

Abstract
Background
Ethanol exposure during gastrulation and early neurulation induces apoptosis within certain embryonic cell populations, leading to craniofacial and neurological defects. There is currently little information about the initial kinetics of ethanol-induced apoptosis, and interest in the ability of endogenous survival factors to moderate apoptosis is growing. Ethanol alters intracellular signaling, leading to cell death in chick embryos, suggesting that apoptosis could occur rapidly and that signaling pathways activated by survival factors might reduce apoptosis.
Methods
Pregnant mice were intubated with 1, 2, or 4 g/kg ethanol on day 7.5 of embryogenesis (E7.5) 1, 3, or 6, hours before harvesting gastrulation-stage embryos. Control animals received maltose/dextran. Blood alcohol concentrations (BAC) were determined by gas chromatography. E7.5 embryos isolated from untreated dams were cultured in vitro for 1 or 3 hr with 0 or 400 mg% ethanol and 0 or 5 nM heparin-binding epidermal growth factor (EGF)-like growth factor (HB-EGF). Apoptosis was quantified using fluorescence microscopy to detect annexin V binding and DNA fragmentation [terminal deoxynucleotidyl transferase-mediated dUTP-X nick end labeling (TUNEL)] in whole-mount or sectioned embryos.
Results
Both annexin V binding and TUNEL were elevated (p<0.05) in embryos exposed in utero to 1 g/kg ethanol for 3 hours, increasing linearly with time and ethanol concentration. Apoptosis increased (p<0.05) in all germ cell layers. Mice treated with 4 g/kg sustained BAC of 400 mg% for nearly 3 hours, significantly increasing apoptosis within the first hour. Cultured embryos exposed to 400 mg% ethanol displayed 2- to 3-fold more TUNEL than vehicle-treated embryos (p<0.05); however, exogenous HB-EGF prevented apoptosis.
Conclusions
Ethanol rapidly produced apoptosis in gastrulation-stage embryos, consistent with induction by intracellular signaling. The ethanol-induced apoptotic pathway was blocked by the endogenous survival factor, HB-EGF. Differences in the expression of survival factors within individual embryos could be partly responsible for variations in the teratogenic effects of ethanol among offspring exposed prenatally.
Keywords: Prenatal Alcohol Exposure, Apoptosis, Embryo, Gastrulation, Annexin V, HB-EGF
PRENATAL EXPOSURE TO ethanol is highly toxic to mammals, manifesting in infants born to alcoholic women as a pattern of growth retardation, characteristic facial anomalies and mental retardation known as fetal alcohol syndrome (FAS) (Hannigan and Armant, 2000; Jones and Smith, 1973). The teratogenic effects of alcohol are dependent on the peak maternal blood alcohol concentration (BAC) and the gestational age of the fetus at the time of exposure. These critical parameters have been more precisely defined in animal models. Birth defects associated with aberrant craniofacial development and neural tube disorders can arise due to fetal ethanol exposure during gastrulation, as shown in a mouse model (Sulik et al., 1981). Evidence suggests that these anomalies are also produced in humans during the equivalent period of development (Clarren and Bowden, 1982; Clarren and Smith, 1978; Ernhart et al., 1987; Friedman, 1982).
Ethanol induces apoptosis in embryonic cells, as it does in other cell populations (Ewald and Shao, 1993; Freund, 1994; Holownia et al., 1997; Kotch and Sulik, 1992b). Whole embryo studies show that gastrulation-stage ethanol exposure causes neural crest losses through enhanced cell death within the neuroepithelium and craniofacial regions of early organogenesis-stage mice (Kotch and Sulik, 1992a, 1992b) and chicks (Cartwright and Smith, 1995a, 1995b; Smith and Debelak-Kragtorp, 2005). Patterns of ethanol-induced apoptosis have been distinguished in mouse embryos between embryonic days 7 (E7.0) and E11.0 (Dunty et al., 2001). A teratogenic dose of ethanol administered i.p. on E7.0 results 16 hours later in a noticeable increase in Nile blue sulfate dye uptake throughout the ectoderm and neuroectoderm, suggesting a specific pattern of cell death in alcohol-exposed embryos. This experiment demonstrates that acute exposure to ethanol can have a significant impact during gastrulation and neurulation. Differences between the clinical impact of acute and chronic prenatal exposure to ethanol derive from the specific cellular effects, combined with the opportunity for affected tissues to recover and repair themselves. Even acute exposure to ethanol, if it occurs at a critical period of development, can have a profound impact on the offspring.
While previous studies have documented the induction of apoptosis at the gastrulation stage by assessing embryos 12 to 16 hours after ethanol exposure, there is no information available about the immediate effects on the apoptotic pathway. The mobilization of intracellular Ca2+ within seconds of ethanol administration is a contributory factor in apoptosis of emerging neural crest cells in the chick embryo (Debelak-Kragtorp et al., 2003; Garic-Stankovic et al., 2005). Early during apoptosis there is a random redistribution of phosphatidylserine from the inner leaflet of the plasma membrane, which is followed somewhat later by genomic DNA fragmentation due to internucleosomal cleavage (Allen et al., 1997). These biochemical events have been observed in cortical neurons isolated from rat E16 to E17 fetuses exposed to ethanol for 2 and 12 hours (Ramachandran et al., 2003). We hypothesize that, due to the ability of ethanol to rapidly perturb endogenous signal transduction, apoptosis is initiated within the first several hours after exposure to ethanol. This idea was tested by comparing the dose dependency and time dependency of in utero ethanol exposure on apoptosis of mouse gastrulation-stage embryos during the initial 6 hours after treatment, assessing two early biochemical markers (phosphatidylserine externalization and DNA fragmentation) of the apoptotic pathway.
Intracellular signaling induced by growth factors can inhibit apoptosis and other detrimental effects in embryonic cells exposed to ethanol (Bonthius et al., 2003; de la Monte et al., 2000; Luo et al., 1997; Mitchell et al., 1999). Anti-apoptotic activity in cells subjected to oxidative stress has been reported for numerous cytokines and growth factors, including members of the epidermal growth factor (EGF), insulin-like growth factor (IGF), and transforming growth factor-β families, due to downstream signaling by their receptors (Dhandapani and Brann, 2003; Hong et al., 2001; Ozaki et al., 2003; Suzuki, 2003; Wells, 1999). Notably, heparin-binding EGF-like growth factor (HB-EGF) promotes cell survival during normal development (Chobotova et al., 2005) and inhibits apoptosis caused by ischemia reperfusion injury (El Assal and Besner, 2004). No role for this ubiquitious member of the EGF family in moderating the effects of alcohol during embryogenesis is known. Since HB-EGF is highly expressed during early embryonic development in the reproductive tract of mice (Das et al., 1994) and humans (Leach et al., 1999; Yoo et al., 1997), we cultured gastrulation-stage mouse embryos in vitro to examine the ability of exogenous HB-EGF to ameliorate ethanol-induced apoptosis.
MATERIALS AND METHODS
Embryo Production, In Utero Treatment, and Processing
Gastrulation-stage embryos were obtained from natural matings of C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME) maintained on a 14-hours light cycle, using established methods (Hogan et al., 1994). Nulliparous females judged by vaginal smears to be in estrus were paired with males for mating at the beginning of the dark cycle and checked for vaginal plugs 4 hours later. Pregnancy was estimated to begin at the midpoint of the dark cycle for the staging of embryos (Downs and Davies, 1993). Pregnant mice were treated on E 7.5 by intragastric intubation with a 15% (w/v) solution of ethanol to deliver 1, 2, or 4 g ethanol per kg body weight. Control animals were intubated with a maltose/dextran solution that was isocaloric with the highest ethanol dose. Food and water were removed 1 hour before intubation. Mice were killed 1, 3, or 6 hours after intubation and uteri were dissected to remove decidua swellings through an incision in the antimesometrial wall.
For analysis of sectioned material, concepti were fixed overnight in phosphate-buffered saline (PBS) containing 4% paraformaldehyde and processed for paraffin embedding. For whole-mount assays, concepti were either processed for immediate assessment of apoptosis by fluorescence microscopy or collected from untreated dams for in vitro culture. In either case, the decidua swellings were dissected to expose the fetal membranes. The visceral yolk sac was isolated by removing Reicherts’s membrane and ectoplacental cone.
In Vitro Embryo Culture
Intact visceral yolk sacs were cultured for 1 or 3 hours at 37 °C in a 5% CO2/air incubator using Ham’s F10 medium (Sigma Chemical Co., St. Louis, MO) supplemented with 4 mg/ml BSA, 50 U/ml penicillin, and 100 U/ml streptomycin. Medium was supplemented with or without 400 mg% (w/v) ethanol and 5 nM human recombinant HB-EGF (R&D Systems, Minneapolis, MN), as detailed in the Results section. Culture was conducted in 20-μl drops of medium on a Falcon petri dish (EMD Biosciences, San Diego, CA) flooded with embryo tested mineral oil (Sigma) to prevent ethanol evaporation, as previously reported (Leach et al., 1993). At the conclusion of the culture period, the extraembryonic tissue was removed and embryos were fixed in preparation for whole-mount terminal deoxynucleotidyl transferase-mediated dUTP-X nick end labeling (TUNEL) assay, described below.
Determination of BAC
Nonpregnant female mice intubated with 0.5 to 4.0 g ethanol per kg body weight were bled from the tail vein into capillary tubes at various times up to 4 hours after intubation. No more than two bleedings were obtained from individual mice. Ethanol concentrations were determined by mixing 50 μl of blood with 1 ml butanol and performing headspace gas chromatography, as previously described (Leach et al., 1993).
Whole-Mount Labeling with Annexin V and TUNEL
The extraembryonic tissues of live, isolated visceral yolk sacs were removed, exposing the neuroectoderm lining the inner proamniotic cavity during annexin V labeling. Embryonic tissues were incubated for 15 minutes at room temperature in 10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2, pH 7, containing a 1:20 dilution of biotin-labeled annexin V (Molecular Probes, Eugene, OR). They were next fixed for 30 min in 3% paraformaldehyde prepared in PBS, followed by an overnight incubation at 4 °C in PBS containing 5 mg/ml BSA and 5 μg/ml UltraAvidin-Texas Red (Leinco Technologies, St. Louis, MO). Embryos were then permeabilized for 10 minutes with 0.1% Triton X-100 in PBS and subjected to the TUNEL method using fluorescein-dUTP with a kit from Roche Applied Science (Indianapolis, IN). Embryos were mounted under a coverslip to partially flatten them and imaged by brightfield and epifluorescence microscopy (Leica DM IRB, Wetzlar, Germany) using a Spot Jr. (Diagnostic Research, Sterling Heights, MI) color digital camera. Identical camera settings were used to obtain all fluorescent images. Simple PCI (C-Imaging Corp., Cranberry Township, PA) imaging software was used to generate an annexin V binding index by measuring the number of pixels in each embryo image that were labeled by Texas Red/annexin V, based on a preset fluorescence threshold, divided by the total number of pixels occupied by embryonic tissue. A TUNEL index was determined using the imaging software to count the number of fluorescein/TUNEL-labeled nuclei for each embryo, divided by the total embryo area.
TUNEL Determination with Paraffin-Embedded Embryos
Apoptotic cells within sectioned embryos were identified using a 5-bromodeoxyuridine (BrdU)-based TUNEL protocol adapted from a published procedure (Darzynkiewicz et al., 2002). Whole, paraformaldehyde-fixed decidua swellings containing E7.5 embryos were embedded in paraffin and 5-μm sections, cut on the saggital plane, dewaxed, rehydrated, incubated in proteinase-K (20 μg/ml), and refixed in 10% formalin. Sections were then reacted with terminal deoxynucleotidyl transferase (5 units/μl, Promega) diluted in labeling buffer (80 μM BrdUTP, 1 mM CoCl2, 0.2 M sodium ca-codylate, pH 6.6, 250 μg/ml albumin) for 90 min at 37 °C. BrdU epitopes were detected with antibody G3G4 (Developmental Studies Hybridoma Bank, Iowa City, IA) and visualized using Alexa 488-conjugated donkey anti-mouse immunoglobulin (Molecular Probes). Nuclei were counterstained with 0.1 μg/ml propidium iodide. TUNEL-positive and total nuclei were counted by observers blinded to treatment. Each embryo was examined in its entirety. Cells were scored separately in the neuroectoderm and in the mesendoderm (combined endoderm and mesoderm).
Sampling and Statistical Analysis
For BAC determinations, at least three samples were obtained for each dose/time to determine the average BAC. For the ethanol dose response/time course analysis of TUNEL and annexin V binding in whole-mount embryos, 9 to 10 embryos from 3 to 5 litters were analyzed in each treatment group. For comparison of TUNEL in the neuroectoderm and mesendoderm, the control group consisted of three embryos from different litters and the ethanol-treated group contained six embryos from three litters. Each embryo was completely sectioned and every nucleus in all recovered sections was included in the analysis.
Statistical analyses were conducted using SPSS 13.0 for Windows (SPSS, Inc., Chicago, IL). All TUNEL and annexin V binding data were compared by 1- or 2-way ANOVA. The 1-way ANOVAs were followed by the Student-Newman-Keuls post hoc test. If a 2-way ANOVA identified a significant variable, a Student-independent t-test was used for post hoc comparisons of that variable.
RESULTS
BACs in female mice after administration of a single oral dose of 0.5, 1.0, 2.0, or 4.0 g ethanol per kg body weight were determined by gas chromatography. For all treatments, BACs peaked within 30 minutes of ethanol administration, reaching 40 mg% for 0.5 g/kg, 100 mg% for 1.0 g/kg, 180 mg% for 2.0 g/kg, and 400 mg% for 4.0 g/kg (Fig. 1). Clearance times were dose dependent. The three lowest doses were cleared by 1, 2, or 4 hours, respectively, while the 4.0 g/kg treatment maintained BACs in excess of 200 mg% 4 hours after administration.
Fig. 1.
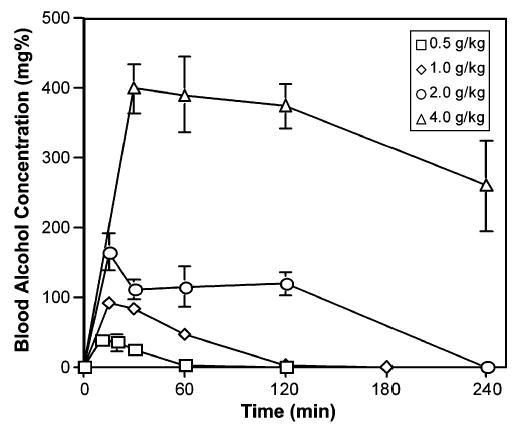
Blood alcohol concentrations (BAC) after intragastric intubation. Female mice were intubated with 0.5 to 4.0 g/kg ethanol and blood was collected from the tail vein at the indicated times. The BAC of each sample was determined by head space gas chromatography. Mean and SE for triplicate samples are shown.
To assess the effect of in utero ethanol treatment on cell death, embryos exposed to ethanol on E7.5 were assayed for both phosphatidylserine externalization (annexin V binding) and DNA fragmentation (TUNEL) using a fluorescent, whole-mount, double-labeling technique. Apoptosis was not readily detected in control embryos by either labeling method (Fig. 2A–2C). Similar regions of ethanol-exposed embryos were labeled by the two procedures, producing fluorescent TUNEL-positive nuclei and annexin V-labeled cell membranes (Fig. 2D–2F). Double labeling revealed that similar populations were detected by both methods. These cells were identified as apoptotic rather than necrotic, as defined by criteria that included the ability to be DNA end labeled by terminal transferase, binding of annexin V, and the presence of pyknotic nuclei (inset in Fig. 2F). Using image analysis to quantify annexin V binding and TUNEL, we determined the ethanol dose dependency and kinetics of apoptosis. The time course for cell death in gastrulation-stage embryos was similar at all three ethanol doses (Fig. 3), although it tended to level off after 3 hours at the highest dose. Increased (p<0.05) numbers of TUNEL-positive cells were detected by 1 hour after administration of 4 g/kg ethanol, and significant increases in both annexin V binding and TUNEL occurred as early as 3 hours at the lower doses (Fig. 3). We conclude that ethanol induces apoptosis in a dose-dependent manner within 1 to 6 hours during gastrulation, a stage associated with alcohol teratogenesis in mice (Sulik et al., 1981).
Fig. 2.
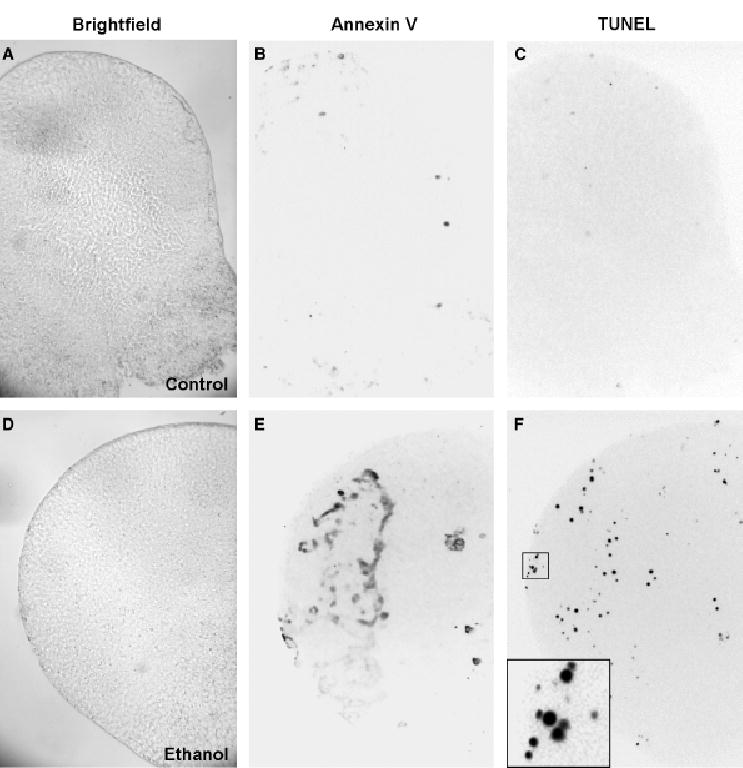
Embryos double labeled for terminal deoxynucleotidyl transferase-mediated dUTP-X nick end labeling (TUNEL) and annexin V binding. Images of embryos tested for annexin V binding and TUNEL are shown in brightfield (A, D) and in inverted images of fluorescence due to bound annexin V (B, E) and TUNEL (C, F). The upper embryo (A–C) is a control and the lower embryo (D–F) was exposed in utero to 4 g/kg ethanol for 3 hours. The boxed region in F is shown at higher magnification in the inset to illustrate TUNEL-positive nuclei, both intact and pyknotic fragments.
Fig. 3.
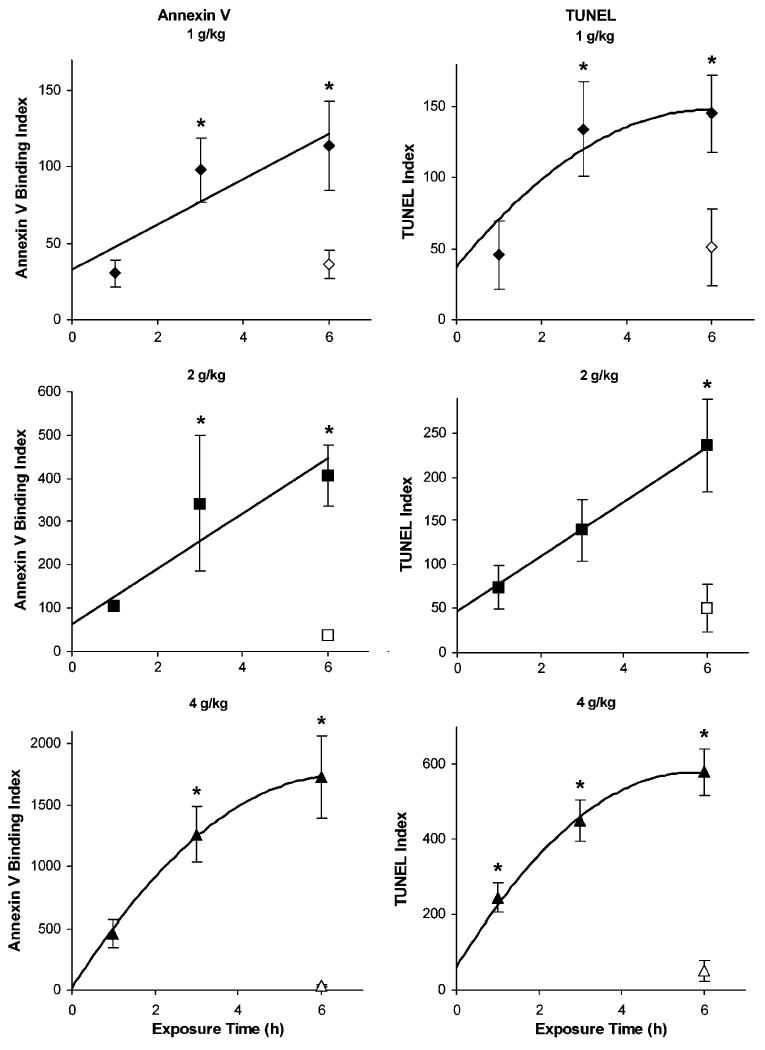
Time dependency and dose dependency of cell death in embryos exposed to ethanol in utero. Annexin V binding (left panels) and terminal deoxynucleotidyl transferase-mediated dUTP-X nick end labeling (TUNEL; right panels) were quantified in E7.5 embryos, as described in the Materials and Methods section, during the first 6 hours after treating pregnant females with 1 (diamonds), 2 (squares), or 4 (triangles) g/kg ethanol. Pregnant dams treated for 6 hours with maltose–dextran served as control embryos (open symbols). Mean and SE are shown. N = 59 to 10 embryos per treatment. *p<0.05 compared to control embryos.
TUNEL-positive nuclei were found throughout the embryonic germ layers by cross-section analysis at the mid-gastrulation stage in pregnant mice treated for 3 hours with 4 g/kg ethanol on E7.5 (Fig. 4). Control embryos from dams treated with isocaloric maltose/dextran solution contained less than 3% TUNEL-positive cells. Cell death increased with ethanol treatment, rising to 13% in the neuroectoderm and 20% in the mesendoderm. A 2-way ANOVA demonstrated a significant effect of ethanol treatment (p<0.0001), but no effect of germ cell type (p = 0.051). The effect of ethanol relative to the corresponding controls was significant for both the neuroectoderm (p=0.0001) and the mesendoderm (p < 0.001) when analyzed by Student’s independent t-test.
Fig. 4.
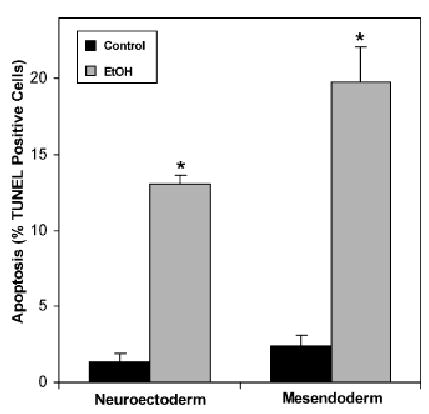
Cell-specific terminal deoxynucleotidyl transferase-mediated dUTP-X nick end labeling (TUNEL) in sectioned embryos exposed to ethanol in utero. TUNEL positive nuclei and propidium iodide counterstained (total) nuclei were counted in the entirety of endoderm plus mesoderm regions (mesendoderm) and in the neuroectoderm cells of embryos from dams gavaged on E7.5 with 4 g/kg ethanol (EtOH) or isocaloric maltose/dextran (control) 3 hours before embryo collection. Mean and SE are shown. N = 3 embryos for control. N = 6 embryos for ethanol. *p<0.005 compared to the respective control. See text for specific significance levels.
The influence of the survival factor HB-EGF on ethanol-induced embryotoxicity was investigated by establishing an experimental model for embryo exposure. E7.5 visceral yolk sacs were cultured in vitro for 1 or 3 hour and then assessed for apoptosis within the embryonic germ layers using the fluorescent whole-mount TUNEL technique. The TUNEL index for control embryos cultured for 1 hour was similar to embryos freshly isolated and nearly doubled (p<0.05) after 3 hours of culture (Fig. 5). Cell death was compared in embryos cultured in medium containing 400 mg% ethanol. This concentration was chosen based on the peak BAC obtained during intubation with 4 g/kg ethanol (Fig. 1). Ethanol treatment increased the TUNEL index 3-fold (p<0.05) above the respective control at 1 hour of culture and 2-fold (p<0.05) above the respective control at 3 hours (Fig. 5), indicating that embryonic cell death was induced by ethanol in this experimental model. Addition of 5 nM HB-EGF during the ethanol treatment significantly reduced the TUNEL index at both culture times, demonstrating that HB-EGF is capable of inhibiting apoptosis induced by ethanol in gastrulation-stage mouse embryos.
Fig. 5.
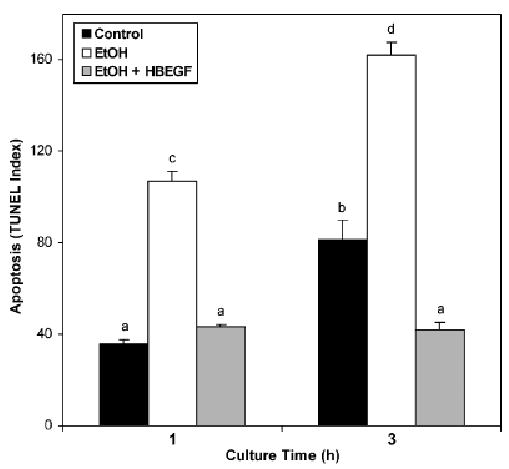
Heparin-binding epidermal growth factor (EGF)-like growth factor (HB-EGF) amelioration of embryonic cell death induced by exposure to ethanol in vitro. Intact visceral yolk sacs were isolated from nontreated dams on E7.5 and cultured for 1 or 3 hour in Ham’s F10 medium alone (control), medium containing 400 mg% ethanol (EtOH), or medium containing 400 mg% ethanol and 5 nM HB-EGF (EtOH+HB-EGF). The embryonic portion of the yolk sacs was then isolated, fixed, permeabilized, and assayed for terminal deoxynucleotidyl transferase-mediated dUTP-X nick end labeling (TUNEL) by the fluorescence microscopy procedure detailed in the Materials and Methods section. Mean and SE are shown. N = 4 to 5 embryos per treatment. Bars labeled with nonmatching letters were significantly different.
DISCUSSION
Embryonic cells at the gastrulation stages were highly susceptible to ethanol-induced cell death, as assessed using two independent measures of apoptosis in a double-labeling approach. Heightened apoptosis during embryonic development has been suggested as a possible mechanism underlying the teratogenic effects of ethanol (Ikonomidou et al., 2000; Kotch and Sulik 1992a; Smith, 1997; Smith and Debelak-Kragtorp, 2005), as demonstrated in pregnant C57BL/6J mice treated acutely with ethanol on E7 (Dunty et al., 2001). We have suggested that ethanol rapidly activates an intracellular biochemical pathway that induces apoptosis in embryonic cells. This hypothesis is based on evidence of ethanol-induced Ca2+ signaling in preimplantation mouse embryos and gastrulating chicken embryos (Armant, 1996; Debelak-Kragtorp et al., 2003; Garic-Stankovic et al., 2005). In the present study, significant levels of apoptosis were observed within 1 hour of ethanol exposure on GD 7.5, using an alcohol dose of 4 g/kg. Lower doses (1 to 2 g/kg) of ethanol produced a linear increase in cell death over a 6-hour period that reached significance after 3 hours, while apoptosis was rarely observed in control embryos. Dunty et al. (2001) also noted a negligible amount of endogenous apoptosis in vehicle-exposed embryos at this stage of development. A similar pattern of cell death could be reproduced in vitro by culturing visceral yolk sac embryos isolated on E7.5. Cell death assessed by TUNEL was low to moderate after 1 to 3 hours of culture, but increased significantly in medium containing 400 mg% ethanol, a dose comparable to the BAC attained in mice intubated with 4 g/kg ethanol. The observation of a significant increase in TUNEL after 1 hour suggests a good correlation with exposure to 4 g/kg in utero. The in vitro model provides a useful experimental system for investigating the mechanism of ethanol-induced apoptosis and other cellular responses to this teratogen in gastrulation-stage mouse embryos. Our results support the view that cell death induced by ethanol in gastrulating mouse embryos is a rapid process, consistent with a mechanism mediated by intracellular signaling molecules that activate the apoptosis pathway.
The kinetics of apoptosis in E7.5 mouse embryos suggest that we have observed the initial wave of apoptosis caused by prenatal ethanol exposure. Morphological examination of cell death 3 hours after embryos were exposed in utero to 4 g/kg ethanol revealed apoptotic cells in all three germ layers, although sampling size was much lower than in the time course experiment. There was no significant effect of cell type on apoptosis levels, but the effect of ethanol was highly significant in a 2-way ANOVA. TUNEL-positive cells were frequently pyknotic and annexin V binding paralleled TUNEL both temporally and spatially, providing evidence for cell death through apoptosis. Identical cells were not always labeled by both methods because phosphotidylserine externalization and DNA fragmentation occur separately within the apoptotic pathway (Allen et al., 1997). The distribution of apoptotic cells could be quite different at later stages of embryogenesis as dead cells are removed and possibly replaced through proliferation within each germ layer. Therefore, it is not surprising that Dunty et al. (2001) found cell death to be restricted to the ectoderm when embryos were examined 16 hours after exposure. Two waves of apoptosis have been observed in the chick embryo, with the first peak 6 hours after ethanol exposure and a second higher peak in neural crest 18 to 24 hours after treatment (Debelak and Smith, 2000).
Survival factors produced by the embryo or entering the embryonic tissues from outside could play an important role in determining the extent and location of apoptosis after ethanol exposure. Our in vitro model provided a useful experimental system in which to test this hypothesis. HB-EGF, a member of the EGF family of growth factors, ligates ErbB1 (EGF receptor) and ErbB4, members of the EGF receptor family of receptor tyrosine kinases (Riese and Stern, 1998). The levels of HB-EGF and its receptors during gastrulation are not known, but paracrine signaling by HB-EGF, which accumulates in the endometrium (Das et al., 1994; Leach et al., 1999; Yoo et al., 1997), is possible. Ethanol, through its ability to mobilize intracellular Ca2+ in embryonic cells (Armant, 1996; Debelak-Kragtorp et al., 2003; Garic-Stankovic et al., 2005), may induce autocrine secretion of HB-EGF. Signaling by Ca2+ downstream of G-protein-coupled receptors is well known to activate metalloproteinases that cleave the extracellular domain from proHB-EGF, a transmembrane protein, permitting it to ligate adjacent receptors (Prenzel et al., 1999). HB-EGF activation of ErbB receptors is known to stimulate survival pathways, including phosphoinositol 3-kinase/Akt (Fang et al., 2001). Using a recombinant protein identical to the secreted form of HB-EGF, we have shown that HB-EGF can completely ameliorate cell death in gastrulation-stage embryos cultured in medium with a high concentration of ethanol. At 3 hours of culture, apoptosis in embryos exposed to ethanol and HB-EGF was below that of controls, suggesting that HB-EGF also prevented apoptosis caused by in vitro culture conditions.
The appropriate stage-specific and cell-type-specific expression of survival factors, their cognate receptors, and downstream pathways are all required for protection of embryonic cells from ethanol-induced apoptosis. Fetal brain cells exposed to alcohol are protected from apoptosis in the presence of brain-derived neurotrophic factor, fibroblast growth factor-2, or nerve growth factor, but not glial cell line-derived neurotrophic factor (GDNF), IGF-1, or EGF (Bonthius et al., 2003; Mitchell et al., 1999). However, in a homogeneous GDNF-responsive neuroblastoma cell line, GDNF reverses the rapid induction of apoptosis by ethanol in conjunction with reduced JNK phosphorylation (McAlhany et al., 2000). In cerebellar granule cells, a mechanism involving protein kinase G activation downstream of nitric oxide attenuates ethanol-induced apoptosis (Bonthius et al., 2003). In ethanol-treated PNET2 immature neuronal cells, apoptosis is reversed by IGF-1 through reduced p53 and JNK, with elevated levels of phosphoinositol 3-kinase (de la Monte et al., 2000).
HB-EGF is capable of downstream signaling to prevent apoptosis in intestinal cells (El Assal and Besner, 2004), and now appears to have a comparable function in ethanol-exposed mouse embryos at the gastrulation stage. Our findings suggest that the delivery of HB-EGF or other effective survival factors to the conceptus, or stimulation of their endogenous production, might reduce some of the adverse effects of prenatal alcohol exposure. HB-EGF can be added to an expanding list of growth factors and compounds (e.g., activity-dependent neuroprotective protein, activity-dependent neurotrophic factor, octanol, GM1 ganglioside) that protect against ethanol embryotoxic effects (Chen et al., 1996, 2001; Spong et al., 2001; Wilkemeyer et al., 2003).
Acknowledgments
The authors would like to thank Michael Kruger, Wayne State University, for help with statistical analysis and Dr. Karen Downs, University of Wisconsin, for helpful advice on the production and culture of postimplantation mouse embryos.
Footnotes
Supported by Grant AA12057 (to DRA) and Grants AA11085 and ES09090 (to SMS) from the National Institutes of Health.
References
- Allen RT, Hunter WJ, Agrawal DK. Morphological and biochemical characterization and analysis of apoptosis. J Pharmacol Toxicol Methods. 1997;37:215–228. doi: 10.1016/s1056-8719(97)00033-6. [DOI] [PubMed] [Google Scholar]
- Armant DR. Ethanol-induced acceleration of preimplantation embryonic development. In: Abel EL, editor. Fetal Alcohol Syndrome: From Mechanism to Prevention. CRC Press; Boca Raton, FL: 1996. pp. 1–26. [Google Scholar]
- Bonthius DJ, Karacay B, Dai D, Pantazis NJ. FGF-2, NGF and IGF-1, but not BDNF, utilize a nitric oxide pathway to signal neurotrophic and neuroprotective effects against alcohol toxicity in cerebellar granule cell cultures. Brain Res Dev Brain Res. 2003;140:15–28. doi: 10.1016/s0165-3806(02)00549-7. [DOI] [PubMed] [Google Scholar]
- Cartwright MM, Smith SM. Increased cell death and reduced neural crest cell numbers in ethanol-exposed embryos: partial basis for the fetal alcohol syndrome phenotype. Alcohol Clin Exp Res. 1995a;19:378–386. doi: 10.1111/j.1530-0277.1995.tb01519.x. [DOI] [PubMed] [Google Scholar]
- Cartwright MM, Smith SM. Stage-dependent effects of ethanol on cranial neural crest cell development: partial basis for the phenotypic variations observed in fetal alcohol syndrome. Alcohol Clin Exp Res. 1995b;19:1454–1462. doi: 10.1111/j.1530-0277.1995.tb01007.x. [DOI] [PubMed] [Google Scholar]
- Chen SY, Wilkemeyer MF, Sulik KK, Charness ME. Octanol antagonism of ethanol teratogenesis. FASEB J. 2001;15:1649–1651. doi: 10.1096/fj.00-0862fje. [DOI] [PubMed] [Google Scholar]
- Chen SY, Yang B, Jacobson K, Sulik KK. The membrane disordering effect of ethanol on neural crest cells in vitro and the protective role of GM1 ganglioside. Alcohol. 1996;13:589–595. doi: 10.1016/s0741-8329(96)00073-0. [DOI] [PubMed] [Google Scholar]
- Chobotova K, Karpovich N, Carver J, Manek S, Gullick WJ, Barlow DH, Mardon HJ. Heparin-binding epidermal growth factor and its receptors mediate decidualization and potentiate survival of human endometrial stromal cells. J Clin Endocrinol Metab. 2005;90:913–919. doi: 10.1210/jc.2004-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarren SK, Bowden DM. Fetal alcohol syndrome: A new primate model for binge drinking and its relevance to human ethanol teratogenesis. J Pediatr. 1982;101:819–824. doi: 10.1016/s0022-3476(82)80340-5. [DOI] [PubMed] [Google Scholar]
- Clarren SK, Smith DW. The fetal alcohol syndrome. N Engl J Med. 1978;298:1063–1067. doi: 10.1056/NEJM197805112981906. [DOI] [PubMed] [Google Scholar]
- Darzynkiewicz Z, Bedner E, Smolewski P. In situ detection of DNA strand breaks in analysis of apoptosis by flow- and laser-scanning cytometry. In: Didenko VV, editor. In Situ Detection of DNA Damage: Methods and Procedures. Humana Press; Totowa, NJ: 2002. pp. 69–77. [DOI] [PubMed] [Google Scholar]
- Das SK, Wang XN, Paria BC, Damm D, Abraham JA, Klagsbrun M, Andrews GK, Dey SK. Heparin-binding EGF-like growth factor gene is induced in the mouse uterus temporally by the blastocyst solely at the site of its apposition: a possible ligand for interaction with blastocyst EGF-receptor in implantation. Development. 1994;120:1071–1083. doi: 10.1242/dev.120.5.1071. [DOI] [PubMed] [Google Scholar]
- de la Monte SM, Ganju N, Banerjee K, Brown NV, Luong T, Wands JR. Partial rescue of ethanol-induced neuronal apoptosis by growth factor activation of phosphoinositol-3-kinase. Alcohol Clin Exp Res. 2000;24:716–726. [PubMed] [Google Scholar]
- Debelak KA, Smith SM. Avian genetic background modulates the neural crest apoptosis induced by ethanol exposure. Alcohol Clin Exp Res. 2000;24:307–314. [PubMed] [Google Scholar]
- Debelak-Kragtorp KA, Armant DR, Smith SM. Ethanol-induced cephalic apoptosis requires phospholipase C-dependent intracellular calcium signaling. Alcohol Clin Exp Res. 2003;27:515–523. doi: 10.1097/01.ALC.0000056615.34253.A8. [DOI] [PubMed] [Google Scholar]
- Dhandapani KM, Brann DW. Transforming growth factor-beta: a neuroprotective factor in cerebral ischemia. Cell Biochem Biophys. 2003;39:13–22. doi: 10.1385/CBB:39:1:13. [DOI] [PubMed] [Google Scholar]
- Downs KM, Davies T. Staging of gastrulating mouse embryos by morphological landmarks in the dissecting microscope. Development. 1993;118:1255–1266. doi: 10.1242/dev.118.4.1255. [DOI] [PubMed] [Google Scholar]
- Dunty WCJ, Chen SY, Zucker RM, Dehart DB, Sulik KK. Selective vulnerability of embryonic cell populations to ethanol-induced apoptosis: implications for alcohol-related birth defects and neurodevelopmental disorder. Alcohol Clin Exp Res. 2001;25:1523–1535. [PubMed] [Google Scholar]
- El Assal ON, Besner GE. Heparin-binding epidermal growth factor-like growth factor and intestinal ischemia-reperfusion injury. Semin Pediatr Surg. 2004;13:2–10. doi: 10.1053/j.sempedsurg.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Ernhart CB, Sokol RJ, Martier S, Moron P, Nadler D, Ager JW, Wolf A. Alcohol teratogenicity in the human: a detailed assessment of specificity, critical period, and threshold. Am J Obstet Gynecol. 1987;156:33–39. doi: 10.1016/0002-9378(87)90199-2. [DOI] [PubMed] [Google Scholar]
- Ewald SJ, Shao H. Ethanol increases apoptotic cell death of thymocytes in vitro. Alcohol Clin Exp Res. 1993;17:359–365. doi: 10.1111/j.1530-0277.1993.tb00776.x. [DOI] [PubMed] [Google Scholar]
- Fang L, Li G, Liu G, Lee SW, Aaronson SA. p53 induction of heparin-binding EGF-like growth factor counteracts p53 growth suppression through activation of MAPK and PI3K/Akt signaling cascades. EMBO J. 2001;20:1931–1939. doi: 10.1093/emboj/20.8.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund G. Apoptosis and gene expression: perspectives on alcohol-induced brain damage. Alcohol. 1994;11:385–387. doi: 10.1016/0741-8329(94)90022-1. [DOI] [PubMed] [Google Scholar]
- Friedman JM. Can maternal alcohol ingestion cause neural tube defects? J Pediatr. 1982;101:232–234. doi: 10.1016/s0022-3476(82)80129-7. [DOI] [PubMed] [Google Scholar]
- Garic-Stankovic A, Hernandez MR, Chiang PJ, Debelak-Kragtorp KA, Flentke GR, Armant DR, Smith SM. Ethanol triggers neural crest apoptosis through the selective activation of a pertussis toxin-sensitive G-protein and a phospholipase Cβ-dependent Ca2+transient. Alcohol Clin Exp Res. 2005;29:1237–1246. doi: 10.1097/01.alc.0000172460.05756.d9. [DOI] [PubMed] [Google Scholar]
- Hannigan JH, Armant DR. Alcohol in pregnancy and neonatal outcome. Semin Neonatol. 2000;5:243–254. doi: 10.1053/siny.2000.0027. [DOI] [PubMed] [Google Scholar]
- Hogan BL, Beddington RS, Constantini F, Lacy E. Manipulating the Mouse Embryo. A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1994. [Google Scholar]
- Holownia A, Ledig M, Menez JF. Ethanol-induced cell death in cultured rat astroglia. Neurotoxicol Teratol. 1997;19:141–146. doi: 10.1016/s0892-0362(96)00226-7. [DOI] [PubMed] [Google Scholar]
- Hong F, Kwon SJ, Jhun BS, Kim SS, Ha J, Kim SJ, Sohn NW, Kang C, Kang I. Insulin-like growth factor-1 protects H9c2 cardiac myo-blasts from oxidative stress-induced apoptosis via phosphatidylinositol 3-kinase and extracellular signal-regulated kinase pathways. Life Sci. 2001;68:1095–1105. doi: 10.1016/s0024-3205(00)01012-2. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K, Price MT, Stefovska V, Horster F, Tenkova T, Dikranian K, Olney JW. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:1056–1060. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- Jones KL, Smith DW. Recognition of the fetal alcohol syndrome in early infancy. Lancet. 1973;2:999–1001. doi: 10.1016/s0140-6736(73)91092-1. [DOI] [PubMed] [Google Scholar]
- Kotch LE, Sulik KK. Experimental fetal alcohol syndrome: proposed pathogenic basis for a variety of associated facial and brain anomalies. Am J Med Genet. 1992a;44:168–176. doi: 10.1002/ajmg.1320440210. [DOI] [PubMed] [Google Scholar]
- Kotch LE, Sulik KK. Patterns of ethanol-induced cell death in the developing nervous system of mice; neural fold states through the time of anterior neural tube closure. Int J Dev Neurosci. 1992b;10:273–279. doi: 10.1016/0736-5748(92)90016-s. [DOI] [PubMed] [Google Scholar]
- Leach RE, Khalifa R, Ramirez ND, Das SK, Wang J, Dey SK, Romero R, Armant DR. Multiple roles for heparin-binding epidermal growth factor-like growth factor are suggested by its cell specific expression during the human endometrial cycle and early placentation. J Clin Endocrinol Metab. 1999;84:3355–3363. doi: 10.1210/jcem.84.9.5980. [DOI] [PubMed] [Google Scholar]
- Leach RE, Stachecki JJ, Armant DR. Development of in vitro fertilized mouse embryos exposed to ethanol during the preimplantation period: Accelerated embryogenesis at subtoxic levels. Teratology. 1993;47:57–64. doi: 10.1002/tera.1420470110. [DOI] [PubMed] [Google Scholar]
- Luo J, West JR, Pantazis NJ. Nerve growth factor and basic fibroblast growth factor protect rat cerebellar granule cells in culture against ethanol-induced cell death. Alcohol Clin Exp Res. 1997;21:1108–1120. [PubMed] [Google Scholar]
- McAlhany RE, Jr, West JR, Miranda RC. Glial-derived neurotrophic factor (GDNF) prevents ethanol-induced apoptosis and JUN kinase phosphorylation. Brain Res Dev Brain Res. 2000;119:209–216. doi: 10.1016/s0165-3806(99)00171-6. [DOI] [PubMed] [Google Scholar]
- Mitchell JJ, Paiva M, Walker DW, Heaton MB. BDNF and NGF afford in vitro neuroprotection against ethanol combined with acute ischemia and chronic hypoglycemia. Dev Neurosci. 1999;21:68–75. doi: 10.1159/000017368. [DOI] [PubMed] [Google Scholar]
- Ozaki M, Haga S, Zhang HQ, Irani K, Suzuki S. Inhibition of hypoxia/reoxygenation-induced oxidative stress in HGF-stimulated antiapoptotic signaling: Role of PI3-K and Akt kinase upon rac1. Cell Death Differ. 2003;10:508–515. doi: 10.1038/sj.cdd.4401172. [DOI] [PubMed] [Google Scholar]
- Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, Ull-rich A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999;402:884–888. doi: 10.1038/47260. [DOI] [PubMed] [Google Scholar]
- Ramachandran V, Watts LT, Maffi SK, Chen J, Schenker S, Henderson G. Ethanol-induced oxidative stress precedes mitochondrially mediated apoptotic death of cultured fetal cortical neurons. J Neurosci Res. 2003;74:577–588. doi: 10.1002/jnr.10767. [DOI] [PubMed] [Google Scholar]
- Riese DJ, Stern DF. Specificity within the EGF family/ErbB receptor family signaling network. Bioessays. 1998;20:41–48. doi: 10.1002/(SICI)1521-1878(199801)20:1<41::AID-BIES7>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Smith SM. Alcohol-induced cell death in the embryo. Alcohol Health Res World. 1997;21:287–295. [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Debelak-Kragtorp KA. Neural crest and alcohol exposure. In: Miller MW, editor. The Developing Brain: Lessons Learned from Alcohol and Nicotine Exposures. Oxford University Press; New York, NY: 2005. (in press) [Google Scholar]
- Spong CY, Abebe DT, Gozes I, Brenneman DE, Hill JM. Prevention of fetal demise and growth restriction in a mouse model of fetal alcohol syndrome. J Pharmacol Exp Ther. 2001;297:774–779. [PubMed] [Google Scholar]
- Sulik KK, Johnston MC, Webb MA. Fetal alcohol syndrome: embryogenesis in a mouse model. Science. 1981;214:936–938. doi: 10.1126/science.6795717. [DOI] [PubMed] [Google Scholar]
- Suzuki YJ. Growth factor signaling for cardioprotection against oxidative stress-induced apoptosis. Antioxid Redox Signal. 2003;5:741–749. doi: 10.1089/152308603770380043. [DOI] [PubMed] [Google Scholar]
- Wells A. EGF receptor. Int J Biochem Cell Biol. 1999;31:637–643. doi: 10.1016/s1357-2725(99)00015-1. [DOI] [PubMed] [Google Scholar]
- Wilkemeyer MF, Chen SY, Menkari CE, Brenneman DE, Sulik KK, Charness ME. Differential effects of ethanol antagonism and neuroprotection in peptide fragment NAPVSIPQ prevention of ethanol-induced developmental toxicity. Proc Natl Acad Sci USA. 2003;100:8543–8548. doi: 10.1073/pnas.1331636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo HJ, Barlow DH, Mardon HJ. Temporal and spatial regulation of expression of heparin-binding epidermal growth factor-like growth factor in the human endometrium: A possible role in blastocyst implantation. Dev Genet. 1997;21:102–108. doi: 10.1002/(SICI)1520-6408(1997)21:1<102::AID-DVG12>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]