

Abstract
Tel is an Ets transcription factor that is the target of chromosome translocations in lymphoid and myeloid leukemias and in solid tumors. It contains two functional domains, a pointed oligomerization domain and a DNA-binding domain. Retroviral transduction of a wild-type Tel cDNA into a clonal subline of NIH3T3 fibroblasts resulted in a striking morphologic change: at confluency, the cells reorganized into a specific “bridge-like” pattern over the entire surface of the culture dish, and started migrating, thereby leaving circular holes in the monolayer. Thereafter, formation of cellular cords became apparent. This sequence of events was inhibited by coating the culture dishes with fibronectin and collagen IV. Retroviral transduction of Tel into MS1 endothelial cells reproduced the aggregation phenotype, but not the cellular cord formation. Tel-mutagenesis showed that both the pointed domain and the DNA-binding domain of Tel are required for the morphologic change. Other Ets family genes, Fli-1 and Ets-1 that are both endogenously expressed in endothelial cells, could not induce this morphologic change. Exogenous Tel expression is associated with transcriptional upregulation of entactin/nidogen, Smad5, Col3a1, CD44 and fibronectin, and downregulation of Col1a1 and secretory leukocyte protease inhibitor. Interestingly, Tel, Smad5, fibronectin, Col1a1 and Col3a1 all have essential roles during vascular development.
Keywords: Tel, differential gene expression, ECM, adhesion, vasculogenic mimicry
Introduction
Tel/ETV6 is a nuclear phosphoprotein, discovered when studying the t(5;12) present in a subgroup of CMML patients. The N-terminal part of Tel, containing the pointed protein interaction domain (PNT), was fused to the C-terminal part of the PDGFRβ [1–3]. Subsequently, Tel was found to be involved in many more translocations in both myeloid and lymphoid leukemia, but also in solid tumors. One class of translocations is characterized by the fusion of the N-terminus of Tel, including the PNT to the C-terminal domains of the tyrosine kinases harboring a kinase moiety. In addition to PDGFRβ, Tel was found to be fused to Abl in the t(9;12) (q34;p13), to Jak2 in another t(9;12) (p24;p13), and to the neurotrophin 3 receptor (NTRK3/TRKC) [1,4–9]. The latter fusion protein was found in cases of congenital fibrosarcoma, mesoblastic nephroma and myeloid leukemia. In another category of translocations, Tel is fused to transcription factors. The fusion of the N-terminal half of Tel to AML-1, resulting from a t(12;21) is seen in about 15% to 25% of cases of childhood pre-B cell leukemia [10–13]. The Tel-AML-1 fusion is often accompanied by loss of the remaining wild-type Tel allele [14–16]. In the t(12;22) and t(4;12), both associated with myelodysplasia, MN1- and BTL-genes, respectively, were fused to Tel [17,18]. Both fusions harbor the DNA-binding domain (DBD) of Tel and therefore make up a unique class of Tel fusion proteins. Another Tel-translocation linked to myelodysplasia, fuses the N-terminus of Tel, excluding the PNT domain and the DBD, to the entire MDS1/EVI-1 or EVI-1 proteins [19]. Recently, in a case of acute myeloid leukemia, the N-terminus of Tel was found fused to the homeobox protein CDX2 [20].
Despite the plethora of characterized translocations involving the Tel gene, the physiology of Tel remains largely unknown. Tel is ubiquitously expressed and encodes two protein isoforms (p50 and p60), generated by usage of the first or second start codon of the open reading frame. Tel contains three putative MAPK-sites, of which one is unique to the longer isoform. To date, no Tel-target genes have been identified. A Tel knock-out (KO) study revealed that the gene product is essential during murine development. Defects in angiogenesis of the yolk sac and intra-embryonic apoptosis were apparent [21]. As the Tel-/- embryos died around E10.5, the same research group generated mouse chimaeras using Ter-/- ES cells. They showed that Tel has an essential role in the homing of the hematopoietic system from fetal liver to bone marrow, but is dispensable for the development of the hematopoietic system [22]. A yeast two-hybrid screen using most of the Tel-protein as a bait and a library derived from hematopoietic stem cells, showed interaction of Tel with Fli-1 and with itself [23]. The latter was also found by Jousset et al. (1997) [2]. However, their study showed that not all PNT-containing Ets factors, such as Ets-1, Erg-2 and GABPα, interact with Tel.
To identify possible Tel-target genes, we retrovirally transduced mouse fibroblast with the Tel cDNA. As this resulted in a striking morphologic change of the fibroblasts, we studied this phenomenon more in depth. We found that this phenotypic change was highly Tel-specific and required both its PNT and DBD domain. We identified differentially expressed genes encoding proteins involved in extracellular matrix (ECM) build-up. We discuss the similarity of the phenotype to that of endothelial cells that differentiate to tubes in vitro, and its potential physiological relevance for tumorigenesis and vasculogenic mimicry in particular.
Materials and Methods
Constructs
Construction of MN1-Tel type I, Tel-ΔPNT, VP16-Tel, Tel-DBD mutant (R396L;R399L) and wild-type Tel constructs are described by Buijs et al. (submitted). Site-directed mutagenesis was performed using the Mutagene™ kit (BioRad) according to the manufacturer's directions. The following oligonucleotides were used: CCTCGAGCGCTCAGGATcGAG (M43C), ACACCTCCAGAGgccCCAGTGCCGAG (S22A) and ACACCTCCAGAGgaaCCAGTGCCGAG (S22E). A second Tel-DBD mutant (R396K;R399K) was made using overlap-polymerase chain reaction (PCR) and the oligonucleotides: GAGAAAATGTCCaaaGCCCTGaaaCACTACTACA (sense) and TGTAGTAGTGtttCAGGGCtttGGACATTTTCTC (antisense). Tel-mutants were subcloned into the pSRα-TK/CD8 retroviral vector (see Buijs et al., submitted). Full-length cDNA encoding Ets-1 (J. Ghysdael), Fli-1 (S. Baker) and Scl/Tal-1 (R. Baer) were also subcloned in pSRα-TK/CD8.
Cell Culture and Retroviral Transduction
NIH3T3-UCLA cells were obtained from O. Witte [24,25] and MS1 cells were purchased from ATCC. NIH3T3-UCLA cells are a clonal derivative of mouse fibroblasts obtained by the 3T3 protocol. NIH3T3-UCLA cells differ from NIH3T3 in that they adhere loosely to the substratum and that they are spindle-shaped. Cells were cultured in Dulbecco's modified Eagle's medium (DMEM)/10% fetal calf serum (FCS) containing penicillin/streptomycin. Retroviral supernatants were prepared by transfecting 2x106 293T cells, seeded in a 10-cm dish, with 4 µg of ecotropic helper plasmid (Psi2) [25] and 4 µg of pSRα-retroviral construct using Lipofectamine-Plus (Gibco). Virus-containing supernatant was harvested 24, 43, 48, 53 and 70 hours after the start of transfection, filtered through 0.45 µm filters and frozen at -80°C. For retroviral transduction, cells were seeded at 2x105 in a 10-cm dish or at 5x104/well in a six-well plate and incubated for 4 to 6 hours in the presence of 8 µg/ml of polybrene, using 4.5 or 1 ml of viral supernatant, respectively.
Indirect Immunofluorescence
Retrovirally transduced cells, seeded on microscope slides, were fixed in 3.5% paraformaldehyde for 20 minutes, washed 3 times for 10 minutes with phosphate-buffered saline (PBS) and incubated in blocking buffer (2% non-fat dry milk) containing 0.2% Triton X-100 for 30 minutes. Cells were then incubated for 1 hour with polyclonal antisera, diluted in blocking buffer containing 0.2% Triton X-100. The polyclonal antiserum, raised against a C-terminal Tel-peptide was immuno-affinity purified on a peptide column before use (Buijs et al., submitted). Antisera raised against Fli-1, fibronectin and Scl/Tal-1 were obtained from Santa Cruz, Sigma Chemical Co. and R. Baer (U.T. Southwestern), respectively. Slides were washed three times for 5 minutes in PBS and subsequently incubated with a donkey-anti-rabbit/Cy3 labeled secondary antibody (Amersham). Digital images were acquired using a SPOT™-camera (Diagnostic Instruments Inc.).
Differential Expression Analysis
Differential expression was studied in a systematic fashion using ATLAS cDNA arrays (Clontech), Mouse GDA filters (Genome Systems) and oligonucleotide chips (Affymetrix). Total RNA was isolated from vector- and Tel-transduced cells using Trizol (Gibco) and processed for labeling and hybridization to the DNA chips or filters according to the manufacturer's instructions. Significant differential expression was assessed electronically (DNA chips) or by visual examination of autoradiographs (filters) or PhosphorImager (Molecular Dynamics) scans. IMAGE clones representing putative differentially expressed clones were purchased from Genome Systems and their identity was verified by restriction digests and DNA sequencing. A Smad5 probe, used for in situ hybridization [26], was obtained from A. Zwijsen and D. Huylebroeck (Leuven, Belgium). Inserts were amplified by PCR using a T3/T7 or an M13 primer set, or generated by restriction enzyme digestion. The resulting fragments were then purified from the reaction mixtures (QiaQuick and QiaExll, respectively; Qiagen). Probes were prepared by random-primed labeling using 32P-dATP or 32P-dCTP and diluted in UltraHyb (Ambion) at 1x106 cpm/ml. Membranes were hybridized for 20 hours at 42°C in UltraHyb (contains 50% formamide) and washed at high stringency (0.1% sodium dodecyl sulfate (SDS), 0.1xSSC at 65°C). The hybridization results were quantified using a PhosphorImager (Molecular Dynamics). The expression level of actin mRNA was used to normalize data.
Results
A subline of mouse NIH3T3 fibroblasts (NIH3T3-UCLA) [24,25] was transduced with retroviruses encoding the wild-type Tel cDNA and with a vector control. At confluency, the Tel-transduced cells underwent a morphologic change (Figure 1A, 4–6), whereas the vector-transduced cells remained as a flat monolayer (Figure 1A, 1–3). This became apparent at day 5 postinfection (pi) at which the monolayer of Tel-transduced cells was no longer flat, but showed relief. We termed this phenomenon “bridging.” On day 6 pi, the bridging became more extensive and holes started to appear in the monolayer. On day 7 pi, the holes were much bigger and the formation of cellular cords, tightly associated strings of cells, started. The latter structures seemed to result from the compaction of the cells forming “bridges.” From day 7 pi onwards, most of the cells were present in aggregates that were still attached to the dish, or in floating cellular cords still connected to adherent patches of cells. The cells in the cords were alive as they failed to stain with trypan blue. Moreover, after trypsinization of the aggregates and reseeding at subconfluency, almost all cells reattached to the culture dish, grew to confluency and repeated the sequence of events described above.
Figure 1.

Transduction of NIH3T3-UCLA fibroblasts with a Tel retroviral construct results in cellular cord formation. 5x104 Fibroblasts were seeded in six-well plates and transduced with retroviruses containing vector sequences (1–3) or containing Tel coding sequences (4–9). (A) The panels show monolayers at day 5 (A.1 and 4), day 6 (A.2 and 5) and day 7 (A.3 and 6) after transduction. Cellular cord formation became apparent on day 7 pi., for Tel-infected cells only. Phenotypic changes became apparent on day 5, at confluency, when “bridging” of cells started. On day 6, holes started to appear and from day 7 on, cellular cord formation started. (B) Tel-transduced cells were seeded on bacterial-type culture dishes or on dishes coated with fibronectin at various concentrations (7:0.1 µg/ml; 8: 0.5 µg/ml and 9: 2.5 µg/ml). Aggregation and cellular cord formation were only observed in dishes coated with low amounts of fibronectin.
The observations pointed toward an increased cell-cell adhesion, or decreased cell substrate binding of the Tel-transduced versus control cells. We therefore tested whether the phenotype was subject to differences in cell-substrate binding. Tel-transduced and vector-transduced control cells were seeded on bacterial tissue culture dishes that were coated with different concentrations of fibronectin. The Tel-induced phenotype was still apparent when plates were coated with low concentrations of fibronectin (0.1 and 0.5 µg/ml) (Figure 1B). Higher coating densities (higher than 2.5 µg/ml) inhibited the morphologic change. Cells did not spread or grow on the non-coated plates. Coating of the plates with collagen type IV produced the same results as with fibronectin (data not shown).
To map the domains of Tel that were needed to induce this phenotype, cells were retrovirally transduced with mutants of Tel or with Tel fusion genes (Table 1). To study whether the phenotype was Tel-specific or whether it was a general feature of PNT-domain containing Ets factors, we used two other Ets family members that contain a PNT domain and that are expressed in endothelial cells, Ets-1 and Fli-1 [27,28]. Finally, we also tested the possible involvement of Scl/Tal-1 in this process since this bHLH transcription factor is essential for angiogenesis in the yolk sac during mouse development [29]. Retroviruses harbouring the respective cDNA were prepared and used to infect NIH3T3-UCLA cells. We tested the infection efficiency, ectopic expression, subcellular distribution and induction of the morphologic changes for all constructs. Transfection efficiencies were assessed by immunofluorescence and found to be over 95% for all constructs. Western blot analysis revealed that exogenous protein levels were highly elevated over endogenous levels (Figure 2A). Only VP16-Tel and MN1-Tel were expressed moderately. The first panel (Figure 2A) shows a long exposure of the blot, visualizing endogenous Tel (first lane) and VP16-Tel. The subcellular localization of most ectopically expressed proteins was nuclear (Figure 2B). The cells expressing the highest levels of exogenous Tel and most of the Tel-mutants showed both cytoplasmic and nuclear localization (exemplified in Figure 2B, 2: Tel-transduced cells). Deletion of the PNT domain of Tel, or replacement of the two highly conserved Arg residues (Arg396,399) [30] with Leu or Lys residues in the DBD, resulted in complete (R/L) or partial (R/K and ΔPNT) cytoplasmic localization (Figure 2B, 9–11). The MN1-Tel protein localized to the nucleus, but was also found in speckles in both cytoplasm and nucleus (Figure 2B, 12). The phenotypic change was not observed for Tel-mutants that lacked the PNT domain or that contained Arg396,399 point mutations (Table 1). In contrast, all Tel-proteins containing both intact domains (Tel, M1C, S22A, S22E, S22A;M43C, S22E;M43C, M43C; Figure 2, 2–8) induced the morphologic change with similar kinetics and to a similar extent as wild-type Tel. Expression of fusions of the transactivating domains of VP16 or MN1 to the N-terminus of the Tel protein did not result in a phenotypic change at confluency (Figure 2B, 12 and 13). Also retroviral transduction of the transcription factors Ets-1, Fli-1 and Scl/Tal-1 did not induce the phenotypic change (Figure 2B, 14 and 15). Ets-1, however, did induce a limited amount of “bridging,” but the bridges were thinner and smaller than those observed for Tel (data not shown) and never progressed into cord formation. These results showed that the observed phenotype is highly specific for Tel and that the two functional domains of Tel are required.
Table 1.
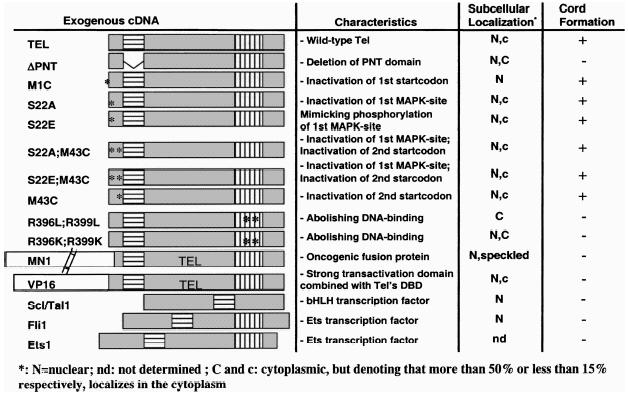
Figure 2.

Analysis of expression levels and subcellular localization of Tel, Tel mutants, Tel homologues and Scl/Tal-1. NIH3T3-UCLA cells were transduced with the various retroviruses and replated 3 days pi. (A) Cells were harvested and subjected to Western blot analysis. (B) Cells were seeded 3 days pi on microscope slides and processed for immunofluorescent observation. The presence and subcellular localization of the exogenously expressed proteins was studied by indirect immunofluorescence. Polyclonal antisera were used to detect endogenous Tel (1), exogenous wild-type Tel (2), Tel-M1C (p50) (3), Tel-M43C (p60) (4), Tel-S22A (5), Tel-S22E (6), Tel-S22A;M43C (7), Tel-S22E; M43C (8), Tel-R396L;R399L (9), Tel-R396K;R399K (10), Tel-ΔPNT (11), MN1-Tel (12), VP16-Tel (13), Fli-1 (14) and Scl/Tal-1 (15). Subcellular localization is summarized in Table 1 by “N” stands for nucleus, and “C” or “c” for cytoplasm. “C” and “c” denote that the majority, respectively, the minority of the exogenous protein is expressed in the cytoplasm.
The changes in morphology induced by exogenous Tel-expression closely resembled tube formation of endothelial cells [31,32]. Since Tel plays a role during angiogenesis in the yolk sac [21], we analyzed whether the observed cellular cords were hollow tubes, resembling those formed by primary endothelial cell explants. Cellular cord structures were therefore dissected from the culture dish and prepared for electron microscopy (EM). Cross-sections of large cellular cords (Figure 3A and B) revealed that the inner part of the cords is not hollow, but filled with fibroblasts. Examination of small cords of only one to two cells thick at any given cross section was inconclusive, since in some sections, they appeared hollow and in others not. Figure 3C shows the outer cell layer of a large cellular cord at high magnification (x80,000), consisting of thin and long cells. Polarized deposition of a basement membrane was observed (Figure 3C, arrows), only at the outer side of the cells that line the cord.
Figure 3.

Large cellular cords do not contain a lumen. NIH3T3-UCLA cells, transduced with Tel-expressing retrovirus, were prepared for EM. (A) Cross-sections revealed that the inner part of the cord is not hollow, but filled with fibroblasts. Picture was taken using light microscopy; original magnification, x200. (B) EM picture of the outer layer of the cellular cord; original magnification, x2500. (C) Original magnification, x80,000. Arrows indicate the deposited extracellular matrix.
Although Tel did not induce these fibroblasts to induce bona fide tubes, we tested if it would induce such structures in endothelial cells. We therefore retrovirally transduced Tel into the mouse endothelial cell line MS1. At confluency, these cells showed significant “bridging” when compared to the vector-infected control cells (Figure 4, 1 vs 2), but no further phenotypic changes were observed during the following days. When the cells were left in their conditioned medium for a week or longer, cell death was observed in both cultures, as judged by the presence of floating cells. Surprisingly, Tel-transfected endothelial cells did not show a random depletion of the monolayer. Circular and half-circular patches of cells were observed (Figure 4, 3 and 4), reminiscent of the holes formed in the monolayers of Tel-transduced NIH3T3-UCLA cells. The cells within these circular patches were thus protected from cell death for a longer time. As extracellular matrices are known to deliver survival and proliferative signals to the surrounding cells through cell adhesion molecules [33,34], an observed non-random deposition of ECM could explain the observed Tel-specific phenomenon.
Figure 4.

Tel induces aggregation of endothelial cells. MS1 cells (5x104/well) were seeded in six-well plates and transduced with retroviruses containing vector sequences (1 and 3) or containing Tel coding sequences (2 and 4). Cells were photographed at 9 days pi (1 and 2), or at 14 days pi (3 and 4). Tel-transduced cells clearly show bridging (2), whereas vector-transduced cells (1) remain as a flat and confluent monolayer. When the medium becomes depleted, the monolayer of vector-transduced cells loses cells at random (3), whereas the Tel-transduced cells are not lost in a randomized fashion (4): cells that are present in aggregates stay alive longer than those that do not form cell aggregates.
Fibronectin is produced by many cell types and is a major constituent of basement membranes. Coating of culture dishes with fibronectin also suppressed the Tel-induced morphologic change (Figure 1, 9). We therefore studied fibronectin expression in Tel- and vector-transduced NIH3T3-UCLA cells. Transfected cells were grown on glass slides for 3 days and their fibronectin content was visualized by indirect immunofluorescence. In several independent experiments, we consistently observed a lower level of intracellular and fibrillar fibronectin in vector-transduced cells than in Tel-transduced cells (Figure 5). The increase of fibronectin levels in Tel-transduced cells corroborated with a modest, but consistent increase of 50% of fibronectin mRNA on Northern blots of Tel-transduced cells over that of control cells (Figure 6).
Figure 5.

Analysis of Tel-induced fibronectin upregulation. Vector- (V) and Tel-transduced (TEL) NIH3T3-UCLA cells were seeded on microscope slides and indirect immunofluorescence was performed using a polyclonal antiserum raised against fibronectin. Images for “V” and “Tel” panels were acquired at fixed exposure times using a SPOT™ camera.
Figure 6.

Northern blot analysis of Tel-regulated genes. (A) Total RNA (15 µg/lane) derived from vector- and Tel-transduced NIH3T3-UCLA cells was loaded on a denaturing (formaldehyde) 1% agarose gel and blotted onto positively charged nylon membrane. Northern blot strips were incubated with 32P-labeled probes for 20 h, washed at high stringency (0.1% SDS, 0.1xSSC at 65°C) and analyzed in a PhosphorImager. Representative results of at least three independent experiments are shown. (B) Effect of exogenous Ets-1 and Fli-1 in NIH3T3-UCLA cells and of Tel in MS1 cells on the identified differentially expressed genes. Samples were processed as in A.
Since the occurrence of the cellular cords depended on the presence of Tel molecules harboring the wild-type PNT domain and DBD, we embarked on a systematic search for differentially expressed genes. Total RNA from Tel- and vector control-transduced NIH3T3-UCLA cells was used to prepare 32P-labeled cDNA for filter hybridizations and biotinylated cRNA for oligonucleotide chip hybridizations. About 1 in 100 of the spotted cDNA showed a differential hybridization signal. After Northern blot verification, this number was reduced to about 1 in 500, at least for the clones we analyzed. A similar frequency of differentially expressed genes was observed when searching for putative p53 target genes using the SAGE technique [35]. We limited our Northern blot analysis of putatively differentially expressed genes to those that supposedly function in ECM build-up or cell adhesion. Six differentially expressed genes were found in this way: entactin/nidogen, Col3A1, Smad5, CD44, Col1A1 and secretory leukocyte protease inhibitor (SLPI) (Figure 6A). The latter two were found to be downregulated by Tel, the others upregulated. The differential levels of expression were found to be modest, but consistent, ranging between 1.3- and 3.0-fold.
Since Tel induced cellular aggregation in NIH3T3-UCLA cells and MS1 cells, we assessed by Northern blot analysis whether Tel also influenced expression of the seven identified differentially expressed genes in MS1 cells. In addition, we studied the effect of Ets-1 and Fli-1 overexpression in NIH3T3-UCLA cells on these seven genes (Figure 6B). In MS1 cells, only Smad5 and fibronectin expression was detectable. These genes did not show differential expression, excluding their involvement in the Tel-induced aggregation. Both Ets-1 and Fli-1 induced differential expression of several of the genes under study. However, neither Ets-1 nor Fli-1 mimicked Tel's effect closely.
Discussion
Exogenous expression of the Tel protein in NIH3T3-UCLA fibroblasts resulted in a striking change in morphology that became apparent only after cells had reached confluency. Infection of the fibroblasts with viruses harboring cDNA that encode mutants of Tel, showed that both the DBD and the PNT domain were required to induce the phenotypic change. This suggests that both protein-protein interactions and DNA binding by Tel are important for this effect. Fusion of polypeptides with transactivating properties to the N-terminus of Tel, such as VP16 and MN1, abolished the phenotype. Together with the fact that the Ets family genes Fli-1 and Ets-1 or the bHLH transcription factor Scl/Tal-1 failed to induce an appreciable level of cellular cord formation, these observations make it likely that up- or downregulation of specific Tel target genes is causally involved in generating the phenotype. Whether any of the seven identified differentially expressed genes directly contributes to the phenotypic change remains to be determined. Although the observed differential expression was small, it should be noted that translation typically produces 103 to 104 polypeptides of a single mRNA molecule (R.A. Young, personal communication). This amplification is fine-tuned by posttranscriptional and posttranslational modifications, further enhancing or quenching the translational amplification effect. Thus, on the one hand, small differences in mRNA levels can cause a considerable difference in protein levels. Conversely, differential mRNA expression does not necessarily correlate linearly with differential protein expression. We observed this for fibronectin, where immunofluorescence analysis indicated a more than two- to three-fold upregulation in fibronectin protein levels, but Northern blot analysis revealed only a 1.4-fold increase in mRNA level. The presence of endogenous nuclear Tel (see Figure 2, 1) might be responsible for the fact that we only see modest differences in expression levels: overexpression of Tel can only show differences in target gene expression against a background level of expression by endogenous Tel.
Most of the differentially expressed genes we identified, have been shown to be important players in ECM composition and cell adhesion, but their function does not directly provide an explanation for the observed phenotype. However, gene ablation studies in mice revealed that Tel, fibronectin, Smad5, Col1a1 and Col3a1 have important and non-redundant roles in angiogenesis, at least during murine development. We will therefore only discuss these differentially expressed genes.
Fibronectin is a major ECM component, involved in cell migration, differentiation and growth [36]. The targeted disruption of the murine fibronectin gene resulted in embryonic lethality around E10.5 and showed several phenotypic abnormalities, one of which is defective yolk sac angiogenesis [37]. Using immunofluorescence, the Tel-induced upregulation of fibronectin was observed in NIH3T3-UCLA cells, regular NIH3T3 cells and in primary mouse embryo fibroblasts (L.V.R. and G.G., unpublished observations). This suggests that Tel might have a physiological role in upregulating fibronectin in vivo. Although the aggregation phenotype was inhibited by high coating densities of fibronectin, it is unlikely that the aggregation is due solely to fibronectin upregulation. We anticipate that more cell adhesion molecules and extracellular matrix molecules will be involved. Since fibronectin expression has tumor suppressor activity [38], its downregulation by partial or complete loss of Tel might contribute to the formation of tumors of fibroblast origin. This possibility is exemplified by a published case of congenital fibrosarcoma in which one Tel allele was fused to the neurotrophin 3 receptor, whereas the remaining allele was inactivated [8]. The loss of both Tel alleles in combination with a presumptive constitutive tyrosine kinase signaling by Tel-TRKC3 will contribute to fibrosarcoma development. This hypothesis endows Tel with a tumor suppressor function, and it will be interesting to see if (partial) loss of Tel function remains restricted to a few isolated cases or whether it is a more general phenomenon in mesenchymal tumors.
Smad5 has been identified as a downstream effector of BMP signaling [39]. Interestingly, the recently reported Smad5 KO phenotype shows similarities to the Tel KO phenotype as well as to those of transforming growth factor (TGF)-βRII and TGF-β1 and endoglin [40–44]. Smad5 and Tel KO mice show a defect in yolk sac angiogenesis and display mesenchymal apoptosis. These genetic data, combined with our in vitro data suggest that Smad5 might be a downstream target of Tel.
The TGF-β pathway is known to enhance ECM production, suggesting that Tel possibly functions in this pathway. The Tel-induced upregulation of ECM components underscores this possibility, in particular in light of the fact that the fibronectin KO phenotype also shows an angiogenic yolk sac defect [37]. However, we have data that argue against a common TGF-β/Tel pathway. Addition of purified TGF-β1 to NIH3T3-UCLA cells, transduced with Tel-encoding retrovirus or a control retrovirus, did not induce or accelerate aggregation and cord formation. Also, Tel does not seem to increase expression of TGF-β1, -2 or -3 in fibroblasts, as measured by RPA (L.V.R. and G.G., unpublished observations).
The expression levels of the fibrillar collagens Col1a1 and Col3a1 were specifically altered by exogenous Tel expression as Col4a1 or Col5a2 expression was not affected (L.V.R., W.D. and G.G., unpublished observations). Col1a1 and Col3a1 deficiencies can result in several disease states, such as Ehlers-Danlos syndromes VII and IV which are characterized by fragile blood vessels and skin. Targeted disruption of both genes in mice results in lethal phenotypes, caused by rupture of major blood vessels. Col1a1-deficient mice die at E13, whereas Col3a1 KO mice die around birth [45,46]. Moreover, Col1a1 deficiency during murine development resulted in mesenchymal cell death, a phenotype that was also observed in Smad5- and Tel-deficient mice [21,45]. The specific effect of Tel on Col1a1 and Col3a1 expression levels might therefore be of physiological significance, at least during embryonic development.
The study of Tel KO mice and of chimaeras derived from Tel-/- ES cells, showed that Tel has essential non-redundant functions in yolk sac angiogenesis and homing of the fetal liver hematopoietic system to the postnatal bone marrow [21,22]. Therefore, the lack of Tel expression in the mesenchymal cell layer surrounding the endothelial cells may result in insufficient deposition of ECM components that provide a substrate for the endothelial cells to adhere to. Similarly, insufficient deposition of cell adhesion substrate in the postnatal bone marrow stroma might underlie the hematopoietic homing defect. As CD44 has been implicated in homing of T lymphocytes, it becomes relevant to study whether it is a direct target gene of Tel and whether it is involved in homing of the hematopoietic system during development.
Microscopic examination of the Tel-induced phenotypic change in NIH3T3-UCLA cells showed it to be similar to tube formation of primary endothelial cells in vitro [47]. Cells clustered, formed holes in the monolayer and cellular cords appeared elevated above the substratum. Interestingly, coating the tissue culture dishes with fibronectin and collagen IV, abolished these effects [31]. The similarity in ultrastructure between cellular cord formation in our cell line and endothelial tube formation in vitro might reflect a partial overlap in transcriptional programs between the two types of cells. This is also suggested by the Tel-induced aggregation of both the endothelial MS1 cell line and the NIH3T3-UCLA fibroblasts. Whether Tel's target genes are both necessary and sufficient for the phenotypic change is an interesting question. A comparison between changes in gene expression levels in NIH3T3-UCLA fibroblasts undergoing cellular cord formation and primary endothelial cells undergoing tube formation could reveal shared features in the induced transcriptional programs. We could detect expression of only fibronectin and Smad5 in the MS1 cells. In these cells, Tel did not induce differential expression for these two genes, indicating that they take no part in the Tel-induced aggregation. Ets-1 and Fli-1 did induce differential expression for most of the seven genes in NIH3T3-UCLA fibroblasts. However, neither of them showed a similar pattern of differential expression as Tel. It is thus possible that a specific combination of the identified differentially expressed genes is causally involved in the observed cellular cord formation.
It was recently shown that a tumor does not rely entirely on endothelial cells for its blood supply [48]. Highly metastatic uveal melanomas formed patterned networks of interconnected channels, consisting of extracellular matrix and melanoma cells, which guided red blood cells. This process was aptly termed “vasculogenic mimicry.” These channels could be reconstituted in vitro in 3D-matrices and were shown to conduct fluid. Poorly invasive melanomas did not form these channels. The presence of these networks in the tumor samples correlated with poor prognosis in patients (Refs. [48,49] and references therein). It also correlated with a dedifferentiation of the melanocyte to a pluripotent cell: cells stained for both vimentin and cytokeratin and they expressed endothelial-specific genes (e.g., TIE-1). Additionally, several extracellular matrix components including fibronectin, were upregulated when highly metastatic versus poorly metastatic melanomas were compared. In this work, we observed deposition of basement membrane at the outside of large extracellular cords, a function uncharacteristic of fibroblasts. We also found fibronectin expression upregulated, and Western blot analysis showed the presence of both vimentin and cytokeratins in the NIH3T3-UCLA cells (L.V.R. and G.G., unpublished observations), whereas cytokeratins are not normally expressed in fibroblasts. In addition, when one seeds NIH3T3-UCLA cells on Matrigel, a 3D-matrix that has many features of a basement membrane, they form a tesselated pattern of interconnected cells just as endothelial cells do (L.V.R. and G.G., unpublished observations). These data point to a dedifferentiated phenotype of NIH3T3-UCLA cells. It is very likely that many more cell lines have pluripotent features, such as ECV-304 that is derived from a human bladder tumor (note in Ref. [49]). Also, highly aggressive astrocytomas isolated from patients, showed a phenotype in a culture dish that is similar to that of Tel-overexpressing NIH3T3-UCLA cells (E. Holland, personal communication). Marker analysis further indicated that these astrocytomas had reverted to a more progenitor-like cell type. This conversion correlated with increased aggressiveness of the tumor.
The highly specific role that Tel plays in induction of cellular cord formation in NIH3T3-UCLA cells and aggregation of MS1 cells, together with Tel's role in angiogenesis during murine development, warrants further study of Tel's involvement in tumorigenesis and vasculogenic mimicry.
Acknowledgements
We are grateful to T. Golub and R. Martinez for performing the oligonucleotide chip experiments. We thank J. Ghysdael and S. Baker for gifts of Ets-1 and Fli-1 cDNA and A. Zwijsen and D. Huylebroeck for a mouse Smad5 probe. We also thank R. Baer for the Scl/Tal-1 plasmid and antisera and G. Oliver for critically reading of the manuscript. Electron microscopy analysis was performed by the St. Jude EM-core facility (D. Davis, P. Brown and G. Murti), with special thanks to P. Brown for an in-depth analysis of the pictures.
Abbreviations
- ECM
extracellular matrix
- DBD
DNA-binding domain
- PNT
pointed domain
- pi
postinfection
Footnotes
This work was supported in part by NCI grant CA72996-03, the cancer center (CORE) support grant CA-21765 and by the American Lebanese Syrian Associated Charities (ALSAC) of St. Jude Childrens's Research Hospital.
References
- 1.Golub T, Barker G, Lovett M, Gilliland D. Fusion of PDGF receptor beta to a novel ets-like gene, tel, in chronic myelomonocytic leukemia with t(5;12) chromosomal translocation. Cell. 1994;77:307–316. doi: 10.1016/0092-8674(94)90322-0. [DOI] [PubMed] [Google Scholar]
- 2.Jousset C, Carron C, Boureux A, TranQuang C, Oury C, Dusanter-Fourt I, Charon M, Levin J, Bernard O, Ghysdael J. A domain of TEL conserved in a subset of ETS proteins defines a specific oligomerization interface essential to the emitogenic properties of the TEL-PDGFRβ oncoprotein. EMBO J. 1997;16:69–82. doi: 10.1093/emboj/16.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Poirel H, Oury C, Caron C, Duprez E, Laabi Y, Romana S, Tsapis A, Machauffe M, LeConiat M, Berger R, Ghysdael J, Bernard O. The TEL gene products: nuclear phosphoproteins with DNA binding properties. Oncogene. 1997;14:349–357. doi: 10.1038/sj.onc.1200829. [DOI] [PubMed] [Google Scholar]
- 4.Papadopoulos P, Ridge SA, Boucher CA, Stocking C, Wiedemann LM. The novel activation of ABL by fusion to an ets-related gene. Cancer Res. 1995;55:34–38. [PubMed] [Google Scholar]
- 5.Peeters P, Raynaud S, Cools J, Wlodarska I, Grosgeorge J, Philip P, Monpoux F, Van Rompaey L, Baens M, Van den Berghe H, Marynen P. Fusion of TEL, the ETS-variant gene 6 (ETV6), to the receptor-associated kinase JAK2 as a result of t(9;12) in a lymphoid and t(9;15;12) in a myeloid leukemia. Blood. 1997;90:2535–2540. [PubMed] [Google Scholar]
- 6.Lacronique V, Boureux A, Valle V, Poirel H, Quang C, Mauchauffe M, Berthou C, Lessard M, Berger R, Ghysdael J, Bernard O. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science. 1997;278:1309–1312. doi: 10.1126/science.278.5341.1309. [DOI] [PubMed] [Google Scholar]
- 7.Knezevich S, Garnett M, Pysher T, Beckwith J, Grundy P, Sorensen P. ETV6-NTRK3 gene fusions and trisomy 11 establish a histogenetic link between mesoblastic nephroma and congenital fibrosarcoma. Cancer Res. 1998;58:5046–5048. [PubMed] [Google Scholar]
- 8.Knezevich S, McFadden D, Tao W, Lim J, Sorensen P. A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat Genet. 1998;18:184–187. doi: 10.1038/ng0298-184. [DOI] [PubMed] [Google Scholar]
- 9.Eguchi M, Eguchi-Ishimae M, Tojo A, Morishita K, Suzuki K, Sato Y, Kudoh S, Tanaka K, Setoyama M, Nagamura F, Asano S, Kamada N. Fusion of ETV6 to neurotrophin-3 receptor TRKC in acute myeloid leukemia with t(12;15) (p13;q25) Blood. 1999;93:1355–1363. [PubMed] [Google Scholar]
- 10.Romana SP, Machauffe M, Le Coniat M, Chumakov I, Le Paslier D, Berger R, Bernard OA. The t(12;21) of acute lymphoblastic leukemia results in a Tel-AML1 gene fusion. Blood. 1995;85:3662–3670. [PubMed] [Google Scholar]
- 11.Golub TR, Barker GF, Bohlander SK, Hiebert SW, Ward DC, Bray-Ward P, Morgan E, Raimondi SC, Rowley JD, Gilliland DG. Fusion of the TEL gene on 12p13 to the AML1 gene on 21q22 in acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 1995;92:4917–4921. doi: 10.1073/pnas.92.11.4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shurtleff SA, Buijs A, Behm FG, Rubnitz JE, Raimondi SC, Hancock ML, Chan GC, Pui C, Grosveld G, Downing JR. TEL/AML1 fusion resulting from a cryptic t(12;21) is the most common genetic lesion in pediatric ALL and defines a subgroup of patients with an excellent prognosis. Leukemia. 1995;9:1985–1989. [PubMed] [Google Scholar]
- 13.Romana SP, Poirel H, Leconiat M, Flexor MA, Mauchauffe M, Jonveaux P, Macintyre EA, Berger R, Bernard OA. High frequency of t(12;21) in childhood B-lineage acute lymphoblastic leukemia. Blood. 1995;86:4263–4269. [PubMed] [Google Scholar]
- 14.Romana S, Mauchauffe M, LeConiat M, Chumakov I, Paslier DL, Berger R, Bernard O. The t(12;21) of acute lymphoplastic leukemia results in a Tel-AML1 gene fusion. Blood. 1995;85:3662–3670. [PubMed] [Google Scholar]
- 15.Raynaud S, Mauvieux L, Cayuela JM, Bastard C, Bilhou-Nabera C, Debuire B, Bories D, Boucheix C, Charrin C, Fière D, Gabert J. Brief Communication: TEL/AML1 fusion gene is a rare event in adult acute lymphoblastic leukemia. Leukemia. 1996;10:1529–1530. [PubMed] [Google Scholar]
- 16.Takeuchi S, Seriu T, Bartram C, Golub T, Reiter A, Miyoshi I, Gilliland D, Koeffler H. TEL is one of the targets for deletion on 12p in many cases of childhood B-lineage acute lymphoblastic leukemia. Leukemia. 1997;11:1220–1223. doi: 10.1038/sj.leu.2400743. [DOI] [PubMed] [Google Scholar]
- 17.Buijs A, Sherr S, van Baal S, van Bezouw S, van der Plas D, Geurts van Kessel A, Riegman P, Lekanne Deprez R, Zwarthoff E, Hagemeijer A, Grosveld G. Translocation (12;22) (p13;q11) in myeloproliferative disorders results in fusion of the ETS-like TEL gene on 12p13 to the MN1 gene on 22q11. Oncogene. 1995;10:1511–1519. [PubMed] [Google Scholar]
- 18.Cools J, Bilhou-Nabera C, Wlodarska I, Cabrol C, Talmant P, Bernard P, Hagemeijer A, Marynen P. Fusion of a novel gene, BTL, to ETV6 in acute myeloid leukemias with a t(4;12) (q11–q12;p13) Blood. 1999;94:1820–1824. [PubMed] [Google Scholar]
- 19.Peeters P, Wlodarska I, Baens M, Criel A, Selleslag D, Hagemeijer A, Van den Berghe H, Marynen P. Fusion of ETV6 to MDS1/EVI1 as a result of t(3;12) (q26;p13) in myeloproliferative disorders. Proc Natl Acad Sci USA. 1997;94:1852–1856. [PubMed] [Google Scholar]
- 20.Chase A, Reiter A, Burci L, Cazzaniga G, Biondi A, Pickard J, Roberts I, Goldman J, Cross N. Fusion of ETV6 to the caudal-related homeobox gene CDX2 in acute myeloid leukemia with the t(12;13) (p13;q12) Blood. 1999;93:1025–1031. [PubMed] [Google Scholar]
- 21.Wang L, Kuo F, Fujiwara Y, Gilliland D, Golub T, Orkin S. Yolk sac angiogenic defect and intra-embryonic apoptosis in mice lacking the Ets-related factor TEL. EMBO J. 1997;16:4374–4383. doi: 10.1093/emboj/16.14.4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang L, Swat W, Fujiwara Y, Gilliland D, Golub T, Orkin S. The TEL/ETV6 gene is required specifically for hematopoiesis in the bone marrow. Genes Dev. 1998;12:2392–2402. doi: 10.1101/gad.12.15.2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwiatkowski B, Bastian L, Bauer T, Tsai S, Zielinska-Kwiatkowska A, Hickstein D. The ets family member Tel binds to the Fli-1 oncoprotein and inhibits its transcriptional activity. J Biol Chem. 1998;273:17525–17530. doi: 10.1074/jbc.273.28.17525. [DOI] [PubMed] [Google Scholar]
- 24.May WA, Gishizky MI, Lessnick SL, Lunsford LB, Lewis BC, Delattre O, Zucman J, Thomas G, Denny CT. Ewing sarcoma 11;22 translocation produces a chimeric transcription factor that requires the DNA-binding domain encoded by FLI1 for transformation. Proc Natl Acad Sci USA. 1993;90:5752–5756. doi: 10.1073/pnas.90.12.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muller AJ, Young JC, Pendergast A, Pondel M, Landau NR, Littman DR, Witte ON. BCR first exon sequences specifically activate the BCR/ABL tyrosine kinase oncogene of Philadelphia chromosome-positive human leukemias. Mol Cell Biol. 1991;11:1785–1792. doi: 10.1128/mcb.11.4.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meersseman G, Verschueren K, Nelles L, Blumenstock C, Draft H, Wuytens G, Remacle J, Kozak C, Tylzanowski P, Niehrs C, Huylebroeck D. The C-terminal domain of Mad-like signal transducers is sufficient for biological activity in the Xenopus embryo and transcriptional activation. Mech Dev. 1997;61:127–140. doi: 10.1016/s0925-4773(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 27.Maroulakou IG, Papas TS, Green JE. Differential expression of ets-1 and ets-2 proto-oncogenes during murine embryogenesis. Oncogene. 1994;9:1551–1565. [PubMed] [Google Scholar]
- 28.Melet F, Motro B, Rossi D, Zhang L, Bernstein A. Generation of a novel Fli-1 protein by gene targeting leads to a defect in thymus development and a delay in Friend virus-induced erythroleukemia. Mol Cell Biol. 1996;116:2708–2718. doi: 10.1128/mcb.16.6.2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Visvader J, Fujiwara Y, Orkin S. Unsuspected role for the T-cell leukemia protein SCL/tal-1 in vascular development. Genes Dev. 1998;12:473–479. doi: 10.1101/gad.12.4.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kodandapani R, Pio F, Ni C-Z, Piccialli G, Clemsz M, McKercher S, Maki RA, Ely KR. A new pattern for helix-turn-helix recognition revealed by the PU.1 ETS-domain-DNA complex. Nature. 1996;380:456–460. doi: 10.1038/380456a0. [DOI] [PubMed] [Google Scholar]
- 31.Ingber D, Folkman J. Mechanochemical switching between growth and differentiation during fibroblast growth factor-stimulated angiogenesis in vitro: role of extracellular matrix. J Cell Biol. 1989;109:317–330. doi: 10.1083/jcb.109.1.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bischoff J. Approaches to studying cell adhesion molecules in angiogenesis. Trends Cell Biol. 1998;5:69–74. doi: 10.1016/s0962-8924(00)88949-7. [DOI] [PubMed] [Google Scholar]
- 33.Meredith J, Fazeli B, Schwartz M. The extracellular matrix as a cell survivor factor. Mol Biol Cell. 1993;4:953–961. doi: 10.1091/mbc.4.9.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen C, Mrksich M, Huang S, Whitesides G, Ingber D. Geometric control of cell life and death. Science. 1997;276:1425–1428. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- 35.Polyak K, Xia Y, Zweier J, Kinzler K, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 36.Hynes R. Fibronectins. New York: Springer-Verlag; 1990. [Google Scholar]
- 37.George E, Georges-Labouesse E, Patel-King R, Rayburn H, Hynes R. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development. 1993;119:1079–1091. doi: 10.1242/dev.119.4.1079. [DOI] [PubMed] [Google Scholar]
- 38.Ruoslahti E. Integrins as signaling molecules and targets for tumor therapy. Kidney Int. 1997;51:1413–1417. doi: 10.1038/ki.1997.193. [DOI] [PubMed] [Google Scholar]
- 39.Heldin C, Miyazono K, Kijke Pt. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- 40.Chang H, Huylebroeck D, Verscheren K, Guo Q, Matzuk M, Zwijsen A. Smad5 knockout mice die at mid-gestation due to multiple embryonic and extraembryonic defects. Development. 1999;126:1631–1642. doi: 10.1242/dev.126.8.1631. [DOI] [PubMed] [Google Scholar]
- 41.Yang X, Castilla L, Xu X, Li C, Gotay J, Weinstein M, Liu P, Deng C. Angiogenesis defects and mesenchymal apoptosis in mice lacking SMAD5. Development. 1999;126:1571–1580. doi: 10.1242/dev.126.8.1571. [DOI] [PubMed] [Google Scholar]
- 42.Oshima M, Oshima H, Taketo M. TGF-beta receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev Biol. 1996;179:297–302. doi: 10.1006/dbio.1996.0259. [DOI] [PubMed] [Google Scholar]
- 43.Dickson M, Martin J, Cousins F, Kulkarni A, Karlsson S, Akhurst R. Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knockout mice. Development. 1995;121:1845–1854. doi: 10.1242/dev.121.6.1845. [DOI] [PubMed] [Google Scholar]
- 44.Li D, Sorensen L, Brooke B, Urness L, Davis E, Taylor D, Boak B, Wendel D. Defective angiogenesis in mice lacking endoglin. Science. 1999;284:1534–1537. doi: 10.1126/science.284.5419.1534. [DOI] [PubMed] [Google Scholar]
- 45.Lohler J, Timpl R, Jaenisch R. Embryonic lethal mutation in mouse collagen 1 gene causes rupture of blood vessels and is associated with erythropoietic and mesenchymal cell death. Cell. 1984;38:597–607. doi: 10.1016/0092-8674(84)90514-2. [DOI] [PubMed] [Google Scholar]
- 46.Liu X, Wu H, Byrne M, Krane S, Jaenisch R. Type III collagen is crucial for collagen 1 fibrillogenesis and for normal cardiovascular development. Proc Natl Acad Sci U S A. 1997;94:1852–1856. doi: 10.1073/pnas.94.5.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Folkman J, Haudenschild C. Angiogenesis in vitro. Nature. 1980;288:551–556. doi: 10.1038/288551a0. [DOI] [PubMed] [Google Scholar]
- 48.Maniotis A, Folberg R, Hess A, Seftor E, Gardner L, Pe'er J, Trent J, Meltzer P, Hendrix M. Vascular channel formation by human melanoma cells in vivo and in vitro. Am J Pathol. 1995;155:739–752. doi: 10.1016/S0002-9440(10)65173-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bissell M. Tumor plasticity allows vaculogenic mimicry, a novel form of angiogenic switch. Am J Pathol. 1999;155:675–679. doi: 10.1016/S0002-9440(10)65164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]