

Abstract
The long QT syndrome (LQTS) is a cardiac disorder characterized by prolongation of the QT interval on electrocardiograms (ECGs), syncope and sudden death caused by a specific ventricular tachyarrhythmia known as torsade de pointes. LQTS is caused by mutations in ion channel genes including the cardiac sodium channel gene SCN5A, and potassium channel subunit genes KCNQ1, KCNH2, KCNE1, and KCNE2. Little information is available about LQTS mutations in the Chinese population. In this study, we characterized 42 Chinese LQTS families for mutations in the two most common LQTS genes, KCNQ1 and KCNH2. We report here the identification of four novel KCNQ1 mutations and three novel KCNH2 mutations. The KCNQ1 mutations include L191P in the S2–S3 cytoplasmic loop, F275S and S277L in the S5 transmembrane domain, and G306V in the channel pore. The KCNH2 mutations include L413P in transmembrane domain S1, E444D in the extracellular loop between S1 and S2, and L559H in domain S5. The location and character of these mutations expand the spectrum of KCNQ1 and KCNH2 mutations causing LQTS. Excitement, exercises, and stress appear to be the triggers for developing cardiac events (syncope, sudden death) for LQTS patients with KCNQ1 mutations F275S, S277L, and G306V, and all three KCNH2 mutations L413P, E444D and L559H. In contrast, cardiac events for an LQTS patient with KCNQ1 mutation L191P occurred during sleep or awakening from sleep. KCNH2 mutations L413P and L559H are associated with the bifid T waves on ECGs. Inderal or propanolol (a beta blocker) appears to be effective in preventing arrhythmias and syncope for an LQTS patient with the KCNQ1 L191P mutation.
Keywords: Long QT Syndrome; LQTS; cardiac arrhythmia; KCNQ1; KVLQT1; KCNH2; HERG; potassium channel; mutation; torsade de pointes; sudden death; ion channel; IKs, IKr
INTRODUCTION
The long QT syndrome (LQTS; MIM# 192500; 152427) is a cardiac disorder characterized by an abnormality in cardiac repolarization that leads to prolongation of the QT interval, T wave changes, and torsade de pointes (TdP) on the surface electrocardiograms (ECG) (Schwartz et al., 1975; Moss et al., 1991; Roden, 1998; Vincent et al., 1998; Zhang et al., 2000). LQTS causes syncope and sudden cardiac death, usually in the young. The incidence of sudden cardiac deaths in untreated patients approaches 50% over 10 years (Roden and Spooner, 1999). Arrhythmogenic syncope and sudden death often occur in association with acute physical, emotional or auditory arousal, or during sleep (Schwartz et al. 2001). The syncopal episodes were frequently misinterpreted as a seizure disorder. The prevalence rate of LQTS in the United States is estimated at 1 in 5,000 (Roden and Spooner, 1999), but the rate in China is not clear. Molecular genetic studies have established that LQTS is caused by mutations in several cardiac ion channel genes (reviewed by Wang et al., 2002). LQTS has been reported on chromosome 11q15.5 (LQT1), 7q35-36 (LQT2), 3p21 (LQT3), 4q25-27 (LQT4), 21q22 (LQT5), and 21q22 (LQT6). Five LQTS genes have been cloned or identified as the cardiac sodium channel gene SCN5A (LQT3) and potassium channel subunit genes KCNQ1 (LQT1), KCNH2 (LQT2), KCNE1 (LQT5), and KCNE2 (LQT6).
LQTS-causing mutations have been reported in patients and families from the Europe, North America, and Japan. Few genetic studies have been carried out with LQTS patients and families from China. In this report, we studied 42 LQTS families from China for mutations in KCNQ1 and KCNH2, the two most common LQTS genes (Wang et al., 1996; Splawski et al., 2000). We identified four disease-causing mutations in KCNQ1 and three in KCNH2 in a Chinese population. The mutation findings were correlated to clinical phenotype.
METHODS
Identification and clinical evaluation of LQTS patients
LQTS patients were identified from medical clinics throughout China. Informed consent was obtained according to the standards established by the People’s Hospital of Peking University and the United Hospital Committee on Clinical Investigations. Family members were evaluated by family history, physical examinations, and 12-lead electrocardiography. Historical data (the presence of syncope, number of syncopal episodes, age of onset of symptoms, and occurrence of sudden death) and the length of QTc were obtained. Phenotypic criteria used were as follows: (1) individuals without any symptoms and with QTc of ≤410 ms were classified as unaffected; (2) symptomatic individuals with QTc of ≥450 ms and asymptomatic individuals with QTc of ≥470 ms were classified as affected, and (3) symptomatic individuals with QTc of ≤440 ms and asymptomatic individuals with QTc between 410 and 460 ms were classified as uncertain (Schwartz et al., 1993).
DNA isolation and mutational analysis
Genomic DNA was isolated from peripheral blood lymphocytes using the DNA Isolation Kits for Mammalian Blood according to the manufacturer’s instruction (Roche Biochemical, Inc.). The exons and exon-intron boundaries of KCNQ1 and KCNH2 were PCR-amplified and sequenced for identifying LQTS-causing mutations. PCR primers were designed as described (Wang et al., 1996; Splawski et al., 1998). Mutations were confirmed by cloning the PCR products into pBLueScript SK (+) using the T-vector cloning kit (Promega) and by sequencing several independent clones, or by RFLP (restriction fragment length polymorphism) analysis.
RESULTS
KCNQ1 mutations in the Chinese population
We characterized 42 families with LQTS from a Chinese population. We screened LQTS-causing mutations in KCNQ1 and KCNH2 with all probands with LQTS. As shown in Figure 1 and Table 1, four disease-causing mutations in KCNQ1 were identified in four independent LQTS families.
Figure 1.
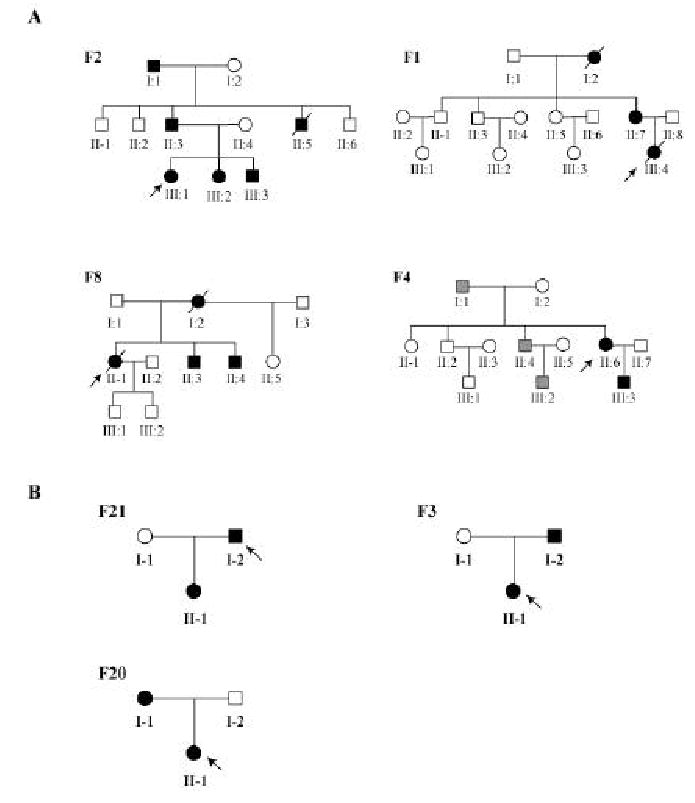
(A) Chinese families with KCNQ1 mutations. Kindred F2, mutation L191P; F1, F275S; F8, S277L; F4, G306V. (B) Chinese families with KCNH2 mutations. Kindred F21, mutation L413P; F3, E444D; F20, L559H. Males are denoted by squares and females are indicated by circles. Affected individuals are shown as filled symbols, normal individuals as empty symbols, and individuals with uncertain diagnosis of LQTS based on ECGs are stippled. The probands are indicated by arrows, and the deceased individuals are shown with “/”.
Table 1.
KCNQ1 Mutations in Chinese LQTS Families
| Kindred | Nucleotide Change* | Codon* Change | Protein Domain | Clinical Characteristics of Mutation Carriers | Trigger for Arrhythmias |
|---|---|---|---|---|---|
| F2 | 572T>C | L191P | Intracellular loop between S2 and S3 | I:1, QTc 480–500 ms; syncope, 1 episode
II:3, QTc 600 ms; syncope, 3 episodes; inverted T-wave (V1–V5), appearance of U-wave II:5, sudden death at age of 2 years III:1, proband: QTc 580 ms; TdP, syncope, 2 episodes (early morning sleeping) III:2, QTc 480 ms III:3, QTc 520 ms |
Sleeping or Awakening from sleep |
| F1 | 824T>C | F275S | S5 | I:2, QTc 470 ms; TdP; sudden death triggered by excitement
II:7, QTc 580 ms; history of palpitation III:4, proband: QTc 530 ms; syncope; sudden death at age of 19 years triggered by skiing, stress, and excitement |
Excitement
Exercise Stress |
| F8 | 830C>T | S277L | S5 | I:2, sudden death.
II:1, proband, QTc 680 ms; sudden death II:3, QTc 470 ms; no symptom. II:4, QTc 470 ms; no symptom. |
Exercise
Excitement |
| F4 | 917G>T | G306V | Pore | I:1, QTc 540 ms; no symptom.
II:4, QTc 600 ms; no symptom. II:6, proband, QTc 560 ms; syncope III:3, QTc 500 ms; no symptom |
Exercise
Excitement |
Positions of nucleotides and codons for KCNQ1 are based on NM-000218 and AF000571, respectively. TdP, torsade de pointes.
The proband of kindred F2 contained a T to C change at nucleotide position 572 (572T>C) (Figure 1A). This mutation leads to substitution of a leucine residue by a proline residue at codon 191 (L191P) in the intracellular domain between transmembrane domains S2 and S3 of the KCNQ1 channel. Mutation L191P was present in affected family members only, and was not present in unaffected family members or in 100 normal unrelated individuals (data not shown), suggesting that it is a pathogenic mutation. The proband in kindred F2 (individual III:1, Figure 1A) developed an episode of syncope while awakening in an early morning at the age of 29 years, and recovered from syncope shortly. A few hours later, she developed the second episode of syncope that lasted for 18s. Her QTc was 580 ms. After admitting to the hospital, she developed torsade de pointes and another round of syncope (19 seconds) that required cardioversion. X-ray, echocardiography, and blood chemistry were normal. The patient was prescribed with a beta blocker (Inderal, propanolol, 70 mg/day), and the medication appears to be effective because the patient has not developed syncope since 1999. The proband’s father’s ECGs showed QTc of 600 ms, inverted T waves at leads V1 to V5, and the U wave (Table 1), and he developed three episodes of syncope. Individual I:1 has QTc of 480–500 ms, and developed one episode of syncope. The paternal uncle (II:5, Figure 1A, F2) died suddenly at the age of 2 years (Table 1). Two other individuals in the family were also diagnosed with LQTS with QTc of 480 ms and 520 ms, respectively, but did not develop syncope (III:2 and III:3, Figure 1A, Table 1).
Mutational analysis in kindred F1 identified a T to C change at nucleotide position 824 (824T>C), which leads to substitution of a phenylalanine residue by a serine residue at codon 275 (F275S) (Figure 1A; Table 1). The F275S mutation is located in transmembrane domain S5. Mutation F275S co-segregated with affected family members only, and was not present in unaffected family members or 100 normal unrelated individuals (data not shown). The proband in kindred F1 (individual III:4, Figure 1A, F1) developed syncope in a crowded after-skiing gathering. ECGs showed QTc of 530 ms and detected torsade de pointes and ventricular fibrillation that required cardioversion. One day later, the patient died from ventricular tachycardia and ventricular fibrillation. Individual I:2 in the family also died from arrhythmia that was triggered by excitement.
The third KCNQ1 mutation occurs in exon 6 and changes nucleotide 830C into T (830C>T), which results in substitution of the serine residue at codon 277 by leucine (S277L). This mutation was observed in LQTS patients in family F8 (Figure 1A), but not in normal individuals in the family and 100 normal controls (data not shown). Mutation S277L is a non-conservative change and it is located in transmembrane domain S5. These results suggest that S277L is a pathogenic mutation. The proband in F8 (individual II:1, Figure 1A) died suddenly one year after enrolled in this study. The proband’s mother also died suddenly. Exercises and excitement are the trigger for sudden deaths in the family (Table 1).
The fourth KCNQ1 mutation also occurs in exon 6 and results in substitution of the glycine residue at codon 306 by valine (G306V) (Figure 1A, F4). Mutation G306V was present in another affected family member in kindred F4, but not in normal individuals in the family and 100 normal controls (data not shown). It is located in the pore region of the KCNQ1 channel. The proband in F4 (individual II:6) developed syncope during exercises and excitements (Table 1).
In addition to the four novel disease-causing mutations, we also identified two other changes in KCNQ1: a C to T change in intron 11 of KCNQ1 (IVS11+18C>T) in one of 42 patients and an A to G change at nucleotide position of 873 (873A>G or S291S) in another patient. The first change occurs in an intron and the second change is an unclassified variant that did not result in any amino acid substitution.
KCNH2 mutations in the Chinese population
In KCNH2, we identified three novel mutations: L413P in transmembrane domain S1, E444D in the extracellular loop between domains S1 and S2, and L559H in domain S5 (Figure 1B and Table 2). All three KCNH2 mutations were observed only in the LQTS patients in corresponding families (Figure 1B), but not in the normal family members and 100 normal controls. These data provide genetic evidence that L413P, E444D, and L559H are mutations. Mutations L413P and L559H in families F21 and F20 are non-conservative changes in critical domains (S1 and S5, respectively) of the KCNH2 channel. The proband in F21 developed syncope and torsade de pointes due to exercises and excitement (Table 2). The proband in F20 also developed syncope due to exercises and excitement (Table 2). The ECG patterns for the patients with KCNH2 mutations L413P and L559H mutation are characterized by the typical bifid T wave on leads V2–V4 (Figure 2). E444D is a conservative amino acid substitution, and similar conservative mutations have been identified in KCNQ1 (E261D, V310I) and in KCNH2 (V630L) (http://www.serum.dk/en/forskning/lqtsdb). Both patients with KCNH2 mutation E444D in kindred F3 developed syncope triggered by exercises and excitement (Table 2).
Table 2.
KCNH2 Mutations in Chinese LQTS Families
| Kindred | Nucleotide Change* | Codon* Change | Protein Domain | Clinical Characteristics of Mutation Carriers | Trigger for Arrhythmias |
|---|---|---|---|---|---|
| F21 | 1421T>C | L413P | S1 | I:2, QTc 440 ms; syncope, TdP
II:1, QTc 480 ms; no symptom |
Exercise, excitement |
| F3 | 1332G>T | E444D | Extracellular loop between S1 and S2 | II:1, QTc 600 ms; Syncope, TdP
I:2, QTc 520 ms; syncope symptom |
Exercise, excitement |
| F20 | 1676T>A | L559H | S5 | II:1, QTc 570 ms; syncope
I:1, QTc 480 ms; no symptom |
Exercise, excitement |
Positions of codons and nucleotides for KCNH2 are based on NM_000238. TdP, torsade de pointes.
Figure 2.
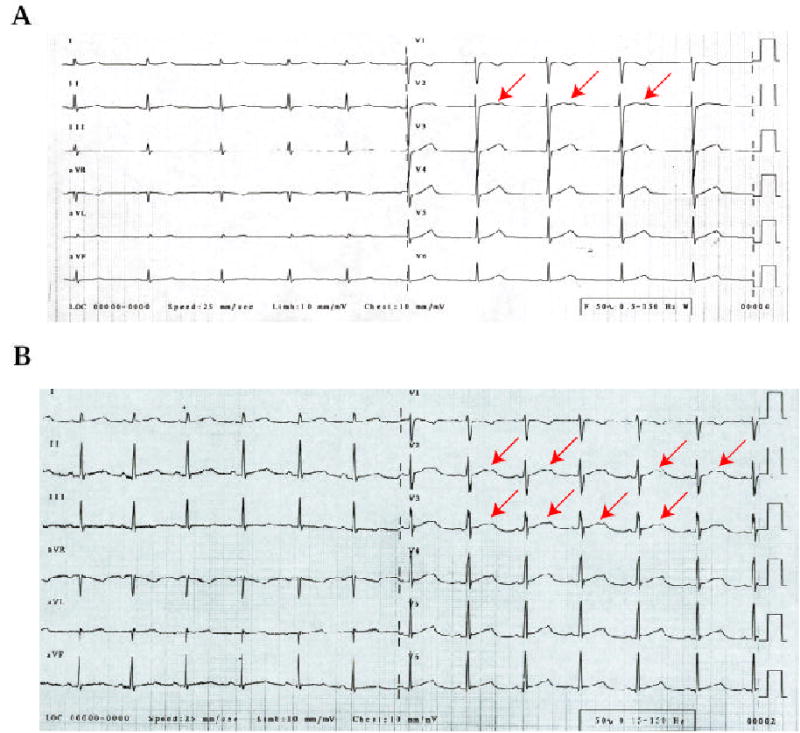
Typical bifid T waves associated with KCNH2 mutations in Chinese families with LQTS. (A) ECG for a patient with KCNH2 mutation L413P in kindred F21. (B) ECG for a patient with KCNH2 mutation L559H in kindred F20. The bifid T waves are indicated by arrows.
Finally, in one of 42 LQTS patients we detected a C to T polymorphism at the nucleotide position of 1467 (1467C>T) in KCNH2 that does not result in any amino acid substitution (I489I) and appears to be a polymorphism (http://pc4.fsm.it:81/cardmoc/).
DISCUSSION
We have identified four KCNQ1 mutations (L191P, F275S, S277L, G306V) and three KCNH2 mutations (L413P, E444D, L559H) in seven of 42 Chinese families with LQTS. These data indicate that LQTS in the Chinese population is caused by similar genes to ones identified in the Caucasian and Japanese populations. All seven mutations appear to be novel, therefore, they expand the spectrum of KCNQ1 and KCNH2 mutations causing LQTS. Due to the sheer size of the Chinese population (1.4 billion people), the finding of LQTS mutations suggests that mutation studies and genetic testing for LQTS will benefit many individuals. Presymptomatic diagnosis will be particularly important for individuals from families with a history of sudden death.
The triggers of developing cardiac events (syncope, sudden death) for LQTS patients are under active investigation. Clinical characterization of the four LQTS families with KCNQ1 mutations suggests that the triggers of developing cardiac events for LQTS patients with KCNQ1 mutations include both excitement/exercise/stress (mutations F275S, S277L, G306V), and sleep or awakening from sleep (mutation L191P). The triggering factors for cardiac events for the probands carrying KCNH2 mutations L413P, E444D, and L559H are exercises and excitements. It has been reported that LQTS patients with mutations in the cardiac sodium channel gene SCN5A experience cardiac events during sleep or awakening from sleep (Schwartz et al., 2001). It is interesting to note that the LQTS patient with KCNQ1 mutation L191P developed syncope while awakening in an early morning.
LQT1 and LQT2 are the two most common types of LQTS, as mutations in KCNQ1 and KCNH2 account for 42% and 45% of the identified LQTS mutations in North America, Europe, and Japan, respectively (Splawski et al., 2000). We, however, were able to detect KCNQ1 or KCNH2 mutations in only 10% (4/42) and 7% (3/42) of LQTS families in the Chinese population, respectively. The true prevalence rate of KCNQ1 and KCNH2 mutations in the Chinese population may be slightly higher due to our small sample size. Future studies with a large sample size of LQTS families will reveal the true prevalence rate of KCNQ1 and KCNH2 mutations in the Chinese population.
Footnotes
Grant sponsors: The Cardiovascular Institute and the United Hospital, Wuhan, China; The National Natural Science Foundation of China (NNSFC); Grant number: 30170381; Grant sponsor The American Heart Association Ohio-Affiliate; Grant number: Grant-in-Aid 0051205B.
Communicated by Mark H. Paalman
References
- Moss AJ, Schwartz PJ, Crampton RS, Tzivoni D, Locati EH, MacCluer J, Hall WJ, Weitkamp L, Vincent GM, Garson A., Jr The long QT syndrome. Prospective longitudinal study of 328 families. Circulation. 1991;84:1136–1144. doi: 10.1161/01.cir.84.3.1136. [DOI] [PubMed] [Google Scholar]
- Roden DM. Mechanisms and management of proarrhythmia. Am J Cardiol. 1998;82:49I–57I. doi: 10.1016/s0002-9149(98)00472-x. [DOI] [PubMed] [Google Scholar]
- Roden DM, Spooner PM. Inherited long QT syndromes: A paradigm for understanding arrhythmogensis. J Cardiovasc Electrophysiol. 1999;10:1664–1683. doi: 10.1111/j.1540-8167.1999.tb00231.x. [DOI] [PubMed] [Google Scholar]
- Schwartz PJ, Periti M, Malliani A. The long Q-T syndrome. Am Heart J. 1975;89:378–390. doi: 10.1016/0002-8703(75)90089-7. [DOI] [PubMed] [Google Scholar]
- Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circulation. 1993;88:782–784. doi: 10.1161/01.cir.88.2.782. [DOI] [PubMed] [Google Scholar]
- Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, Denjoy I, Guicheney P, Breithardt G, Keating MT, Towbin JA, Beggs AH, Brink P, Wilde AA, Toivonen L, Zareba W, Robinson JL, Timothy KW, Corfield V, Wattanasirichaigoon D, Corbett C, Haverkamp W, Schulze-Bahr E, Lehmann MH, Schwartz K, Coumel P, Bloise R. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95. doi: 10.1161/01.cir.103.1.89. [DOI] [PubMed] [Google Scholar]
- Splawski I, Shen J, Timothy KW, Vincent GM, Lehmann MH, Keating MT. Genomic structure of long QT syndrome genes: KVLQT1, HERG, and KCNE1. Genomics. 1998;51:86–97. doi: 10.1006/geno.1998.5361. [DOI] [PubMed] [Google Scholar]
- Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, Moss AJ, Schwartz PJ, Towbin JA, Vincent GM, Keating MT. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178–1185. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- Vincent GM. The molecular genetics of the long QT syndrome: genes causing fainting and sudden death. Annu Rev Med. 1998;49:263–274. doi: 10.1146/annurev.med.49.1.263. [DOI] [PubMed] [Google Scholar]
- Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, de Jager T, Schwartz PJ, Toubin JA, Moss AJ, Atkinson DL, Landes GM, Connors TD, Keating MT. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nature Genet. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- Wang Q, Pyeritz RE, Seidman C, Basson C. Genetics of myocardial and vascular disease. Textbook of Cardiovascular Medicine. In: Topol EJ, editor. 2nd ed. New York, NY: Lippincott Williams & Wilkins; 2002. pp. 1967–1990. [Google Scholar]
- Zhang L, Timothy KW, Vincent GM, Lehmann MH, Fox J, Giuli LC, Shen J, Splawski I, Priori SG, Compton SJ, Yanowitz F, Benhorin J, Moss AJ, Schwartz PJ, Robinson JL, Wang Q, Zareba W, Keating MT, Towbin JA, Napolitano C, Medina A. Spectrum of ST-T-wave patterns and repolarization parameters in congenital long-QT syndrome: ECG findings identify genotypes. Circulation. 2000;102:2849–55. doi: 10.1161/01.cir.102.23.2849. [DOI] [PubMed] [Google Scholar]