

Abstract
The analysis of tandem mass (MS/MS) data to identify and quantify proteins is hampered by the heterogeneity of file formats at the raw spectral data, peptide identification, and protein identification levels. Different mass spectrometers output their raw spectral data in a variety of proprietary formats, and alternative methods that assign peptides to MS/MS spectra and infer protein identifications from those peptide assignments each write their results in different formats. Here we describe an MS/MS analysis platform, the Trans-Proteomic Pipeline, which makes use of open XML file formats for storage of data at the raw spectral data, peptide, and protein levels. This platform enables uniform analysis and exchange of MS/MS data generated from a variety of different instruments, and assigned peptides using a variety of different database search programs. We demonstrate this by applying the pipeline to data sets generated by ThermoFinnigan LCQ, ABI 4700 MALDI-TOF/TOF, and Waters Q-TOF instruments, and searched in turn using SEQUEST, Mascot, and COMET.
Keywords: analysis platform, open XML, proteomics
Introduction
Liquid chromatography tandem mass spectrometry (LC-MS/MS) has been used extensively to identify and quantify proteins in a sample (Mann and Aebersold, 2003). Experiments typically involve the collection of thousands of MS/MS spectra generated from a proteolytically treated sample. Subsequent analysis includes assigning peptides to the spectra, validating the peptide assignments to remove incorrect results, determining relative quantitation ratios between two or more samples (in the case of heavy and light isotopically labeled samples), and inferring protein identifications from the assigned peptides. We have previously described a set of open source tools that perform many of these steps: PeptideProphet validates peptides assigned to MS/MS spectra (Keller et al, 2002a), XPRESS (Han et al, 2001) and ASAPRatio (Li et al, 2003) quantitate peptides and proteins in differentially labeled samples, Pep3D enables a view of the raw spectral data (Li et al, 2004), and ProteinProphet infers sample proteins (Nesvizhskii et al, 2003). These analysis tools provide a standardized way of interpreting MS/MS data. For example, accurate probabilities provided by PeptideProphet and ProteinProphet serve as guides for interpretation of peptide and protein identifications, respectively, and enable the prediction of false positive error rates that can be used as objective criteria for the comparison of data sets generated by different researchers. Applying these tools in a uniform manner to a wide variety of data sets, generated by any type of mass spectrometer and assigned peptides using any search engine, would be of great benefit.
A major obstacle to uniform proteomic analysis has been the great heterogeneity of data formats at three distinct levels: different mass spectrometers output their raw spectral data in different proprietary formats, alternative methods that assign peptides to MS/MS spectra output their results in a variety of formats, and different methods to infer protein identifications from lists of peptides output their results in different formats. We describe in this work an analysis platform, the Trans-Proteomic Pipeline, which makes use of open XML file formats for storage of data at the raw data, peptide, and protein levels. This platform enables a uniform analysis of MS/MS spectra generated from a variety of different instruments, and assigned peptides using a variety of different database search programs, including COMET, a newly described method. We demonstrate the utility of the Trans-Proteomic Pipeline by applying it to data sets generated by ThermoFinnigan LCQ, ABI 4700 MALDI-TOF/TOF, and Waters Q-TOF instruments, and searched in turn using SEQUEST, Mascot, and COMET.
Results and discussion
Trans-Proteomic Pipeline
The Trans-Proteomic Pipeline for analysis of MS/MS data is illustrated in Figure 1. MS/MS spectra generated from a sample are used in high throughput to identify and quantify peptides and proteins. During this process, three different XML file formats are used to store data. The first format, mzXML, provides an OS and architecture independent file format for the standardized representation of MS data from various vendors and instruments and removes the burden of having to support multiple native formats from the developers of analytical applications (Pedrioli et al, 2004). By converting all native binary to mzXML and using mzXML as the start of our analysis pipeline (Figure 1, #1), the same downstream software tools, specifically the database search, relative quantitation software, and raw spectral data viewer, can be used in each case in a uniform manner regardless of the machine used to generate the data. The second format, pepXML, stores the results of assigning peptides to MS/MS spectra and subsequent peptide-level analyses. Once search results are written or converted to pepXML (Figure 1, #2), they can uniformly be subjected to peptide-level applications and viewed without regard to the method used to assign peptides. The third format, protXML, stores protein identifications inferred from input lists of peptides, and their subsequent protein-level analysis. Once protein identifications are converted to protXML (Figure 1, #3), protein-level analyses such as protein quantitation can proceed, and data viewed, without regard to the method used to infer protein identification.
Figure 1.

Trans-Protoemic Pipeline using open XML file formats at three steps: (1) raw spectral data generated by different mass spectrometers; (2) peptide assignments using different search engines; and (3) protein identifications using different methods of inference. Asterisk indicates that PeptideProphet must be specialized for each search engine.
The Trans-Proteomic Pipeline is currently applicable to MS/MS data generated by mass spectrometers from the five vendors for which mzXML converters are available, to peptide identifications made by SEQUEST, Mascot, and COMET, and to protein identifications made by ProteinProphet. The pipeline was used in this study to analyze in a uniform manner MS/MS data sets generated by ThermoFinnigan LCQ, ABI 4700 MALDI-TOF/TOF, and Waters Q-TOF instruments, and searched in turn using SEQUEST, Mascot, and COMET.
ThermoFinnigan LCQ data
The HaloICAT LCQ data set was generated on a ThermoFinnigan LCQ mass spectrometer with combined complex samples of Halobacterium proteins labeled with light or heavy ICAT reagent, digested with trypsin, and avidin affinity purified. These spectra were searched with SEQUEST, Mascot, and COMET using a human protein sequence database appended with Halobacterium protein sequences. Therefore, all search results corresponding to human proteins were inferred to be incorrect, whereas those corresponding to Halobacterium proteins were inferred to be correct, with the exception of the 5% of incorrect peptide assignments that correspond by chance to a Halobacterium protein.
Validation of search results
PeptideProphet is a component of the Trans-Proteomic Pipeline that validates search results by computing for each a probability that it is correct in the data set. Figure 2A shows that for all three search engines, the probabilities computed by PeptideProphet are accurate assessments of the likelihood that search results in the data set are correct. Thus, approximately 50% of all results assigned a probability 0.5 were actually correct, and approximately 90% of all results assigned a probability 0.9 were actually correct. These probabilities can be used to discriminate between correct and incorrect results within a data set. Figure 2B shows the number of results for each search engine passing a minimum probability threshold to achieve a false positive error rate of 2.5% predicted by PeptideProphet, an objective criterion by which to compare the different search engines. It is evident that SEQUEST at this error rate conferred 60% more results than Mascot or COMET for this data set. Similar results were observed using minimum probability thresholds conferring false positive error rates throughout the 0–10% range. This reflects not only the discrimination of each search engine, but also the additional discrimination conferred in each case by PeptideProphet using peptide properties in addition to search engine scores. It should be emphasized that these results were observed for searches performed under a defined set of conditions (see Materials and methods), and that each engine may perform better or worse under an alternative set. In fact, this analysis could be used to optimize the set of search parameters for each search engine.
Figure 2.

Trans-Protoemic Pipeline analysis of LC-MS/MS data sets. (A) Accuracy of PeptideProphet-computed peptide probabilities for HaloICAT LCQ data set in sliding window of 50 search results. (B) Numbers of search results for HaloICAT LCQ data set filtered at a minimum PeptideProphet probability to achieve a predicted 2.5% error rate. The inset shows the numbers using Mascot results with probabilities adjusted by SearchCombiner to take into account the results of SEQUEST and COMET applied to the same data set. (C) Numbers of ProteinProphet identifications for HaloICAT LCQ data set filtered at a minimum ProteinProphet probability to achieve a predicted 2.5% error rate. Each asterisk indicates an incorrect protein identification. (D) Numbers of ProteinProphet identifications for Serum MALDI-TOF/TOF data set filtered at a minimum ProteinProphet probability to achieve a predicted 2.5% error rate. (E) Numbers of ProteinProphet identifications for Yeast Q-TOF data set filtered at a minimum ProteinProphet probability to achieve a predicted 2.5% error rate.
The amount of overlap between the search results of the three methods is also shown in Figure 2B. Some assigned peptides were observed only with one search engine, predominantly with SEQUEST. However, many more of the results (46%) were obtained identically for all three search engines. In contrast, incorrect results were much less often observed by more than one search engine. For example, only 5% of the incorrect results in this data set were seen by more than one search engine, and 0.1%, with all three. This suggests that using multiple search engines in combination can help distinguish between correct and incorrect results.
A program, SearchCombiner, can be run following PeptideProphet to modify peptide probabilities using the results of other searches performed on the same data set. It assigns a score to each search result in a data set reflecting whether or not the peptide was also observed with each other search engine. It then determines the fraction of correct and incorrect results with each score value and uses that information to appropriately adjust the computed probabilities. For example, Figure 2B (inset) shows the results of applying SearchCombiner to adjust the PeptideProphet probabilities computed for the Mascot search results, taking into account the search results of SEQUEST and COMET. It is evident that the adjustment to the Mascot probabilities increased their discrimination, conferring 13% more results at a predicted 2.5% error rate relative to the unadjusted (original PeptideProphet) probabilities. Specifically, the probabilities of peptide assignments also observed with SEQUEST and COMET were increased by an appropriate amount for the data set, whereas those of assignments observed with Mascot alone were decreased.
Protein inference
ProteinProphet is a component of the Trans-Proteomic Pipeline used to infer protein identifications based on peptides assigned to MS/MS spectra and their computed probabilities of being correct. It was applied separately to the HaloICAT LCQ data set search results of SEQUEST, Mascot, and COMET, as well as to the combined search results for all three search engines using the probabilities computed by SearchCombiner reflecting the results of the other engines. The protein probabilities computed by ProteinProphet were accurate in each case (see Supplementary information). Figure 2C shows the amount of overlap among protein identifications inferred from results of each search engine and the combined results, passing a minimum probability threshold to achieve a false positive error rate of 2.5% predicted by ProteinProphet. As at the peptide level, SEQUEST conferred more protein identifications than the other two engines, and 60% of all protein identifications were observed using all three database search programs. Interestingly, the greatest number of protein identifications was obtained using the combined results, 10% more than SEQUEST on its own.
It is worth noting the importance of adjusting the peptide probabilities of each search engine to reflect the results of the other search engines before running ProteinProphet on the combined search results. Since incorrect results (indicated by asterisks in Figure 2C) are rarely observed by more than one search engine, the inclusion of all search results without adjustment to their peptide probabilities would result in a number of incorrect identifications exceeding the predicted 2.5% (i.e. four instead of the predicted two for the HaloICAT LCQ data set). The adjustment, however, penalizes results observed by only a single search engine, ensuring accurate protein probabilities for the combined search results.
Peptide and protein quantitation
The Trans-Proteomic Pipeline has two components that compute relative quantitation of peptides and proteins based upon isotopic labeling methods, XPRESS and ASAPRatio. At the peptide level, they reconstruct the LC elution profiles of the heavy and light labeled precursor ions to compute a relative quantitation ratio. At the protein level, they combine together the quantitation ratios of peptides corresponding to each protein. ASAPRatio differs from XPRESS at the peptide level by using ratios of peptides in different charge states to compute an average ratio and error, and at the protein level by computing P-values that protein ratios in the data set are statistically significant.
The HaloICAT LCQ data set, labeled with ICAT reagent, was analyzed for quantitation information using both XPRESS and ASAPRatio following database search with SEQUEST, Mascot, or COMET. In cases in which the same peptide was assigned to a spectrum by more than one search engine, identical peptide quantitation was computed in the case of each engine by both quantitation programs. This apparently trivial result explicitly demonstrates that quantitation software in the context of the Trans-Proteomic Pipeline can analyze data uniformly, without regard to the method used to assign peptides to MS/MS spectra. At the protein level, quantitation is expected to be more variable since protein identifications in each data set may correspond with different subsets of identified peptides that contribute quantitation information. Nevertheless, ASAPRatio quantitation for each protein agreed within 1 standard deviation error among all the data sets in which the protein was identified.
ABI 4700 MALDI-TOF/TOF and Waters Q-TOF data
The Trans-Proteomic Pipeline was also applied to the Serum MALDI-TOF/TOF data set generated on an ABI 4700 MALDI-TOF/TOF instrument, and the Yeast Q-TOF data set generated on a Waters Q-TOF instrument run in ESI ionization mode. Unlike the HaloICAT LCQ data set, the correct and incorrect results in these data sets are not known. Nevertheless, PeptideProphet peptide probabilities and ProteinProphet protein probabilities enable the prediction of false positive error rates that serve as objective criteria for comparison. Much overlap was observed in each case among results at the peptide and protein levels for the three search engines. Figures 2D and E show the amount of overlap among protein identifications for the two data sets inferred from results of each search engine and the combined results, passing a minimum probability threshold to achieve a false positive error rate of 2.5% predicted by ProteinProphet. It is evident that greater than 66% of protein identifications are common to all three search engines. Mascot conferred 20% more protein identifications than SEQUEST on the Serum MALDI-TOF/TOF data set, and a comparable number on the Yeast Q-TOF data set. Nevertheless, combining the results of all three search engines (using adjusted peptide probabilities computed by SearchCombiner) led to an additional 10% of protein identifications in both data sets, including some not observed with any search engine on its own.
General utility of Trans-Proteomic Pipeline
The XML-based Trans-Proteomic Pipeline enables analysis of MS/MS data to proceed uniformly without regard to the mass spectrometer type used to generate the spectra, or search engine used to assign peptides. This facilitates comparison of the results of applying different search engines to the same data set, and data exchange among groups. We demonstrated this using the Trans-Proteomic Pipeline to analyze in a consistent manner data sets generated by three different mass spectrometer types and assigned peptides using three different search engines, including COMET described in this work. This uniform analysis enabled objective comparison of results at fixed predicted false positive error rates. Overall, COMET performed comparably to both SEQUEST and Mascot. More than half of all identifications were observed with all three search engines, while some were observed exclusively by each. SEQUEST conferred the greatest number of results for the HaloICAT LCQ data set, and Mascot conferred the greatest number for the Serum MALDI-TOF/TOF data set. In addition, we showed how results from multiple engines applied to the same data set can be used together to derive a greater number of protein identifications.
The mzXML, pepXML, and protXML formats are open and freely available at http://sourceforge.net/projects/sashimi/, the latter two under the Trans-Proteomic Pipeline package. The pepXML format can easily be extended to new applications for assigning peptides to MS/MS spectra. In the near future, we are planning to convert results of the search engine ProbID (Zhang et al, 2002) to pepXML, and modify X!Tandem to output its results directly in pepXML format. There are plans for Mascot to optionally output its results directly in pepXML as well.
The pepXML format can store in a single document the results of multiple searches performed on a data set of spectra. It can thus be used for iterative search strategies, as well as for combining together the results of different search engines applied to the same data set. The format can easily be extended to new peptide-level analyses such as iTRAQ quantitation (Applied Biosystems) and multiengine metavalidation such as SearchCombiner. This merely requires defining a schema for analysis-specific XML elements that store summary and result information in place of the wildcard elements in pepXML. In a similar manner, protXML can easily be extended to new protein-level analyses. In the future, we envision a variety of analysis tools that utilize these formats. Data from different groups generated by various programs can thus be easily validated, viewed, and subjected to additional analyses.
Materials and methods
mzXML file format
The mzXML file format has been developed to represent native mass spectral data in an XML framework. This development facilitates open access to the raw spectral data on multiple computing platforms without the need for proprietary data access libraries. Whereas native formats can only be accessed through vendor supplied programming interfaces or dynamic linked libraries often limited to select platforms and operating systems (usually only Microsoft Windows), the mzXML format enables development of analytical software for other platforms. While it is possible for most of the instruments to export native data into a clear text ASCII representation (e.g. SEQUEST.dta, Mascot.mgf, etc.), these formats only contain enough information for using them in the MS/MS peptide assignment step. The intent of the mzXML format is not to completely replace the native formats, but rather to provide an operational representation of information that is required in the analysis of the majority of LC-MS/MS-based proteomics experiments, including database searching, de novo sequencing, quantification using stable isotopic labeling, and quantification of LC-MS traces. The format is flexible and can host any type of data, from highly processed (e.g. centroided, deisotoped, peak-picked, etc.) to essentially raw. The use of a scan indexing technique and of specialized parsers provides a random access, efficient way for software applications to interface with the format. In addition, the format is designed to be extensible in order to account for future innovations in the MS and proteomics fields.
The mzData format has recently been finalized by the Human Proteome Organization Mass Spectrometry Standards Working Group (HUPO PSI-MS) and is proposed to represent the peak list data that are input into the database search engines. Support by MS/MS search engines for reading data in either mzXML or mzData has just begun to be implemented. Therefore, MS/MS spectra are kept in the native flat file formats (SEQUEST.dta or Mascot generic) or are translated from mzXML into these formats using suitable and available translators.
pepXML file format
pepXML is an open file format for storage of peptide assignments to MS/MS spectra and their subsequent peptide-level analysis. There are currently many different applications that assign peptides to MS/MS spectra, including de novo sequencing programs, database search software such as SEQUEST and Mascot, as well as combinations of de novo sequencing and database search (Taylor and Johnson, 1997; Sadygov et al, 2004; Eng et al, 2005). Once peptide assignments to MS/MS spectra are made, they can be validated, quantified, used to infer protein identifications, and subjected to additional analyses such as comparison of the results of applying different peptide assignment methods to the same data set, or comparison of results obtained in different data sets. Many of these peptide-level analyses are performed without regard to the method used to assign peptides to spectra. However, they cannot easily be applied to the results of each application in a uniform manner because each application writes its results in its own proprietary format. For the same reason, it is often difficult for laboratories to exchange data with one another if the data have been generated using different applications, and subjected to different sets of peptide-level analyses. pepXML was developed as an open file format to store the results of peptide assignments to MS/MS spectra and their subsequent peptide-level analysis.
The pepXML format (available at http://sashimi.sourceforge.net/schema_revision/pepXML/pepXML_v18.xsd; see Supplementary information) can store in a single document the results of multiple searches, for example using different search options and/or different search engines, on a data set of one or more LC/MS/MS runs (i.e. each run corresponding to a particular raw data file). The results for each run are maintained together in an msms_run_summary element with references to the original mzXML file (raw data) and details regarding the mass spectrometer used to generate the MS/MS spectra (taken from the mzXML file) and the proteolytic enzyme applied to the sample prior to MS/MS. The msms_run_summary contains a search_summary element for each database search applied to the run, with information regarding the search engine and settings such as search database, peptide modifications, and enzyme and sequence search constraints. More than one search_summary element can be associated with a single msms_run_summary when multiple searches are performed on the same data set, for example using different search conditions or search engines.
The msms_run_summary additionally contains spectrum_query elements, one for each MS/MS spectrum. It includes spectrum name, precursor ion charge and mass, as well as a search_result element for each search applied to the run. Within the search_result are search_hit elements, those peptides assigned to the spectrum, ranking from 1 on up. We currently only store the top ranking hit in pepXML, while maintaining a reference to a search result flat file that contains all lower ranking hits, if available. In the case of multiple searches performed on the same data set, each search_result is linked to its corresponding search_summary element by a common search_id attribute value. Each search_hit has information regarding the assigned peptide, such as its sequence, calculated neutral mass, and mass difference with respect to the precursor ion. It also stores optional information such as the number of termini consistent with sample enzymatic cleavage, number of missed enzymatic cleavages, corresponding proteins in the database, and number of matched ions. Additional information regarding the assigned peptide, such as pI or hydrophobicity, can be added as generic parameter elements. Finally, database search score values are stored as search_score elements that can easily be defined for any method that assigns peptides to MS/MS spectra.
Peptide modification information is stored explicitly in pepXML as a modification_info element (see Supplementary information). This is important for it avoids arbitrary search engine-defined encodings that can vary from search to search (i.e. *, #, @), and can accommodate an unlimited number of modifications. Modified amino acids at any position from 1 to peptide length are indicated by a mod_aminoacid_mass element containing a position and mass attribute, while terminal modifications are indicated by optional modified_nterm_mass and modified_cterm_mass attributes. The optional modified_peptide attribute is used primarily for display purposes, allowing different users to easily customize their view of the modifications.
The pepXML format contains the results of subsequent peptide-level analyses applied to each search hit, such as PeptideProphet validation and XPRESS and ASAPRatio quantitation. Each analysis contributes an analysis_summary element within the root element with details about the analysis, such as name, date, version, and program settings. In addition, the analysis results for each search_hit are stored there as analysis_result elements. Unique identifiers are used to link each particular analysis_result with its corresponding analysis_summary (Supplementary information). Wildcard elements in the analysis_summary and analysis_result elements are used for details specific to each analysis, and facilitate extensibility of the format to new analyses. For example, any new peptide-level analysis can supply a schema and namespace for its own analysis-specific summary and result elements. A pepXML document in which such elements are inserted in place of the wildcards will then be validated as long as it references the namespace of the analysis along with that of pepXML.
Storing the analysis information in the same file as the search results has the advantage that files can easily be filtered (data removed) and exchanged among researchers while retaining the appropriate analysis information. In addition, data sets, even those using different software to assign peptides, and/or subjected to different peptide-level analyses, can easily be merged. For example, combining any two data sets together only requires grouping together their analysis_summary elements, grouping together their msms_run_summary elements, and re-indexing their spectrum_query elements. The unique identifiers ensure that the relationships between analysis results and analysis summaries are maintained for all runs.
The pepXML file format has a similar objective to the aims set out by the Human Proteome Organization Proteomics Standard Initiative (HUPO PSI) in formulating mzIdent (Orchard et al, 2005). We have presented pepXML and protXML at a HUPO PSI meeting and are working closely with them to minimize duplication of effort. The open source search engine X!Tandem (Craig and Beavis, 2004) currently outputs its results in an XML format. The major distinction between that format and pepXML is the general applicability of the latter to different methods for assigning peptides to MS/MS spectra and peptide-level analyses.
Recently, a concise file format for SEQUEST results, SQT, was described (McDonald et al, 2004). The pepXML file format differs from SQT in several ways. pepXML, an XML file format with a defined schema, is easily validated allowing users of data to be sure that files generated by others are properly formed, and thus sound input for analysis software. In addition, pepXML can store results of any method of assigning peptides to spectra and any subsequent peptide-level analysis. The tradeoff, however, is that pepXML undoubtedly results in larger file sizes than SQT, comparable to a corresponding SEQUEST HTML or Mascot DAT file. However, since XML files generally contain a large amount of white space, they are efficiently compressed. For example, a standard gzip compression of a pepXML file resulted in a size savings of 92%.
protXML file format
protXML is a file format for storage of protein identifications inferred from MS/MS data and their subsequent protein-level analysis. There are several different methods to infer protein identifications from database search results. For example, ProteinProphet uses statistical models to combine together the peptide evidence for each protein in order to compute its probability of being present in the sample. Importantly, it apportions identified peptides among all their corresponding proteins in such a way as to derive the simplest set of protein identifications that explain the observed peptides. Another program, QScore, assesses the chance likelihood of protein identifications based upon their size and number of corresponding identified peptides, as well as the effective database and data set sizes (Moore et al, 2002). The database search program Mascot groups together peptides and assigns each protein a score reflecting its confidence (Perkins et al, 1999). These methods output their results in a variety of formats. protXML was developed as an open file format to store the results of protein inference based on MS/MS spectra, and subsequent protein-level analyses.
The protXML file format (available at http://sashimi.sourceforge.net/schema_revision/protXML/protXML_v3.xsd; see Supplementary information) includes a protein_summary_header element that contains information regarding the contributing peptide identifications, and a program_details element that has information on the particular method used for inferring protein identifications, and a wildcard element that can contain details specific for that method. For example, ProteinProphet substitutes in place of the wildcard a proteinprophet_details element containing parameter settings and summary information specific to its analysis. This organization makes the format easily extensible to new programs for inferring protein identifications.
Protein identifications are contained in protein_group elements that store one or multiple protein elements for sets of related protein family members. Assembly of proteins into groups is chiefly aimed at helping biologists organize data in a useful way by associating together related proteins into a single entry, and could be implemented in different ways. ProteinProphet, for example, groups together identified proteins that are highly related, yet have no unshared corresponding peptides, and hence no unambiguous evidence to discriminate among them. Protein elements have attributes for protein name, number of corresponding peptides, peptide coverage, and percent of data set spectrum identifications (Liu et al, 2004). Protein elements may themselves contain elements for annotation and indistinguishable proteins, those that share with them all the identified peptides. They also contain peptide elements for each corresponding peptide assigned to an MS/MS spectrum, with information on the peptide sequence and its properties, including the optional weight attribute to reflect the confidence that the peptide is evidence for that particular protein.
The protXML format contains the results of subsequent protein-level analyses applied to the set of protein identifications, such as XPRESS and ASAPRatio protein quantitation (Supplementary information). Each analysis stores an analysis_summary element in the root element with details about the analysis, such as name, date, version, and program settings. In addition, the analysis results for each protein are stored there as an analysis_result element. Unique identifiers are used to link each particular analysis_result with its corresponding analysis_summary. Wildcard elements in the analysis_summary and analysis_result elements are used for details specific to each analysis, and facilitate extensibility of the format to new analyses. For example, any new protein-level analysis can supply a schema and namespace for its own analysis-specific summary and result elements. A protXML document in which such elements are inserted in place of the wildcards will then be validated as long as it references the namespace of the analysis along with that of protXML.
COMET search engine
Originally developed in 2001, COMET was created to be a high-throughput, scalable, and open source sequence database search program for tandem mass spectrometry data, offering all of the features found in modern database search tools. These include the facility to run on a computer cluster with linear scaling and the ability to search with static or variable modifications with full, semi-, or no enzyme constraint. Although we intended to distribute this tool freely and open source, we are not able to do so until it becomes clear that this would not violate any intellectual property restrictions. We are hopeful this will happen in the near future, given the growing number of commercial and open source MS/MS database search tools now available (Zhang et al, 2002; Craig and Beavis, 2004; Geer et al, 2004).
The score function within COMET is effectively the scalar dot product between two unit vectors representing the input spectrum and fragment ion masses calculated from candidate sequences in the sequence database (Field et al, 2002). The vectors are composed of approximately 1 Da mass bins (1.0005 for monoisotopic masses) to optimize the binning due to the periodicity in peptide isotopic masses (Parker et al, 2004). This dot product score function allows for very fast calculations, as the score is computed based on the simple summation of intensity values that are accessed via a direct lookup of the mass index. However, in order to be more sensitive than simply summing up matched peaks, as a regular dot product effectively accomplishes, the input spectrum is preprocessed such that intensities are modified as follows:
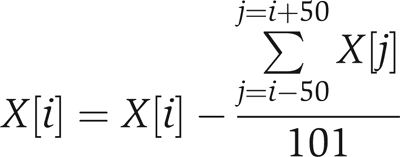
where X is peak intensity, and i and j are spectrum m/z bin values. This modification to the input spectrum allows the simple dot product to effectively take into account noise and unmatched peaks in the input spectrum in addition to fragment ions that do match. This is contrasted with the direct dot product where only matched peaks contribute to the final score and unmatched peaks are ignored. This processing occurs on the input spectrum after peak detection, smoothing, and intensity normalization across the m/z range.
The input spectrum and theoretical spectra are both made into unit vectors prior to the dot product calculation. However, since the input spectrum is further processed as described above, the scale of the dot product is not guaranteed to range from 0.0 to 1.0. Ideally, differences in peptide length, peak counts, and precursor charge state are somewhat mitigated by the unit vector normalization. In practice, the dot product score still does exhibit dependencies on these parameters. A simple Z-score calculation is generated for each peptide, defined as the number of standard deviations away from the mean any peptide score is, where the mean and standard deviations are calculated based on the top 100 unique peptide scores in a search. Uniqueness between peptides is based on the peptides exhibiting a minimum similarity, where similarity is defined as having a minimum percentage, 90%, of similar fragment ions. The program output includes the dot product score (multiplied by 1000 for display purposes) and Z-score for each result.
Pipeline analysis tools
The Trans-Proteomic Pipeline includes open source MS/MS analysis tools developed at the Institute for Systems Biology, adapted to the mzXML, pepXML, and protXML file formats in order to enable uniform analysis without regard to the type of mass spectrometer or search engine used. It includes programs to convert results of SEQUEST, Mascot, and COMET search engines into pepXML. The validation program PeptideProphet, and peptide quantitation programs XPRESS and ASAPRatio were adapted to read and write pepXML. In addition, the protein inference program ProteinProphet was modified to read pepXML as its input and write protXML as its output. Quantitation at the protein level by XPRESS and ASAPRatio (combining together the quantitation values of individual peptides corresponding to a protein) was modified to read both protXML and pepXML, and write protXML. In addition, PeptideProphet was specialized for Mascot and COMET search results using a training data set of spectra (Keller et al, 2002b) generated on a ThermoFinnigan LCQ ion trap mass spectrometer. This specialization involves deriving a discriminant score for each precursor ion charge that combines together relevant scores output by the engine, and defining appropriate parametric representations of the distributions of the discriminant score among correct and incorrect results.
Validated search results from multiple search engines applied to the same data set can be used together to better discriminate between correct and incorrect results. The additional information of whether or not a peptide was also assigned to a spectrum by other search engines can be used to adjust the probabilities computed for that data set on its own. The program SearchCombiner makes such adjustments, appropriate to each data set, by assigning to each result a Boolean vector indicating whether or not the same peptide was also observed with each other search engine. Mixture model distributions for vector values are determined among correct and incorrect results in the 0.1–0.9 initial probability range in each data set, and then used to adjust the initial probabilities. For example, if the learned fraction of correct results with a particular vector value is a, and the learned fraction of incorrect results b, then the initial probability p of a result with that vector value would be adjusted to pa/[pa+(1−p)b]. In this manner, the adjustments to the initial peptide probabilities are learned from each data set in a robust manner, regardless of the search engines used.
Data in pepXML and protXML formats are viewed using stylesheets and XSLT (Extensible Stylesheet Language Transformations). Common Gateway Interface programs PepView and ProtView write stylesheets according to user defined preferences, then in a system call apply the stylesheet to the pepXML and protXML document, respectively, and pipe the resulting HTML to the client browser. In addition, stylesheets can be written to dictate output in tab delimited or XML format. At either the peptide or protein level, a single stylesheet can be applied to data derived using any application, and subjected to any analysis.
Data sets
Three data sets were used in this study: HaloICAT LCQ, Serum MALDI-TOF/TOF, and Yeast Q-TOF. The HaloICAT LCQ data set (Baliga et al, 2002) was generated from complex Halobacterium NRC1 samples labeled with either light (bat− strain) or heavy (bat+ strain) versions of the original ICAT reagent (Gygi et al, 1999), then combined, treated with trypsin, avidin purified, and subjected to ion exchange chromatography and LC-MS/MS on a ThermoFinnigan LCQ mass spectrometer. The resulting 6808 spectra were searched with SEQUEST, Mascot, and COMET against a database containing human and Halobacterium protein sequences, using average masses and a precursor mass tolerance of ±3 Da. These searches were constrained to cysteine-containing peptides without enzyme constraint, and spectra were searched separately for static light and heavy ICAT modifications. The Serum MALDI-TOF/TOF data set (Pan et al, in press) was generated from a human serum sample subjected to solid phase N-glycosylation capture followed by digestion with trypsin, treatment with PNG'aseF glycosidase (Zhang et al, 2003), HPLC, and spotting on a MALDI plate using a microfraction collector. A total of 927 spectra were generated from this sample using an ABI 4700 MALDI-TOF/TOF instrument, and were searched against a human sequence database using monoisotopic masses, a precursor mass tolerance of ±0.4 Da, and a tryptic enzymatic constraint. The Yeast Q-TOF data set (Chen et al, in preparation) consists of 1453 MS/MS spectra generated from an ion exchange fraction of a complex trypsinized yeast sample on a Waters Q-TOF mass spectrometer run in ESI ionization mode. It was searched in an unconstrained manner against a yeast protein sequence database using monoisotopic masses and a precursor mass tolerance of ±3 Da.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Legend to Supplementary Figures
Acknowledgments
We thank John Cottrell for his valuable contributions regarding the pepXML schema, and Patrick Pedrioli and Markus Muller for helpful comments. This work has been funded in part with Federal funds from the National Heart, Lung, and Blood Institute, National Institutes of Health, under contract no. N01-HV-28179.
References
- Baliga N, Pan M, Goo YA, Yi EC, Goodlett DR, Dimitrov K, Shannon P, Aebersold R, Ng WV, Hood L (2002) Coordinate regulation of energy transduction modules in Halobacterium sp. analyzed by a global systems approach. Proc Natl Acad Sci USA 99: 14913–14918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SC, Deutsch EW, Yi EC, Li X-J, Goodlett DR, Aebersold R Improving mass and liquid chromatography based identification of proteins using Bayesian scoring. (manuscript in preparation) [DOI] [PubMed]
- Craig R, Beavis RC (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 12: 1466–1467 [DOI] [PubMed] [Google Scholar]
- Eng J, Martin DB, Aebersold R (2005) Tandem mass spectrometry database searching. In Encyclopedia of Genetics, Genomics, Proteomics and Bioinformatics, Dunn M, Jorde L, Little P, Subramaniam S (eds). John Wiley & Sons, Ltd, ISB 0470849746 [Google Scholar]
- Field HI, Fenyo D, Beavis RC (2002) RADARS, a bioinformatics solution that automates proteome mass spectral analysis, optimises protein identification, and archives data in a relational database. Proteomics 2: 36–47 [PubMed] [Google Scholar]
- Geer LY, Markey SP, Kowalak JA, Wagner L, Xu M, Maynard DM, Yang X, Shi W, Bryant SH (2004) Open mass spectrometry search algorithm. J Proteome Res 3: 958–964 [DOI] [PubMed] [Google Scholar]
- Gygi SP, Rist B, Gerber SA, Turecek F, Gelb M, Aebersold R (1999) Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol 17: 994–999 [DOI] [PubMed] [Google Scholar]
- Han DK, Eng J, Zhou H, Aebersold R (2001) Quantitative profiling of differentiation-induced microsomal proteins using isotope-coded affinity tags and mass spectrometry. Nat Biotechnol 19: 946–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, Nesvizhskii A, Kolker E, Aebersold R (2002a) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem 74: 5383–5392 [DOI] [PubMed] [Google Scholar]
- Keller A, Purvine S, Nsvizhskii A, Stolyar S, Goodlett DR, Kolker E (2002b) Experimental protein mixture for validating tandem mass spectral analysis. OMICS 6: 207–212 [DOI] [PubMed] [Google Scholar]
- Li X-J, Pedrioli PGA, Eng J, Martin D, Yi EC, Lee H, Aebersold R (2004) A tool to visualize and evaluate data obtained by liquid chromatography/electrospray ionization/mass spectrometry. Anal Chem 76: 3856–3860 [DOI] [PubMed] [Google Scholar]
- Li X-J, Zhang H, Ranish JR, Aebersold R (2003) Automated statistical analysis of protein abundance ratios from data generated by stable isotope dilution and tandem mass spectrometry. Anal Chem 75: 6648–6657 [DOI] [PubMed] [Google Scholar]
- Liu H, Sadygov RG, Yates JR III (2004) A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem 76: 4193–4201 [DOI] [PubMed] [Google Scholar]
- Mann M, Aebersold R (2003) Mass spectrometry-based proteomics. Nature 422: 198–207 [DOI] [PubMed] [Google Scholar]
- McDonald WH, Tabb DL, Sadygov RG, MacCoss MJ, Venable J, Graumann J, Johnson JR, Cociorva D, Yates JR III (2004) MS1, MS2, and SQT—three unified, compact, and easily parsed file formats for the storage of shotgun proteomic spectra and identification. Rapid Commun Mass Spectrom 18: 2162–2168 [DOI] [PubMed] [Google Scholar]
- Moore RE, Young MK, Lee TD (2002) Qscore: an algorithm for evaluating SEQUEST database search results. J Am Soc Mass Spectrom 13: 378–386 [DOI] [PubMed] [Google Scholar]
- Nesvizhskii AI, Keller A, Kolker E, Aebersold R (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 75: 4646–4658 [DOI] [PubMed] [Google Scholar]
- Orchard S, Hermjakob H, Binz P-A, Hoogland C, Taylor CFR, Zhu W, Julian RK, Apweiler R (2005) Further steps towards data standardization: The Proteomic Standards Initiative HUPO 3rd annual congress, Beijing 25–27th October, 2004. Proteomics 5: 337–339 [DOI] [PubMed] [Google Scholar]
- Pan S, Zhang H, Rush J, Eng J, Zhang N, Patterson D, Comb MJ, Aebersold R High-throughput proteome-screening for biomarker detection. Mol Cell Proteomics (manuscript submitted) [DOI] [PubMed] [Google Scholar]
- Parker KC, Patterson D, Williamson B, Marchese J, Graber A, He F, Jacobson A, Juhasz P, Martin S (2004) Depth of proteome issues: a yeast isotope-coded affinity tag reagent study. Mol Cell Proteomics 3: 625–659 [DOI] [PubMed] [Google Scholar]
- Pedrioli PGA, Eng JK, Hubley R, Vogelzang M, Deutsch EW, Raught B, Pratt B, Nilsson E, Angeletti R, Apweiler R, Cheung K, Costello CE, Hermjakob H, Huang S, Julian RK Jr, Kapp E, McComb ME, Oliver SG, Omenn G, Paton NW, Simpson R, Smith R, Taylor CF, Zhu W, Aebersold R (2004) A common open representation of mass spectrometry data and its application in a proteomics research environment. Nat Biotechnol 22: 1459–1466 [DOI] [PubMed] [Google Scholar]
- Perkins DN, Pappin DJC, Creasy DM, Cottrell JS (1999) Probability based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20: 3551–3567 [DOI] [PubMed] [Google Scholar]
- Sadygov RG, Cociorva D, Yates JR III (2004) Large-scale database searching using tandem mass spectra: looking up the answer in the back of the book. Nat Methods 1: 195–202 [DOI] [PubMed] [Google Scholar]
- Taylor JA, Johnson RS (1997) Sequence database searches via de novo peptide sequencing by tandem mass spectrometry. Rapid Commun Mass Spectrom 11: 1067–1075 [DOI] [PubMed] [Google Scholar]
- Zhang H, Li X-J, Martin DB, Aebersold R (2003) Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat Biotechnol 21: 660–666 [DOI] [PubMed] [Google Scholar]
- Zhang N, Aebersold R, Schwikowski B (2002) ProbID: a probabilistic algorithm to identify peptides through sequence database searching using tandem mass spectral data. Proteomics 2: 1406–1412 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Legend to Supplementary Figures