberg BA, Cocker ED, Piyawattanametha W, Jung JC, Cheung EL, Schnitzer MJ (2005) Fiber-optic fluorescence imaging. Nat Methods 2: 941–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frykman GK, McMillan PJ, Yegge SA (1988) A review of experimental methods measuring peripheral nerve regeneration in animals. Orthop Clin N Am 19: 209–219 [PubMed] [Google Scholar]
- Hasan MT et al. (2004) Functional fluorescent Ca2+ indicator proteins in transgenic mice under TET control. PLoS Biol 2: e163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmchen F, Denk W (2002) New developments in multiphoton microscopy. Curr Opin Neurobiol 12: 593–601 [DOI] [PubMed] [Google Scholar]
- Jung JC, Mehta AD, Aksay E, Stepnoski R, Schnitzer MJ (2004) In vivo mammalian brain imaging using one- and two-photon fluorescence microendoscopy. J Neurophysiol 92: 3121–3133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krestel HE, Mayford M, Seeburg PH, Sprengel R (2001) A GFP-equipped bidirectional expression module well suited for monitoring tetracycline-regulated gene expression in mouse. Nucleic Acids Res 29: E39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levene MJ, Dombeck DA, Kasischke KA, Molloy RP, Webb WW (2004) In vivo multiphoton microscopy of deep brain tissue. J Neurophysiol 91: 1908–1912 [DOI] [PubMed] [Google Scholar]
- Maskos U, Kissa K, St Cloment C, Brûlet P (2002) Retrograde trans-synaptic transfer of green fluorescent protein allows the genetic mapping of neuronal circuits in transgenic mice. Proc Natl Acad Sci USA 99: 10120–10125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskos U et al. (2005) Nicotine reinforcement and cognition restored by targeted expression of nicotinic receptors. Nature 436: 103–107 [DOI] [PubMed] [Google Scholar]
- Mehta AD, Jung JC, Flusberg BA, Schnitzer MJ (2004) Fiber optic in vivo imaging in the mammalian nervous system. Curr Opin Neurobiol 14: 617–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesenböck G (2004) Genetic methods for illuminating the function of neural circuits. Curr Opin Neurobiol 14: 395–402 [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Shimizu H, Nishijima C, Ueno M, Arakawa Y (2001) Delayed functional recovery by vincristine after sciatic nerve crush injury: a mouse model of vincristine neurotoxicity. Neurosci Lett 304: 5–8 [DOI] [PubMed] [Google Scholar]
- Pan YA, Misgeld T, Lichtman JW, Sanes JR (2003) Effects of neurotoxic and neuroprotective agents on peripheral nerve regeneration assayed by time-lapse imaging in vivo. J Neurosci 23: 11479–11488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin M, Weissleder R (2003) Molecular imaging in drug discovery and development. Nat Rev Drug Discov 2: 123–131 [DOI] [PubMed] [Google Scholar]
- Sawamoto K et al. (2001) Visualization, direct isolation, and transplantation of midbrain dopaminergic neurons. Proc Natl Acad Sci USA 98: 6423–6428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraishi S, Le Quesne PM, Gajree T (1985a) The effect of vincristine on nerve regeneration in the rat. An electrophysiological study. J Neurol Sci 71: 9–17 [DOI] [PubMed] [Google Scholar]
- Shiraishi S, Le Quesne PM, Gajree T, Cavanagh JB (1985b) Morphometric effects of vincristine on nerve regeneration in the rat. J Neurol Sci 71: 165–181 [DOI] [PubMed] [Google Scholar]
- Stoll G, Griffin JW, Li CY, Trapp BD (1989) Wallerian degeneration in the peripheral nervous system: participation of both Schwann cells and macrophages in myelin degradation. J Neurocytol 18: 671–683 [DOI] [PubMed] [Google Scholar]
- Stosiek C, Garaschuk O, Holthoff K, Konnerth A (2003) In vivo two-photon calcium imaging of neuronal networks. Proc Natl Acad Sci USA 100: 7319–7324 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Movie 1
Supplementary Movie 2
Supplementary Movie 3
Supplementary Movie 4
Supplementary Movie 5
Supplementary Movie 6
Supplementary Movie 7
Supplementary Information