

Abstract
There are a number of diseases in which normally soluble proteins associate into regular, insoluble amyloid fibrils. The development of in vitro model systems in which detailed structural, kinetic, and thermodynamic characterization are feasible is of critical importance to our understanding of the amyloid fibril phenomenon. The formation of amyloid fibrils by proteins that are not associated with disease has been recently described, suggesting that this may be a common property of many proteins and not only of the few proteins associated with amyloidoses. The B1 Ig-binding domain of protein G (β1) is an extremely well-characterized model system. We have found that under certain experimental conditions, some variants of β1 form fibrils with high reproducibility. By controlling the stability of the protein—either by mutations or by changing experimental conditions—we are able to modulate the ability of the protein to form fibrils. For all of the variants, we find that the key requirement for fibril formation is to choose conditions in which the population of intermediate conformations present during the unfolding transition is maximized.
The amyloidoses are a group of protein misfolding diseases characterized by the accumulation in extracellular spaces of insoluble fibrillar protein (1–3). The deposition of normally soluble proteins in this insoluble form is believed to lead to cell death and tissue degeneration. To date, 18 different proteins and polypeptides have been identified in disease-associated amyloid deposits. These proteins include the Aβ peptide in Alzheimer's disease, the prion protein (PrP) in the transmissible spongiform encephalopathies, the islet-associated polypeptide in type II diabetes, transthyretin and lysozyme in familial amyloidoses, and other variants of truncated or misprocessed proteins in systemic amyloidoses (4). No sequence or structural similarities are apparent between any of the proteins that display the ability to form amyloids. Despite the dissimilarity of the soluble proteins, the fibrils that they form share a number of common features. The amyloid fibrils are long, straight, unbranched filaments 40–120 Å in diameter, which bind to physiological dyes such as Congo red and thioflavine T and are resistant to protease digestion. X-ray fiber diffraction studies of three transthyretin mutants, apolipoprotein A-I, monoclonal λ Ig light chain, islet-associated polypeptide, amyloid A protein, and a lysozyme mutant show similar x-ray fiber diffraction patterns, indicative of a cross β-structure, with a helical array of β-sheets in which the long axis of the fibril is parallel to the long axis of the helix and perpendicular to the β-strands (5, 6). The β-sheets cannot be distinguished between parallel and antiparallel β-strands (7). The first electron microscopy reconstruction of mutant transthyretin fibrils gave a detailed view of the fibril substructures (8). More recently, image reconstruction cryo-electron microscopy of a Src homology 3 domain fibril provided a detailed model (25-Å resolution) of this structure to be proposed (9). Higher levels of organization have been characterized by atomic force microscopy in which average widths for filaments, protofibrils, and fibrils of Ig light chain and Aβ peptide were determined (10, 11).
The development of in vitro model systems is of critical importance to our understanding of the physicochemical basis of amyloid fibril phenomenon. There has been great progress in our understanding of amyloid phenomenon (1, 12–14); however, the experimental limitations shown by some of the amyloid proteins, for example low solubility and lack of a high-resolution three-dimensional structure, have precluded comprehensive structural and thermodynamic characterization of both their soluble and fibrillar states (15). The formation of amyloid fibrils by a number of proteins unlinked to disease has been recently described (16–20). In addition, the discovery of prion-like proteins in yeast and fungi (21, 22) suggests that the ability to form fibrils may be a common property of many, if not all, proteins under appropriate conditions.
There is increasing evidence that amyloid fibrils do not develop directly from the native conformation, but from precursors that appear to be only partly folded (16, 23–26).
Our goal is to systematically explore the structural and thermodynamic requirements for fibril formation and in particular to investigate the importance of the population of intermediate states Our model system is a collection of variants of the B1 domain of Streptococcal IgG-binding protein G (β1), a 56-residue monomeric domain that is extremely well characterized structurally, thermodynamically, and kinetically (27–36). Our variants are built around the central parallel β-strands of β1 (Fig. 1A). These variants originally were designed for an experimental study of the energetics of aromatic-glycine residue interactions between β-strands (35), motivated by the results of statistical surveys of cross-strand pairing (37, 38). A number of mutations were incorporated at positions 4, 8, 15, and 44 (host site), and different residue combinations were introduced at positions 6 and 53 (guest site). The wild type (WT) protein and a protein with the WT residues in positions 6 and 53 (I6T53) also were studied. The serendipitous observation of fibril-like structures in one of the variants (G6Y53) led us to the investigation of amyloid formation presented here.
Figure 1.

(A) Schematic representation of β1, showing the position numbers where the mutations and the context host-site were introduced with a list of variants used for this study. WT is a variant of β1 with a mutation in position 2 (T2Q) that prevents the cleavage of the N-terminal methionine (23). All of the other variants include the same mutation. The sequence of WT (T2Q) is: NH3+-MTYKLILNGKTLKGETTTEAVDAATAEKVFKQYANDNGVDGEWTYDDATKTFTVTE-COO- where the residues in bold are the positions in which the mutations have been introduced. (B) Refolding recovery of A6A53 and I6T53 at increasing pHs: pH 5.2 (⋄), pH 6.9 (•), pH 8.0 (○), pH 9.0 (▫), and pH 11.0 (▴). The buffer used was 10 mM sodium acetate, 10 mM sodium borate, and 10 mM sodium citrate. (C) Thermal denaturation fraction refolded at pH 5.2 for all variants. WT (x), I6T53 (▾), A6Y53 (•), A6F53 (▪), F6A53 (⋄), A6A53 (○), F6G53 (▴), and G6Y53 (▫). The buffer used was 50 mM sodium acetate. (D) Thermal denaturation refolding at pH 9.0 for all variants. WT (x), I6T53 (▾), A6Y53 (•), A6F53 (▪), F6A53 (⋄), A6A53 (○), F6G53 (▴), and G6Y53 (▫). The buffer used was 10 mM sodium acetate, 10 mM sodium borate, and 10 mM sodium citrate. MRE was used because the fraction folded was impossible to measure because of the lack of unfolded baselines in most of the variants. (E) Electron micrographs showing the fibrils formed during irreversible denaturation for all variants. (F) Thermal denaturation of variant F6G53 at pH 9.0 (•), pH 9.0 in the presence of 1 M Na2SO4 (⋄), refolding at pH 9.0 (▴), and pH 9.0 in the presence of 1 M Na2SO4 (○). The buffer used was 10 mM sodium acetate, 10 mM sodium borate, and 10 mM sodium citrate.
Materials and Methods
Protein Preparation.
Protein expression and purification was performed following published procedures (31). Briefly, ion exchange chromatography (Q-Sepharose) in a gradient of NaCl in 20 mM Tris⋅HCl buffer, pH 8.0 was followed by gel filtration chromatography (Hi-Load Superdex 75, Amersham Pharmacia) in 50 mM sodium acetate, pH 5.2. Protein purity was checked by PAGE, and the protein concentration was determined from the amino acid analysis performed at the Howard Hughes Medical Institute Biopolymer Laboratory and W.M. Keck Foundation Biotechnology Resource Laboratory at Yale University.
CD Spectroscopy.
CD spectra were recorded on an Aviv 62DS spectrometer (Aviv Associates, Lakewood, NJ). Far UV-CD spectra were obtained in the continuous mode, taking measurements every 1 nm with an averaging time of 5 sec at 277 K. Protein samples were 15–50 μM in 0.2-cm path length cells.
Thermal Denaturations.
The ellipticity at 217 nm was monitored every 2° from 277 K to 363 K, with an equilibration time of 1 min between each temperature point and an averaging time of 30 sec. A 0.2-cm cuvette was used.
The thermal denaturation curves were analyzed according to a two-state transition model. Linear extrapolation of the folded and unfolded baselines based on a minimum of 10 points was performed. The fraction folded (FF) at each temperature was calculated by using the equation
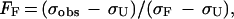 |
1 |
where σobs is the observed ellipticity at 217 nm, and σF and σU are ellipticities of the folded and unfolded states, respectively, derived from the extrapolated baselines. The fraction unfolded (FU) is therefore given by
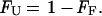 |
2 |
Chemical Denaturations.
The ellipticity at 217 nm was monitored as a function of concentration of guanidinium hydrochloride (GuHCl) at 277 K. Stock solutions of 8 M GuHCl were purchased from Pierce. Sodium acetate was added as a salt to a final concentration of 50 mM and the pH was adjusted to pH 5.2.
Denaturation curves were obtained by mixing two different protein stock solutions in the absence of denaturant (folded) and the second in the presence of the appropriate concentration of GuHCl (unfolded). Different volumes of the two stock solutions were mixed to change the denaturant concentration, while keeping the protein concentration constant. Initial and final concentrations of GuHCl were confirmed by refractometry by using the equation
 |
3 |
where ΔN = difference in refractrometry between the sample and the buffer reference.
Chemical denaturation curves were analyzed by using nonlinear least squares to fit the data directly to the following equation (40),
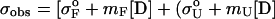 |
4 |
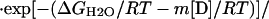 |
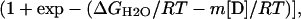 |
where σobs is the observed ellipticity at 217 nm, mF and σoF are the slope and the intercept of the folded baselines, respectively, mU and σoU are the slope and the intercept of the unfolded baselines, respectively, [D] is the molarity of the denaturant, ΔGH2O is the free energy of folding in the absence of denaturant, R is the universal gas constant 1.98 cal mol-1⋅K-1, T is the temperature in kelvin, and m is a constant (41).
The folded and unfolded baselines can be derived from Eq. 4.
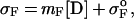 |
5 |
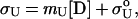 |
6 |
where σF and σU are the ellipticities of the folded and unfolded states at a given denaturant concentration, respectively.
In the transition region, the free energy of folding in the presence of GuHCl is linearly proportional to the concentration of denaturant:
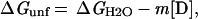 |
7 |
where ΔGunf is the free energy of unfolding at a particular denaturant concentration, and ΔGH2O, m, and [D] are as described above.
The fraction of folded protein present at any concentration of denaturant is given by
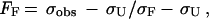 |
8 |
where σobs is the observed ellipticity at 217 nm, and σF and σU are ellipticities of the folded and unfolded states, respectively.
Agitation.
Three hundred microliters of concentrated stocks of each protein (G6Y53, 103 μM; A6Y53, 1,600 μM; A6F53, 883 μM; F6A53, 294 μM; F6G53, 100 μM; A6A53, 110 μM; I6T53, 1,800 μM; and WT, 800 μM) were agitated in 1-ml Eppendorf tubes at 300 rpm for 24 h in different experimental conditions. The formation of visible precipitate began between 3 and 12 h. To confirm reproducibility, three independent experiments were performed for each variant under each condition. We estimated by this protocol approximately 50% of the initial soluble protein was converted to fibrils by hydrolyzing the fibrils and measuring protein concentration.
Seeding.
The seeds were formed by sonicating a dialyzed solution of fibrils prepared by agitation at 338 K and pH 5.2 from all proteins, except I6T53. Electron microscopy indicated that this treatment was effective in fragmenting the fibrils. The formation of fibrils in the presence of 1/200 mole per mole ratio of “seeds”/soluble protein was monitored by CD at 217 nm. The appropriate seeds were added to 22 μM (A6A53) 50 μM (I6T53), 50 μM (A6Y53) in 50 mM sodium acetate and subsequently heated to the experimental temperature (318 K for A6A53, 345 K for I6T53, 338 K for A6Y53). Two different controls were performed: the same concentration of soluble protein in the absence of seeds and the same concentration of seeds in the absence of soluble protein. Both showed no change in mean residue ellipticity (MRE) and no fibril formation.
Electron Microscopy.
Electron microscopy was performed by using a Zeiss JEM-A transmission electron microscope at 80-kV excitation voltage. Six microliters of fibril solution was air-dried for 5 min on a Formvar-coated copper grid. The sample was then negatively stained with 6 μl of 1% phosphotungstic acid solution, pH 7.0.
Congo Red and Thioflavine T Staining.
Aliquots of proteins were air-dried onto glass slides, stained with a saturated solution of Congo red, and examined for birefringence with and optical microscope between cross-polarizers (42). The thioflavine T fluorescence analysis was performed as described in ref. 43. Aliquots (1 or 5 μl) of soluble protein or fibril suspension were added to 3 μM thioflavine T in 50 mM sodium acetate (pH 5.2) to a final volume of 1 ml. Excitation spectra were recorded on a Hitachi F-4500 spectrometer with an excitation wavelength maximum of 450 nm and emission maximum wavelength of 484 nm.
Results
Thermodynamic Properties of the Soluble Form of the Proteins.
The thermodynamic stabilities of the soluble monomeric form of these variants were determined by thermal and chemical denaturation methods. At the pH of maximum stability (5.2), β1 undergoes a two-state, cooperative, reversible denaturation transition both thermally and chemically. The monomeric nature of the variants under these conditions was previously confirmed (35). Thermodynamic parameters for all variants, derived from both chemical and thermal denaturation studies, are shown in Table 1. There was no evidence by electron microscopy of fibril or formation in any of the samples that were denatured either thermally or chemically at pH 5.2.
Table 1.
Thermodynamic parameters for β1 parallel variants determined by chemical and thermal denaturation
| Protein | Tm unfolding, K | Unf. transition range, K | ΔGunf., cal/mol K | Cm |
|---|---|---|---|---|
| WT | 360 | 343 K –>373 | 3,857 ± 210 | 4.6 |
| I6T53 | 345 | 333–363 | 4,697 ± 533 | 3.6 |
| A6Y53 | 334 | 313–343 | 2,661 ± 109 | 1.5 |
| A6F53 | 331 | 313–343 | 1,965 ± 203 | 2.0 |
| F6A53 | 328 | 313–343 | 3,198 ± 142 | 1.7 |
| A6A53 | 319 | 298–338 | 1,306 ± 647 | 1.5 |
| F6G53 | 318 | 298–338 | 1,860 ± 366 | 1.1 |
| G6Y53 | 318 | 298–338 | 1,362 ± 389 | 1.6 |
The variants are listed in decreasing order of melting temperatures. Thermal denaturations were performed in 50 mM sodium acetate buffer, pH 5.2. Protein concentrations ranged from 10 μM to 50 μM. Chemical denaturations were performed with GuHCl at 277 K in 50 mM sodium acetate buffer, pH 5.2. Protein concentrations ranged from 10 μM to 50 μM.
Thermal Denaturation at Different pHs.
When refolding is reversible, we assume a two-state transition from unfolded to folded protein. By contrast, if folding is irreversible, it suggests that the protein is trapped in a different low energy conformation and is unable to return to the native state. Irreversible refolding can be caused by precipitation or aggregation including fibril formation. All variants are partially or fully folded in a pH range from 2.0 to 11.0. We performed thermal denaturations on all of the variants at different pHs to determine the effect of pH on the reversibility of folding and its possible correlation with fibril formation. Fig. 1B shows the refolding curves at increasing pH for variant A6A53 and I6T53. For both variants, increasing the pH above the optimum of pH 5.2 lowers the Tm. I6T53 retains its ability to refold reversibly until pH 11.0, at which point, the reversibility is lost. By contrast, A6A53, which is among the least stable variants, loses all reversibility by pH 6.9. Fig. 1C shows thermal renaturations at pH 5.2, and Fig. 1D shows the thermal renaturations at pH 9.0. In all of the variants, there is a loss of reversibility of folding with increasing pH, but the effect is more dramatic with the less stable variants. There was no visible precipitate evident in any of the samples that display irreversible denaturation. When analyzed by electron microscopy, however, fibril formation and amorphous aggregation was clearly visible (Fig. 1E).
Sodium sulfate has been demonstrated to increase slightly the stability of native β1, but to increase quite significantly the stability of a folding intermediate (33). The presence of 1 M Na2SO4 (herein when we refer to Na2SO4 we mean 1 M Na2SO4) during the thermal denaturation experiments increased the thermal stability of all of the variants both at pH 5.2 and pH 9.0. Fig. 1F shows the refolding of variant F6G53 in the presence and absence of Na2SO4 at pH 9.0. In the presence of Na2SO4, folding reversibility is partly recovered. The change in stability that sodium sulfate induces offers a wider variety of experimental conditions for the formation of amyloid fibrils. We found from our thermal denaturation experiments at increasing pHs that folding reversibility was related to fibril formation, and we explored to what extent fibril formation could be further modulated by using Na2SO4.
Although irreversible thermal denaturation produces fibrils, we also explored conditions under which cleaner preparations of fibrils could be formed in a higher yield.
Methods for Amyloid Fibril Formation.
A unifying feature of the experimental conditions chosen for β1 fibril formation is that the soluble form of the protein is destabilized. Destabilizing conditions could increase the probability of sampling any misfolded states that will drive amyloid fibril formation. We accomplished this by agitating samples at a temperature in the middle of the unfolding transition, and by addition of fibril seeds, conditions that differ completely from the conditions in which the monomeric variants have been characterized. We studied the formation of fibrils as a function of temperature, pH, and ionic strength.
Agitation.
Agitation of soluble protein has been demonstrated to be a successful method for forming amyloid fibrils in vitro. It is not well understood why this method induces the formation of amyloid fibrils although it has been suggested that agitation may cause shearing of the first-formed fibrils, thereby increasing the number of nucleation particles (seeds) and accelerating fibril formation, or that agitation may increase the apparent concentration of protein by removing the amount of protein that sticks to the walls of the tubes. Agitation appears to accelerate fibril formation by these effects, rather than by causing obligatory protein denaturation because fibrils do form in the absence of agitation, only more slowly.
Agitation at pH 5.2 and 338 K results in the formation of precipitate for most variants. Electron micrographs clearly showed that the presence of precipitate was caused by the formation of fibrils (Fig. 2). The most stable proteins (I6T53 and WT) did not form fibrils under these conditions. We performed agitation experiments under different experimental conditions. With the hypothesis that the variants would form fibrils when the agitation experiments are performed under destabilizing conditions a temperature within the unfolding transition range (Table 1; see Table 2, which is published as supplemental material on the PNAS web site, www.pnas.org) was chosen. The F6G53 variant formed fibrils at pH 5.2 and 338 K, in agreement with our prediction that fibril formation conditions should be within the unfolding transition range (for F6G53 at pH 5.2, 298–343 K). Different experimental conditions confirmed our hypothesis (Fig. 1F and Table 3, which is published as supplemental material). We performed an experiment at pH 9.0 and 313 K, conditions in which we observed fibrils (transition range at pH 9.0 is 283 to 323 K). Increasing the temperature to 333 K (outside the unfolding transition range) showed no evidence of fibril formation, consistent with the hypothesis. Addition of Na2SO4 at pH 9.0 moved the transition range to 303–330 K, and, as predicted, fibrils were formed at 313 K, which is within the shifted transition. There was no evidence of aggregation when the experiment was performed at 333 K, Na2SO4, and pH 9.0 because the protein is unfolded under these conditions. For every variant under conditions that map in the unfolding transition fibrils formation is induced by agitation. These results hold when the stability of the variants is modulated either by increasing the pH or by addition of Na2SO4 (Table 2).
Figure 2.

Electron micrographs showing the fibrils formed from agitation experiments.
Nucleation-Dependent Amyloid Formation: Seeding.
Fig. 3A shows an example of the kinetics of fibril formation followed by seeding for A6A53 variant. After 6 h, clusters of precipitate were visible. Two kinetic phases could be distinguished. The first phase (0–600 sec) corresponds to the heating of the sample from 277 K to 318 K. When the experiment started with a sample at 318 K, we observed that the first phase is reduced from 600 to 200 sec (data not shown). The second phase presents a decrease in the β-sheet content soluble signal (MRE value at 217 nm becomes more positive), because the formation of fibrils brings proteins out of solution, reducing the net amount of soluble β-sheet content. An electron micrograph of the precipitate is shown in Fig. 3B.
Figure 3.

(A) Seeding experiment followed by MRE at 217 nm for A6A53 variant at pH 5.2, 318 K, in 50 mM sodium acetate buffer. (Inset) The second phase of the seeding experiment, from 600 s to 21,600 s. (B) Electron micrograph showing the fibrils from auto-seeding experiment for A6A53 variant at pH 5.2 in 50 mM sodium acetate buffer.
An Experiment That Is Relevant to the Concept of Scrapie Prion Infection and the “Species Barrier” for Prion Transmission Is Induction of Fibril Formation by Cross-Seeding.
This experiment tests the ability to form fibrils by using seeds from a variant that forms fibrils with I6T53 that does not form fibrils at pH 5.2 and 338 K. A6Y53 seeds were added and the formation of fibrils was followed by MRE at 217 nm. The MRE values followed the same behavior seen in the auto-seeding experiment (see Fig. 4A, which is published as supplemental material) and electron micrographs confirmed the presence of amyloid fibrils under conditions where these proteins do not spontaneously auto-seed (Fig. 4B). This result suggests that even though I6T53 is unable to nucleate fibrils under these conditions, adding small amounts of A6Y53 seeds nucleates the process and I6T53 can elongate the fibrils.
In addition, we performed a cross-seeding experiment between two proteins that are able to auto-seed. A6Y53-soluble protein was used with F6A53 seeds. The behavior was in agreement with the previous auto- and cross-seeding experiments (data not shown). In cross-seeding experiments in which A6A53-soluble protein was seeded with unrelated fibril seeds from human islet amyloid polypeptide, no evidence of aggregation was found.
These results demonstrate the ability of the variants in our system to cross-seed, suggesting that the seed conformation required for fibril formation is maintained among the environment of the host-guest variants. The situation is different when the seeding is performed with unrelated seeds, suggesting that unrelated fibrils populate different conformations of seeds, analogous to the species barrier in natural systems.
Characterization of Fibrils by Congo Red and Thioflavine T Binding.
The ability of amyloid fibrils to bind Congo red and thioflavine T dyes has been demonstrated (42, 43). Addition of Congo red to samples with fibrils from all of the variants produced the characteristic green birefringence under cross-polarized light, whereas the soluble protein did not (data not shown).
Thioflavine T showed an intensity increase and a fluorescence shift around 430–450 nm in the presence of fibrils and a lower increase in the presence of fibril seeds. The soluble proteins do not bind to thioflavine T and do not show any fluorescence shift (data not shown).
Discussion
Our results suggest that the formation of amyloid fibrils in the β1 model system requires conditions that will populate the ensemble of conformations present during the thermal unfolding transition. We have found four independent means (which can be used in combination) by which to vary the stability of the protein and the population of intermediate states: mutagenesis, pH, temperature, and addition of Na2SO4. These results allowed us to construct a phase diagram and to delineate the circumstances that promote fibril formation.
It had been suggested that the species that initiate fibril formation may be folding intermediates, rather than either the native or unfolded protein for monomeric proteins (17, 23–25) as well as oligomeric proteins (26). The following examples with monomeric proteins illustrate this idea. Booth and colleagues (23) characterized two amyloidogenic variants of lysozyme. The crystal structures of these variants show minimal differences relative to WT lysozyme, but the variants are less stable and their thermal unfolding is less cooperative, with conformations populated at the midpoint of the thermal unfolding that could be considered “molten globule”-like. These mutations cause autosomal dominant heredity amyloidoses, by greatly increasing the propensity of the proteins to form fibrils (23). Amyloid fibril formation was observed in a Src homology 3 domain under acidic conditions. NMR diffusion experiments show that this “A state” amyloidogenic precursor is more compact than the denatured protein, indicative that it is partially folded (17). Hurle and colleagues (25) have found amino acid replacement at specific positions in the variable region of the N-terminal domain of Ig light chain. These positions play a role in maintaining domain structure and stability. The ability of these proteins to aggregate is correlated with the decreased stability. They suggested that these variants decrease the energetic barrier to unfolding and this effect would be expected to increase in population of a non-native state required for amyloid formation. Wall and colleagues (24) described the correlation between thermodynamic stability and fibrillogenicity for human λ6 light chain (Vλ6) and suggested that the potential of a variants to form fibrils is correlated with its reduced thermodynamic stability, which results in an increased population of protein in a non-native, amyloidogenic state.
Physiological ordered protein aggregation processes as diverse as viral coat assembly, microtubule formation, flagellum formation, and sickle-cell fibril formation occur by nucleation-dependent polymerization pathways. Nucleation-dependent aggregation shows characteristic features: (i) No aggregation occurs at protein concentrations below a critical concentration. (ii) There is a lag time before polymerization occurs. (iii) During the lag time, addition of a seed results in immediate aggregation. The presence of seeds will accelerate the formation of fibrils (15).
The prion diseases have been characterized by species barriers that are present for certain directions of infectivity between animals. The concept of scrapie prion (PrPSc) providing a seed to assist the conversion of nascent cellular prion implies that some PrPSc seeds should be more efficient at stabilizing the nascent PrPSc than others in the initial phase of disease propagation (44). The transmission of prions from one species to another generally is accompanied by a prolongation of the incubation time relative to transmissions where the host species is the same.
It has been found that there are three different distant clusters that map the variability of 23 mammalian PrP sequences (21, 45), suggesting that prion sequences do not have a localized region that determines their specificity, but rather involve the overall conformation.
We have shown that our system is able to achieve both auto-seeding and cross-seeding of different β1 variants. The seeding experiments confirm that the β1 system follows the nucleation-propagation mechanism that has been previously proposed for amyloid formation (15). The β1 system also shows specificity in propagation, because a β1 variant is unable to propagate islet amyloid polypeptide seeds.
In conclusion, our results indicate that by changing the stability of the soluble form of β1—either by mutations or by modifying experimental conditions—we are able to modulate the ability of the protein to form fibrils. For all of the variants, we find that the key requirement for fibril formation is to choose conditions in which the population of intermediate conformations present during the unfolding transition is maximized.
Supplementary Material
Acknowledgments
We thank Shae Padrick for the generous gift of islet amyloid polypeptide fibrils for the cross-seeding experiment. We thank Barry Piekos for technical assistance and advice with electron microscopy and Ritu Khurana and Aaron Chamberlain for their advice regarding fibril formation methods. We are grateful to Andrew Miranker, Charles Morgan, Shae Padrick, Charu Chaudhry, Steve Marino, and Gerard Olack for helpful discussions. We acknowledge Regan lab members for thorough manuscript revision and comments. This research was supported by the National Institutes of Health. M.R.-A. is a Human Frontier Science Program long-term fellow, and L.R. is a Dreyfus Teacher-Scholar.
Abbreviations
- PrP
prion protein
- β1
protein G Ig binding B1 domain from Streptococcus
- GuHCl
guanidinium hydrochloride
- MRE
mean residue ellipticity
- WT
wild type
Note
After this manuscript was submitted, an interesting mutational analysis of propensity for amyloid formation by acylphosphatase modulated by trifluoroethanol was published (46).
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.150091797.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.150091797
References
- 1.Kelly J W. Curr Opin Struct Biol. 1996;6:11–17. doi: 10.1016/s0959-440x(96)80089-3. [DOI] [PubMed] [Google Scholar]
- 2.Kelly J W. Curr Opin Struct Biol. 1998;8:101–106. doi: 10.1016/s0959-440x(98)80016-x. [DOI] [PubMed] [Google Scholar]
- 3.Serpell L C, Sunde M, Blake C C. Cell Mol Life Sci. 1997;53:871–887. doi: 10.1007/s000180050107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Westermark P, Araki S, Benson M D, Cohen A S, Frangione B, Masters C L, Saraiva M J, Sipe J D. Amyloid. 1998;6:63–66. doi: 10.3109/13506129908993290. [DOI] [PubMed] [Google Scholar]
- 5.Blake C, Serpell L. Structure (London) 1996;4:989–998. doi: 10.1016/s0969-2126(96)00104-9. [DOI] [PubMed] [Google Scholar]
- 6.Sunde M, Serpell L C, Bartlam M, Fraser P E, Pepys M B, Blake C C F. J Mol Biol. 1997;273:729–739. doi: 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- 7.Serpell L C, Fraser P E, Sunde M. Methods Enzymol. 1999;309:526–536. doi: 10.1016/s0076-6879(99)09036-9. [DOI] [PubMed] [Google Scholar]
- 8.Serpell L C, Sunde M, Fraser P E, Luther P K, Morris E P, Sangren O, Lundgren E, Blake C C F. J Mol Biol. 1995;254:113–118. doi: 10.1006/jmbi.1995.0604. [DOI] [PubMed] [Google Scholar]
- 9.Jiménez J L, Guijarro J I, Orlova E, Zurdo J, Dobson C M, Sunde M, Saibil H R. EMBO J. 1999;18:815–821. doi: 10.1093/emboj/18.4.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ionescu-Zanetti C, Khurana R, Gillespie J R, Petrick J S, Trabachino L C, Minert L J, Carter S A, Fink A L. Proc Natl Acad Sci USA. 1999;96:13175–13179. doi: 10.1073/pnas.96.23.13175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harper J D, Wong S S, Lieber C M, Lansbury P T., Jr Biochemistry. 1999;38:8972–8980. doi: 10.1021/bi9904149. [DOI] [PubMed] [Google Scholar]
- 12.Soto C. J Mol Med. 1999;77:412–418. doi: 10.1007/s001090050371. [DOI] [PubMed] [Google Scholar]
- 13.Wisniewski T, Aucouturier P, Soto C, Frangione B. Amyloid. 1998;5:212–214. doi: 10.3109/13506129809003848. [DOI] [PubMed] [Google Scholar]
- 14.Koo E H, Lansbury P T, Jr, Kelly J W. Proc Natl Acad Sci USA. 1999;96:9989–9990. doi: 10.1073/pnas.96.18.9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harper J D, Lansbury P T., Jr Annu Rev Biochem. 1997;66:385–407. doi: 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- 16.Guijarro J I, Sunde M, Jones J A, Campbell I D, Dobson C M. Proc Natl Acad Sci USA. 1998;95:4224–4228. doi: 10.1073/pnas.95.8.4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chiti F, Webster P, Taddei N, Clark A, Stefani M, Ramponi G, Dobson C M. Proc Natl Acad Sci USA. 1999;96:3590–3594. doi: 10.1073/pnas.96.7.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gross M, Wilkins D K, Pitkeathly M C, Chung E W, Higham C, Clark A, Dobson C M. Protein Sci. 1999;8:1350–1357. doi: 10.1110/ps.8.6.1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.West M W, Wang W, Patterson J, Mancias J D, Beasley J R, Hecht M H. Proc Natl Acad Sci USA. 1999;96:11211–11216. doi: 10.1073/pnas.96.20.11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Litvinovich S V, Brew S A, Aota S, Akiyama S K, Haudenschild C, Ingham K C. J Mol Biol. 1998;280:245–258. doi: 10.1006/jmbi.1998.1863. [DOI] [PubMed] [Google Scholar]
- 21.Prusiner S B. Science. 1997;278:245–251. doi: 10.1126/science.278.5336.245. [DOI] [PubMed] [Google Scholar]
- 22.Wickner R B, Taylor K L, Edskes H K, Maddelein M L, Moriyama H, Roberts B T. Microbiol Mol Biol Rev. 1999;63:844–861. doi: 10.1128/mmbr.63.4.844-861.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Booth D R, Sunde M, Belloti V, Robinson C V, Hutchinson W L, Fraser P E, Hawkins P N, Radford S E, Blake C C F, Pepys M B. Nature (London) 1997;385:787–793. doi: 10.1038/385787a0. [DOI] [PubMed] [Google Scholar]
- 24.Wall J, Schell M, Murphy C, Hrncic R, Stevens F J, Solomon A. Biochemistry. 1999;38:14101–14108. doi: 10.1021/bi991131j. [DOI] [PubMed] [Google Scholar]
- 25.Hurle M R, Helms L R, Li L, Chan W, Wetzel R. Proc Natl Acad Sci USA. 1994;91:5446–5450. doi: 10.1073/pnas.91.12.5446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lai Z, Colón W, Kelly J W. Biochemistry. 1996;35:6470–6482. doi: 10.1021/bi952501g. [DOI] [PubMed] [Google Scholar]
- 27.Gronenborn A M, Filpula D R, Essig N Z, Achari A, Whitlow M, Wingfield P T, Clore G M. Science. 1991;253:657–661. doi: 10.1126/science.1871600. [DOI] [PubMed] [Google Scholar]
- 28.Gallagher T, Alexander P, Bryan P, Gilliland G L. Biochemistry. 1994;33:4721–4729. [PubMed] [Google Scholar]
- 29.Alexander P, Fahnestock S, Lee T, Orban J, Bryan P. Biochemistry. 1992;31:3597–3603. doi: 10.1021/bi00129a007. [DOI] [PubMed] [Google Scholar]
- 30.Alexander P, Orban J, Bryan P. Biochemistry. 1992;31:7243–7248. doi: 10.1021/bi00147a006. [DOI] [PubMed] [Google Scholar]
- 31.Smith C K, Withka J M, Regan L. Biochemistry. 1994;33:5510–5517. doi: 10.1021/bi00184a020. [DOI] [PubMed] [Google Scholar]
- 32.Smith C K, Regan L. Science. 1995;270:980–982. doi: 10.1126/science.270.5238.980. [DOI] [PubMed] [Google Scholar]
- 33.Park S-H, O'Neil K T, Roder H. Biochemistry. 1997;36:14277–14283. doi: 10.1021/bi971914+. [DOI] [PubMed] [Google Scholar]
- 34.Park S-H, Ramachandra Shastry M C, Roder H. Nat Struct Biol. 1999;6:943–947. doi: 10.1038/13311. [DOI] [PubMed] [Google Scholar]
- 35.Merkel J S, Regan L. Folding Des. 1998;3:449–455. doi: 10.1016/s1359-0278(98)00062-5. [DOI] [PubMed] [Google Scholar]
- 36.Merkel J S, Sturtevant J M, Regan L. Structure (London) 1999;7:1333–1343. doi: 10.1016/s0969-2126(00)80023-4. [DOI] [PubMed] [Google Scholar]
- 37.Cootes A P, Curmi P M G, Cunningham R, Donnelly C, Torda A E. Proteins. 1998;32:175–189. [PubMed] [Google Scholar]
- 38.Hutchinson E G, Sessions R B, Thornton J M, Woolfson D N. Protein Sci. 1999;7:2287–2300. doi: 10.1002/pro.5560071106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nozaki Y. Methods Enzymol. 1972;26:43–50. doi: 10.1016/s0076-6879(72)26005-0. [DOI] [PubMed] [Google Scholar]
- 40.Santoro M M, Bolen D W. Biochemistry. 1988;27:8063–8068. doi: 10.1021/bi00421a014. [DOI] [PubMed] [Google Scholar]
- 41.Schellman J A. Biopolymers. 1978;17:1305–1322. doi: 10.1002/bip.1978.360170510. [DOI] [PubMed] [Google Scholar]
- 42.Klunk W E, Pettegrew J W, Abraham D J. J Histochem Cytochem. 1989;37:1293–1297. doi: 10.1177/37.8.2666512. [DOI] [PubMed] [Google Scholar]
- 43.LeVine H D. Protein Sci. 1993;2:404–410. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cohen F E. J Mol Biol. 1999;293:313–320. doi: 10.1006/jmbi.1999.2990. [DOI] [PubMed] [Google Scholar]
- 45.Daggett V. Curr Opin Biotechnol. 1998;9:359–365. doi: 10.1016/s0958-1669(98)80008-6. [DOI] [PubMed] [Google Scholar]
- 46.Chiti F, Taddei N, Bucciantini M, White P, Ramponi G, Dobson C M. EMBO J. 2000;19:1441–1449. doi: 10.1093/emboj/19.7.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}