

Abstract
A combinatorial alanine-scanning strategy was used to determine simultaneously the functional contributions of 19 side chains buried at the interface between human growth hormone and the extracellular domain of its receptor. A phage-displayed protein library was constructed in which the 19 side chains were preferentially allowed to vary only as the wild type or alanine. The library pool was subjected to binding selections to isolate functional clones, and DNA sequencing was used to determine the alanine/wild-type ratio at each varied position. This ratio was used to calculate the effect of each alanine substitution as a change in free energy relative to that of wild type. Only seven side chains contribute significantly to the binding interaction, and these conserved residues form a compact cluster in the human growth hormone tertiary structure. The results were in excellent agreement with free energy data previously determined by conventional alanine-scanning mutagenesis and suggest that this technology should be useful for analyzing functional epitopes in proteins.
Because protein–protein interactions are involved in essentially all cellular processes, a detailed understanding of these interactions is a major goal of modern biology. A large number of high-resolution structures have been solved to reveal the molecular details of many protein–protein interfaces (1–5). Furthermore, a structural genomics effort has been proposed, and the database of protein structures is expected to expand dramatically (6). However, cocrystallization of receptor–ligand complexes is not always possible, and although three-dimensional structures accurately define structural binding epitopes (i.e., residues in direct contact with a ligand), they do not address the energetics of a binding interaction. Thus, a comprehensive understanding of a molecular recognition event also requires elucidation of the functional epitope: the residues within the structural epitope that make energetic contributions to the binding interaction (7, 8).
Site-directed mutagenesis is a powerful tool for probing protein structure and function. Although many different mutagenesis strategies have been used (9–12), alanine-scanning mutagenesis has been particularly successful in systematically mapping functional binding epitopes (13–15). Because substitution with alanine removes all side chain atoms past the β-carbon, the effects of individual alanine mutations can be used to infer the roles of individual side chains. Alanine-scanning mutagenesis provides a detailed map of a protein-binding interface, but the method is laborious. Many mutant proteins must be produced and purified, and the structural integrity and binding constant of each mutant must be assessed separately. As an alternative to conventional alanine-scanning mutagenesis, an in vivo library-based strategy called binomial mutagenesis has been developed (16). This approach is attractive, because a single library simultaneously provides information about many different residues. However, the technique is limited to proteins having a function that can be selected genetically in Escherichia coli and thus is not applicable to most mammalian proteins. Furthermore, in vivo selections cannot distinguish loss of function caused by folding or expression problems rather than genuine loss of favorable binding contacts at a protein–protein interface.
Herein, we describe shotgun scanning, a general combinatorial method for mapping functional epitopes of proteins. Shotgun scanning combines the concepts of alanine-scanning mutagenesis (13–15) and binomial mutagenesis (16) with phage display technology (17, 18). The method is extremely rapid, because many side chains are analyzed simultaneously, and the need for protein purification and biophysical analysis is circumvented. Instead, the energetic contributions of individual side chains are derived from statistical analysis of DNA sequences. We used shotgun scanning to study the binding interaction between human growth hormone (hGH) and its receptor (hGH binding protein, hGHbp). The results were in excellent agreement with previously determined biophysical data (7).
Materials and Methods
Shotgun-Scanning Library Construction.
Phagemid pW1205a was used as the template for library construction. pW1205a is identical to a previously described phagemid (19) designed to display hGH on the surface of M13 bacteriophage as a fusion to the amino terminus of the major coat protein (P8), except for the following changes. First, the hGH–P8 fusion moiety has a peptide epitope flag (amino acid sequence: MADPNRFRGKDLGG) fused to its amino terminus, allowing for detection with an anti-flag antibody. Second, codons encoding residues 41, 42, 43, 61, 62, 63, 171, 172, and 173 of hGH have been replaced by TAA stop codons. The library was constructed by using a previously described method (20). Briefly, pW1205a was used as the template for the Kunkel mutagenesis method (21) with three mutagenic oligonucleotides designed to repair simultaneously the stop codons and introduce mutations at the desired sites.
The mutagenic oligonucleotides had the following sequences: oligo 1 (mutant hGH codons 41, 42, 45, and 48), 5′-ATC CCC AAG GAA CAG RMA KMT TCA TTC SYT CAG AAC SCA CAG ACC TCC CTC TGT TTC-3′; oligo 2 (mutant hGH codons 61, 62, 63, 64, 67, and 68), 5′-TCA GAA TCG ATT CCG ACA SCA KCC RMC SST GAG GAA RCT SMA CAG AAA TCC AAC CTA GAG-3′; oligo 3 (mutant hGH codons 164, 167, 168, 171, 172, 175, 176, 178, and 179), 5′-AAC TAC GGG CTG CTC KMY TGC TTC SST RMA GAC ATG GMT RMA GTC GAG RCT KYT CTG SST RYT GTG CAG TGC CGC TCT-3′. (Note that standard single-letter codes for amino acids are used, and DNA degeneracies are represented by IUB code: K = G/T, M = A/C, N = A/C/G/T, R = A/G, S = G/C, W = A/T, Y = C/T.) The library contained 1.2 × 1011 unique members, and DNA sequencing of the naïve library revealed that 45% of these had incorporated all three mutagenic oligonucleotides. Thus, the library had a diversity of approximately 5.4 × 1010.
Shotgun Library Sorting and Binding Assays.
Phage from the library described above were cycled through rounds of binding selection with hGHbp or anti-hGH monoclonal antibody 3F6.B1.4B1 (22) coated on 96-well Nunc Maxisorp immunoplates as the capture target. Phage were propagated in E. coli XL1-blue with the addition of M13-VCS helper phage (Stratagene). After one (antibody sort) or three (hGHbp sort) rounds of selection, individual clones were grown in a 96-well format in 500 μl of 2YT broth supplemented with carbenicillin (10 μg/ml) and M13-VCS (1010 phage per ml). The culture supernatants were used directly in phage ELISAs (19) to detect phage-displayed hGH variants that bound to either hGHbp or anti-hGH antibody 3F6.B1.4B1 immobilized on a 96-well Maxisorp immunoplate.
DNA Sequencing and Analysis.
Culture supernatant containing phage particles was used as the template for a PCR that amplified the hGH gene and incorporated M13(-21) and M13R universal sequencing primers. The amplified DNA fragment was used as the template in Big-Dye terminator sequencing reactions, which were analyzed on an ABI377 sequencer (PE Biosystems, Foster City, CA). All reactions were performed in a 96-well format.
The program sgcount aligned each DNA sequence against the wild-type (wt) DNA sequence by using a Needleman–Wunch pairwise alignment algorithm, translated each aligned sequence of acceptable quality, and then tabulated the occurrence of each natural amino acid at each position. Additionally, sgcount reported the presence of any sequences containing identical amino acids at all mutated positions (siblings). The antibody sort (175 total sequences) did not contain any siblings, but the hGHbp sort (330 total sequences) contained 16 siblings representing 5 unique sequences. sgcount was written in c and compiled and tested on a Compaq/DEC alpha under Digital Unix 4.0D. The sgcount source code is available (send requests to E-mail: ckw@gene.com) and should compile without modification on most Unix systems.
Shotgun-Scanning Data Analysis.
For each selection, the ratio of wt to alanine at each position was calculated as follows:
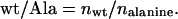 |
We then assumed that wt/Ala = Ka,wt/Ka,Ala, where Ka,wt and Ka,Ala are the association equilibrium constants for hGHbp binding to wt or alanine-substituted hGH, respectively. With this assumption, we calculated a ΔΔG value for the hGHbp selection (ΔΔGbp) and the antibody selection (ΔΔGα) by substituting wt/Ala for Ka,wt/Ka,Ala in the standard equation:
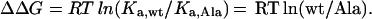 |
In this way, we obtained a measure of each alanine mutant's effect on each selection as a change in free energy relative to that of wt. Finally, we defined the contribution to binding free energy attributable to each side chain (ΔΔGmut-wt) as follows:
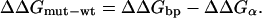 |
Results and Discussion
Shotgun scanning uses combinatorial protein libraries in which residues are ideally allowed to vary only as the wt or alanine (Table 1), although the nature of the genetic code necessitates two other amino acid substitutions for some residues. Because the diversity is limited to only two or four possibilities at each position, current library construction technologies (19) allow the simultaneous mutation of about 20 positions with reasonable probability of complete coverage. The library pool is displayed on filamentous phage particles, and in vitro selections are used to isolate members that are capable of binding to immobilized target ligands (Fig. 1). Selected clones are sequenced, and the occurrence of wt or alanine at each position is tabulated. Depending on the nature of the selected interaction, this information can be used to assess the contribution of each side chain to protein structure and/or function.
Table 1.
The shotgun scanning code
| Amino acid | Shotgun codon* | Shotgun substitutions |
|---|---|---|
| C | KST | A/C/G/S |
| D | GMT | A/D |
| E | GMA | A/E |
| F | KYT | A/F/S/V |
| G | GST | A/G |
| H | SMT | A/H/D/P |
| I | RYT | A/I/T/V |
| K | RMA | A/K/E/T |
| L | SYT | A/L/P/V |
| M | RYG | A/M/T/V |
| N | RMC | A/N/D/T |
| P | SCA | A/P |
| Q | SMA | A/Q/E/P |
| R | SST | A/R/G/P |
| S | KCC | A/S |
| T | RCT | A/T |
| V | GYT | A/V |
| W | KSG | A/W/G/S |
| Y | KMT | A/Y/D/S |
For each amino acid, the appropriate shotgun codon ideally encodes only the wt amino acid or alanine, but the nature of the genetic code necessitates the occurrence of two other amino acids for some shotgun substitutions. Single-letter amino acid and nucleotide abbreviations are used.
DNA degeneracies are represented by IUB code (K = G/T, M = A/C, N = A/C/G/T, R = A/G, S = G/C, W = A/T, Y = C/T).
Figure 1.

Scheme for hGH shotgun scanning. The hGH gene was fused to M13 gene-8, and an hGH library was constructed with 19 mutated positions on three noncontiguous stretches of primary sequence. The library was then phage displayed, resulting in phage particles with hGH variants (blue ovals) displayed on their surface and the cognate hGH genes encapsulated within their coats. For simplicity, we show only two scanned side chains. Mutated positions initially had an approximately even distribution of wt and alanine. After selection for binding to an immobilized ligand (e.g., hGHbp shown in yellow), enrichment for wt side chains was observed for residues that contribute favorably to binding (red side chain). No selection for the wt amino acid was observed for residues that do not contribute to the binding interaction (blue side chain).
Shotgun Scan of hGH.
We used shotgun scanning to study the high-affinity site (site 1) of hGH for binding to the extracellular domain of hGHbp. Crystallographic data were used to identify 19 hGH side chains that become at least 60% buried on binding to hGHbp and together comprise a substantial portion of the structural binding epitope (23). These side chains are located on three noncontiguous stretches of primary sequence, but together they form a contiguous patch in the three-dimensional structure. To ascertain which of these side chains are also part of the functional binding epitope, we constructed a phage-displayed hGH library in which all 19 side chains were simultaneously mutated by using the “shotgun code” (Table 1). The library contained 5.4 × 1010 unique members, and thus, the theoretical diversity for combinatorial mutagenesis at the 19 sites (4.3 × 109) was well represented. The library was subjected to two separate selections. The first selection (antibody selection) isolated variants capable of binding to a monoclonal antibody that binds hGH away from the mutated region but requires native hGH structure (22). The second selection isolated variants capable of binding to hGHbp.
Several hundred binding clones were sequenced from each selection, and the occurrence of wt or alanine was tabulated for each mutated position. At positions that encoded additional side chains (Table 1), our analysis focused entirely on the distribution of wt and alanine. Other observed substitutions were not counted, because we wanted to analyze the effect of removing the wt side chain, and side chains larger than alanine may introduce new interactions that complicate the analysis.
For each selection, the sequence data were used to calculate the ratio of wt to alanine at each position (wt/Ala ratio; Table 2). The wt/Ala ratio is the statistical preference for a wt side chain relative to alanine, and thus, it correlates with a given side chain's contribution to the selected trait (i.e., binding to the antibody or to hGHbp). The wt/Ala ratio for a large, favorable contribution should approach unity (100% enrichment for the wt side chain), whereas the wt/Ala ratio for a large, negative contribution should approach zero (selection against the wt side chain). Because the wt/Ala ratio measures the binding of wt relative to an alanine mutant, we assumed that it is equivalent to the corresponding ratio of equilibrium binding constants. With this single assumption, our statistical data were converted to changes in free energy simply by substituting the wt/Ala ratio for the equilibrium constant ratio in the standard equation: ΔΔG = RT ln(Ka,wt/Ka,Ala) (see Materials and Methods). Thus, for each selection, we calculated the effect of each alanine substitution as a change in free energy relative to that of wt.
Table 2.
Shotgun scanning analysis of hGH
| wt aa | (wt/Ala)bp | (wt/Ala)α | ΔΔGmut-wt, kcal/mol |
|---|---|---|---|
| K41 | 1.81 | 0.77 | 0.5 |
| Y42 | 2.51 | 1.24 | 0.4 |
| L45 | 3.31 | 1.41 | 0.5 |
| P48 | 0.82 | 0.69 | 0.1 |
| P61 | 15.40 | 0.64 | 1.9 |
| S62 | 1.17 | 1.15 | 0.0 |
| N63 | 1.93 | 1.02 | 0.4 |
| R64 | 14.33 | 0.77 | 1.7 |
| T67 | 1.28 | 0.55 | 0.5 |
| Q68 | 2.63 | 1.00 | 0.6 |
| Y164 | 1.48 | 2.08 | −0.2 |
| R167 | 2.77 | 0.82 | 0.7 |
| K168 | 2.42 | 1.75 | 0.2 |
| D171 | 0.99 | 0.49 | 0.4 |
| K172 | 10.54 | 0.75 | 1.6 |
| T175 | 11.15 | 0.78 | 1.6 |
| F176 | 155.50 | 1.89 | 2.6 |
| R178 | 159.00 | 2.03 | 2.6 |
| I179 | 6.27 | 0.62 | 1.4 |
For each of the 19 scanned positions (wt aa), the wt/Ala ratio was determined for the hGHbp selection, (wt/Ala)bp, and the antibody selection, (wt/Ala)α. These data were used to calculate the difference in binding free energy between alanine-substituted and wt hGH for binding to hGHbp (ΔΔGmut-wt), as described in Materials and Methods. Residues in bold have ΔΔGmut-wt > 1.0 kcal/mol.
Although both selections are sensitive to bias in the naïve library, expression biases, and global structural perturbations, only the hGHbp selection is sensitive to the loss or gain of binding energy caused by contacts with the mutated residues in the structural epitope. Thus, the difference between the changes in free energy from the hGHbp selection and the antibody selection should measure ΔΔGmut-wt, the difference in binding free energy between alanine-substituted and wt hGH for binding to hGHbp (Table 2). This parameter measures each deleted side chain's contribution to the functional epitope (7, 8, 13). It is noteworthy that the wt/Ala ratio for the antibody selection was close to 1.0 for all 19 side chains, and thus, it did not have a large effect on the ΔΔGmut-wt calculation. The fact that alanine substitutions did not affect the antibody sort reflects the fact that the mutated side chains are surface residues that do not contribute to the structural stability of hGH. One would expect larger effects for buried residues, because alanine substitutions at such sites may destabilize the hydrophobic core.
Only one side chain (Tyr-64) had a negative ΔΔGmut-wt value, indicating that most side chains in the hGH structural epitope make favorable contacts with hGHbp (Table 2). The individual side chains do not contribute equally to the binding interaction, and in fact, the ΔΔGmut-wt values form two distinct clusters, with one cluster containing side chains with ΔΔGmut-wt < 1.0 kcal/mol and the second cluster containing side chains with ΔΔGmut-wt > 1.0 kcal/mol. The second cluster contains only seven side chains (Pro-61, Arg-64, Lys-172, Thr-175, Phe-176, Arg-178, and Ile-179), and our results indicate that this subset accounts for ≈75% of the total change in binding free energy resulting from the alanine substitutions. These side chains also cluster together in the three-dimensional structure and thus form a compact, functional epitope (Fig. 2). Overall, the shotgun-scanning results are in excellent agreement with the results of conventional alanine-scanning mutagenesis, which identified a similar functional epitope (7). Shotgun scanning identified six of the seven largest binding energy contributors identified by conventional alanine scanning. Only Leu-45 was not identified, but this residue lies on the periphery of the functional epitope defined by alanine scanning (7), and perhaps it is not critical for the binding interaction. A least squares linear fit of our shotgun-scanning ΔΔGmut-wt values versus ΔΔGmut-wt values determined from BIAcore data with individual, purified alanine mutants (7) showed a strong correlation (R = 0.88), with a slope of 1.0 and a y intercept of 0.0 (Fig. 3).
Figure 2.

Shotgun scan of hGH site 1 for binding to the hGHbp. The three-dimensional structure of hGH (23) is shown in space-filling mode. The 19 shotgun-scanned residues are colored, with blue denoting ΔΔGmut-wt < 1.0 kcal/mol and red denoting ΔΔGmut-wt > 1.0 kcal/mol (Table 2). The residues shown in red constitute the functional epitope of hGH site 1. The figure was produced with grasp software (30).
Figure 3.

Correlation between the change in the free energy of binding (ΔΔGmut-wt) calculated for alanine mutants relative to wt for hGH site 1 binding to hGHbp, as determined by shotgun scanning (y axis) or conventional alanine-scanning mutagenesis (x axis). Each point is labeled with the corresponding hGH residue number. The shotgun scanning data are from Table 2, whereas the alanine-scanning data are from Cunningham and Wells (7). The least squares linear fit of the data is shown, with the corresponding equation and R value given at the top left.
Caveats for Statistical Methods.
The discrepancies between shotgun scanning and alanine scanning may be due to nonadditive, cooperative interactions between some residues in the shotgun-scanning library. In particular, although we ignored all substitutions except alanine and wt, it is possible that these additional substitutions skewed the calculated wt frequencies at some positions. However, these nonadditive effects can be addressed by analyzing covariation at mutated sites; such analyses can provide information on intramolecular interactions that cannot be obtained from single-site mutants (16). Also, recent developments in DNA synthesis will make it possible to construct libraries in which any site can be restricted to only alanine or one of the other natural amino acids (24, 25).
The utility of any statistical method depends on the sample size necessary to obtain reliable predictions. Shotgun-scanning libraries are designed to encode ideally only wt or alanine at each scanned position. By analyzing only the wt/Ala ratio, we were able to obtain a tight correlation between ΔΔGmut-wt values measured for purified alanine mutants and ΔΔGmut-wt values calculated from statistical analysis of several hundred DNA sequences. The inclusion of additional substitutions may be useful for some purposes, but it becomes difficult to correlate the data with standard binding constants. For example, Hu et al. (26) reported a combinatorial analysis involving four possible substitutions at multiple positions within a leucine zipper dimer. Statistical probabilities were used to predict preferred side chains at each position and also to detect cooperativity between positions, but statistical data were not converted to free energy values.
The sample size required to determine a side chain's functional significance increases with the side chain's contribution to binding, because the wt/Ala ratio can be determined only if both wt and alanine residues are observed at a given position. For functionally important positions, the wt side chain will dominate in functional clones, and larger sample sizes will be required to detect rare alanine substitutions. Thus, accurate assessment of a given position requires a sample size greater than the ratio of binding affinities for wt relative to the alanine substitution. In practical terms, this requirement means that data sets containing several hundred sequences can be used to determine ΔΔGmut-wt values for an alanine substitution that causes less than a 100-fold change in binding affinity (ΔΔGmut-wt < 3.0 kcal/mol). Alanine substitutions associated with larger ΔΔGmut-wt values will be identified by a complete absence of alanine substitutions in the data set, but the data will only place a lower limit on the magnitude of ΔΔGmut-wt.
Implications of Shotgun Scanning.
Our results address a concern about alanine scanning that was presented by Greenspan and Di Cera (27). They argued that ΔΔGmut-wt values derived from single alanine mutants are a poor measure of individual side chain binding contributions, because cooperative intramolecular interactions could make most large binding interfaces extremely nonadditive. For the first time to our knowledge, we have constructed and analyzed every possible multiple alanine mutant covering a large portion of a structural binding epitope, albeit in a necessarily combinatorial manner. Even in this extremely diverse background, the functional contributions of individual side chains were remarkably similar to their contributions in the fixed wt hGH background. Although nonadditive effects certainly should be considered, it seems that the major contributors of binding energy at the hGH–hGHbp interface act independently, in an essentially additive manner. The results are in good agreement with previous studies that have demonstrated additivity in the hGH site 1 (28) and many other proteins (29).
Shotgun scanning should be widely applicable to the study of protein structure and function. In cases where the structure of a protein–protein complex is known, such as the one reported herein, the structure can be used to identify side chains that are in direct contact with a binding partner. A single library encompassing up to 20 such residues can be used to map the binding energetics rapidly within the structural epitope. In such cases, shotgun scanning is a rapid alternative to conventional alanine scanning.
Shotgun scanning may prove especially useful in cases where the crystal structure is not known, or when structures or models are available only for the uncomplexed binding partners. In such cases, the limited structural information can be used to narrow the search field, but the structural binding epitope cannot be defined reliably. In the extreme case, one may wish to scan every residue in a protein. A complete alanine scan would be daunting, because each mutant would have to be constructed, purified, and analyzed separately. In contrast, a complete shotgun scan is feasible, because the construction and sorting of multiple libraries can be done readily in parallel. For example, a complete shotgun scan of a 200-residue protein could be accomplished with only 10 libraries encompassing 20 contiguous residues each.
Conclusions
Shotgun scanning allows rapid, detailed mapping of functional binding epitopes without protein purification or biophysical analysis. Instead, the binding free energy contributions of individual side chains can be determined from statistical analysis of DNA sequences. The method is applicable to any protein that can be displayed on filamentous phage, provided suitable binding partners are available. Using shotgun scanning in conjunction with structural data, we required only 3 weeks to elucidate the functional epitope within a known structural epitope. The method should also be useful in the absence of structural information, because an entire protein can be scanned with libraries spanning large stretches of contiguous sequence.
Acknowledgments
We thank Andrea Cochran, Kevin Clark, Christian Galindo, Abraham de Vos, Henry Lowman, and James Wells for helpful discussions.
Abbreviations
- hGH
human growth hormone
- hGHbp
hGH binding protein
- wt
wild-type
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.160252097.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.160252097
References
- 1.Wilson D S, Desplan C. Nat Struct Biol. 1999;6:297–300. doi: 10.1038/7524. [DOI] [PubMed] [Google Scholar]
- 2.Gonzalez L, Jr, Scheller R H. Cell. 1999;96:755–758. doi: 10.1016/s0092-8674(00)80585-1. [DOI] [PubMed] [Google Scholar]
- 3.Mackrill J J. Biochem J. 1999;337:345–361. [PMC free article] [PubMed] [Google Scholar]
- 4.Baeuerle P A. Cell. 1998;95:729–731. doi: 10.1016/s0092-8674(00)81694-3. [DOI] [PubMed] [Google Scholar]
- 5.Noel J P. Nat Struct Biol. 1997;4:677–680. doi: 10.1038/nsb0997-677. [DOI] [PubMed] [Google Scholar]
- 6.Montelione G T, Anderson S. Nat Struct Biol. 1999;6:11–12. doi: 10.1038/4878. [DOI] [PubMed] [Google Scholar]
- 7.Cunningham B C, Wells J A. J Mol Biol. 1993;234:554–563. doi: 10.1006/jmbi.1993.1611. [DOI] [PubMed] [Google Scholar]
- 8.Clackson T, Wells J A. Science. 1995;267:383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 9.Chen G, Dubrawsky I, Mendez P, Georgiou G, Iverson B L. Protein Eng. 1999;12:349–356. doi: 10.1093/protein/12.4.349. [DOI] [PubMed] [Google Scholar]
- 10.Warren M S, Benkovic S J. Protein Eng. 1997;10:63–68. doi: 10.1093/protein/10.1.63. [DOI] [PubMed] [Google Scholar]
- 11.Shortle D. J Biol Chem. 1989;264:5315–5318. [PubMed] [Google Scholar]
- 12.Fersht A R. Biochemistry. 1987;26:8031–8037. doi: 10.1021/bi00399a001. [DOI] [PubMed] [Google Scholar]
- 13.Cunningham B C, Wells J A. Science. 1989;244:1081–1085. doi: 10.1126/science.2471267. [DOI] [PubMed] [Google Scholar]
- 14.Matthews B W. FASEB J. 1996;10:35–41. doi: 10.1096/fasebj.10.1.8566545. [DOI] [PubMed] [Google Scholar]
- 15.Wells J A. Methods Enzymol. 1991;202:390–411. doi: 10.1016/0076-6879(91)02020-a. [DOI] [PubMed] [Google Scholar]
- 16.Gregoret L M, Sauer R T. Proc Natl Acad Sci USA. 1993;90:4246–4250. doi: 10.1073/pnas.90.9.4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clackson T, Wells J A. Trends Biotechnol. 1994;12:173–184. doi: 10.1016/0167-7799(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 18.Wells J A, Lowman H B. Curr Opin Biotechnol. 1992;3:355–362. doi: 10.1016/0958-1669(92)90163-d. [DOI] [PubMed] [Google Scholar]
- 19.Sidhu S S, Weiss G A, Wells J A. J Mol Biol. 2000;296:487–495. doi: 10.1006/jmbi.1999.3465. [DOI] [PubMed] [Google Scholar]
- 20.Sidhu, S. S., Lowman, H. B., Cunningham, B. C. & Wells, J. A. (2000) Methods Enzymol., in press. [DOI] [PubMed]
- 21.Kunkel T A, Roberts J D, Zakour R A. Methods Enzymol. 1987;154:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- 22.Jin L, Fendly B M, Wells J A. J Mol Biol. 1992;226:851–865. doi: 10.1016/0022-2836(92)90636-x. [DOI] [PubMed] [Google Scholar]
- 23.de Vos A M, Ultsch M, Kossiakoff A A. Science. 1992;255:306–312. doi: 10.1126/science.1549776. [DOI] [PubMed] [Google Scholar]
- 24.Virnekas B, Ge L, Pluckthun A, Schneider K C, Wellnhofer G, Moroney S E. Nucleic Acids Res. 1994;22:5600–5607. doi: 10.1093/nar/22.25.5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaytan P, Yanez J, Sanchez F, Mackie H, Soberon X. Chem Biol. 1998;5:519–527. doi: 10.1016/s1074-5521(98)90007-2. [DOI] [PubMed] [Google Scholar]
- 26.Hu J C, Newell N E, Tidor B, Sauer R T. Protein Sci. 1993;2:1072–1084. doi: 10.1002/pro.5560020701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greenspan N S, Di Cera E. Nat Biotechnol. 1999;17:936–937. doi: 10.1038/13590. [DOI] [PubMed] [Google Scholar]
- 28.Lowman H B, Wells J A. J Mol Biol. 1993;234:564–578. doi: 10.1006/jmbi.1993.1612. [DOI] [PubMed] [Google Scholar]
- 29.Wells J A. Biochemistry. 1990;29:8509–8517. doi: 10.1021/bi00489a001. [DOI] [PubMed] [Google Scholar]
- 30.Nicholls A, Sharp K A, Honig B. Proteins Struct Funct Genet. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]