

Abstract
Although the human genome has been sequenced, progress in understanding gene regulation in humans has been particularly slow. Many computational approaches developed for lower eukaryotes to identify cis-regulatory elements and their associated target genes often do not generalize to mammals, largely due to the degenerate and interactive nature of such elements. Motivated by the switch-like behavior of transcriptional responses, we present a systematic approach that allows adaptive determination of active transcriptional subnetworks (cis-motif combinations, the direct target genes and physiological processes regulated by the corresponding transcription factors) from microarray data in mammals, with accuracy similar to that achieved in lower eukaryotes. Our analysis uncovered several new subnetworks active in human liver and in cell-cycle regulation, with similar functional characteristics as the known ones. We present biochemical evidence for our predictions, and show that the recently discovered G2/M-specific E2F pathway is wider than previously thought; in particular, E2F directly activates certain mitotic genes involved in hepatocellular carcinomas. Additionally, we demonstrate that this method can predict subnetworks in a condition-specific manner, as well as regulatory crosstalk across multiple tissues. Our approach allows systematic understanding of how phenotypic complexity is regulated at the transcription level in mammals and offers marked advantage in systems where little or no prior knowledge of transcriptional regulation is available.
Keywords: cis-regulatory motifs, correlation, subnetworks, transcription regulation
Introduction
The importance of achieving an accurate quantitative understanding of gene regulation in humans can hardly be overstated. Deregulation of gene expression is a recurring theme in development and progression of several diseases including cancer. The emergence of new experimental platforms that probe transcription globally promises a comprehensive view of these fundamental biological processes in a large number of mammalian systems, in which very little is currently known about their transcriptional regulation. This is, however, possible if such technologies are supplemented with appropriate computational methodologies. A large number of computational approaches have been developed for deciphering cis-regulatory elements in lower eukaryotes by integrating the genome sequence data with global expression profiles (Tompa et al, 2005). Recent evaluation shows that such methods do not generalize to mammals, however (see, for e.g., Figure 1B in Tompa et al, 2005). Multiple factors contribute to this inadequacy. Firstly, the mammalian transcription factor (TF) binding sites are significantly degenerate (Pennacchio and Rubin, 2001; Kel et al, 2003; Wasserman and Sandelin, 2004). Secondly, the role of interactions between TFs in promoter recognition is much more critical (Locker, 2001; Levine and Tjian, 2003). Finally, the multicellular architecture of mammals makes their underlying regulatory networks even more complex (Niehrs and Pollet, 1999).
In this article, we present a computational approach that circumvents the aforementioned limitations and systematically infers active human transcriptional subnetworks anchored on the proximal promoter DNA from genome-wide mRNA profiles. By transcriptional subnetwork, we mean the following triplet: the cis-regulatory motif combination, the direct target genes and the physiological processes regulated by the corresponding TFs. Such subnetworks correspond to segments of global transcriptional networks (Basso et al, 2005). Our algorithm proceeds by correlating the binding strengths of motifs with the expression levels using linear spline functions. Linear splines mimic the switch-like behavior intrinsic to transcriptional regulation and provide a natural framework to model gene regulation mediated by strongly degenerate and interacting cis-control motifs. Active motifs exhibit statistically significant correlation, whereas inactive motifs do not (Bussemaker et al, 2001; Das et al, 2004). Starting from the expression data and DNA sequence, we first identify the potentially active motif combinations by examining their correlation with expression. We next use the inferred mathematical model to determine the putative targets of the TFs binding to such motifs, and perform a functional enrichment analysis on these targets to predict the physiological processes regulated by the TFs. We applied our technique to diverse expression data sets spanning human liver and cell cycle and identified several subnetworks, known and novel, as potentially active, including ones controlled by the canonical regulators like HNF-1 and E2F. Additionally, we demonstrate that the subnetworks are learnt in a condition-specific manner, and thus adaptively. We have presented supportive experimental evidence for some of our novel predictions. Our analysis reveals that this technique has accuracy comparable to that achieved by computational approaches in lower eukaryotes.
Results
Inferring transcriptional subnetworks
Classical approaches to model mammalian transcription are based on clustering genes by similarity in their expression profiles across a large number of conditions (Whitfield et al, 2002; Elkon et al, 2003). Over-represented cis-regulatory motifs are then identified in the promoters of genes in a given cluster. Scores for each position weight matrix (PWM) (Kel et al, 2003), which is a probabilistic representation of TF binding sites, are next enumerated for the corresponding motif. If the PWM score exceeds a certain cutoff, then that gene is inferred as the target of the TF binding to this motif. Such approaches, although useful, have limited applicability however (Bussemaker et al, 2001; Kirmizis and Farnham, 2004). Many genes do not cluster tightly enough that their regulatory motifs can be discovered reliably. Synergy among TFs, critical to mammalian regulatory control, is also difficult to model. Moreover, cutoff scores are often based on predetermined thresholds and background sequences, for which there is not always a clearcut optimal choice (Elkon et al, 2003; Kel et al, 2003). Consequently, such an approach may be biased.
Here we present an approach that does not require clustering. It is based on the fact that gene expression results from integration of various signals onto the promoter DNA, mediated by binding of multiple TFs to the cis-regulatory elements. Thus, the mRNA level of any gene can be construed as a weighted sum of contributions from the active motif combinations. The impact of any motif depends on the binding affinity for its corresponding TF, which in turn, is related to its PWM score (Berg and von Hippel, 1987). Hence, for active motifs, the mRNA levels must be significantly correlated with the PWM scores across genes and vice versa (Conlon et al, 2003). Furthermore, if transcriptional response is sigmoidal (Figure 1A) (Carey, 1998; Veitia, 2003), then the dependence of logarithm of mRNA level on binding affinity has the shape of a linear spline approximately (Das et al, 2004) (Figure 1B), where the transition from constant to linear variation takes place at the gene activation threshold. We fit the PWM scores to the logarithm of expression ratios using linear splines to select the potentially active motifs from a large set of input motifs. The ratio is taken between the observed mRNA level and that in a reference condition. The interacting motif partners, their PWM cutoff scores and weights of their contributions are learnt from the response data. The accuracy of the fitted model is measured by Δχ2, the percent reduction in variance (%RIV) of log ratios. It provides an estimate of how much variation in expression data is explained by the model. Similar approaches have proven very powerful in lower eukaryotes (Bussemaker et al, 2001; Keles et al, 2002; Das et al, 2004; Wang et al, 2005), but could not be applied to mammals owing to strongly degenerate motifs and interactions between motifs. Here we present an algorithm based on correlation with expression that overcomes these difficulties. We first examined a few degenerate mammalian motifs to check if their PWM scores correlate with expression using splines. We indeed found statistically significant correlation (Table I), comparable to the Mcm1 motif in yeast, which has degeneracy similar to that in mammals. The correlation is even more pronounced for cell lines and ChIP-chip data. This prompted us to build a comprehensive approach based on multivariate linear splines to infer the active regulatory subnetworks anchored on the proximal promoter DNA.
Figure 1.

Modeling mammalian transcription with linear splines. (A) Sigmoidal transcriptional response (Carey, 1998). The response is flat below a binding affinity threshold, namely, the gene activation threshold, and varies exponentially above it. It saturates at high binding energies. (B) Example of a linear spline. A linear spline is a piecewise linear function: it is zero below (above) a threshold, termed knot (ξ), and changes linearly above (below) it. Eg refers to the observed mRNA level of gene g, whereas EgC is its mRNA level in the reference sample. Knots are related to gene activation thresholds. All genes with PWM scores Sg>ξ are predicted targets of the motif contributing to this spline, shaded in blue. (C) A schematic view of the key steps in identification of significant motifs and motif pairs using linear splines.
Table 1.
Correlation between PWM scores and expression
| Organism | Experiment | Biological process/tissue | Motif | Average information content | %RIV | P-value |
|---|---|---|---|---|---|---|
| Yeast | Microarray | Cell cycle (G2) | Mcm1 | 0.59 | 4.9 | 3.4 × 10−9 |
| Human | Microarray | Liver | HNF-1 | 0.62 | 2.2 | 1.6 × 10−6 |
| Human | Microarray | Pancreas | C/EBP beta | 0.76 | 2.5 | 2.5 × 10−7 |
| Human | Microarray | Cell cycle (G1/S) | E2F | 1.74 | 5.9 | 1.7 × 10−12 |
| Mouse | Microarray | Liver | HNF-1 | 0.62 | 2.1 | 2.1 × 10−6 |
| Mouse | Microarray | Liver | HNF-4 | 0.60 | 2.0 | 4.1 × 10−6 |
| Human | ChIP-chip | Liver/HNF-1 | HNF-1 | 0.62 | 14.7 | 8.7 × 10−37 |
Comparison of percent reduction in variances (%RIVs) between yeast and mammals is shown. Single linear splines were used to obtain the %RIV. Yeast cell-cycle data were obtained from Spellman et al (1998), liver and pancreas microarray data from Su et al (2004), liver ChIP-chip data for HNF-1 from Odom et al (2004) and human cell-cycle data from Whitfield et al (2002). Mcm1 matrix was obtained from Bussemaker et al (2001); all other matrices were obtained from TRANSFAC and JASPAR databases. P-values were calculated using an F-test (see Materials and methods). Average information content refers to the information content of the position weight matrix, averaged over all the positions (columns). It is 2.0 for non-degenerate words and 0 if all positions were N's (see Materials and methods).
Subnetworks from tissue-specific data
Significant motif combinations
We consider the microarray data obtained from an adult human liver sample (Su et al, 2004) under a normal condition as an example. As these data were recorded for only a single condition, it is not possible to use clustering approach to obtain liver-specific coexpression clusters for regulatory element determination. We obtained the top 1000 genes by variance in expression across tissues (79 total), which were used to build the linear spline model (Supplementary note and Supplementary information). A select set of genes was used to increase the computational speed and reduce noise. Expression ratio of a gene was calculated as the ratio of observed mRNA level in liver to the mean mRNA level across all 79 tissues sampled. We searched all known vertebrate PWMs (521 total: 446 in TRANSFAC, 75 in JASPAR) (Schones et al, 2005) to discover the active motif combinations. Efficient modeling with linear splines requires prioritization of input motifs (Das et al, 2004). However, the scheme used in lower eukaryotes (Das et al, 2004) requires prior knowledge of whether a given motif is present in the promoter of a gene, which is often not known in mammals. Both word counts and PWM scores with default thresholds were used for this purpose in yeast (Das et al, 2004). Such approaches cannot be applied directly to mammals because the binding sites are much more degenerate. The situation is even more acute when many motif matrices are unknown and have to be predicted de novo. To overcome this limitation, we prioritized the input matrices using a single linear spline.
We fitted a single linear spline separately for each PWM that best explains the variation in log expression ratios. Only non-redundant PWMs were retained, sorted by decreasing Δχ2. We find that the distribution of Δχ2 is discontinuous: a few top matrices are well separated from the rest (Supplementary Figure 1). Namely, for liver data, this gap in Δχ2 occurs between HNF-1 and ETF (Supplementary Table I). We used the gap to prepare multiple prioritized matrix sets. For each set, a model was built by additively combining spline contributions from multiple PWMs and their products using the multivariate adaptive regression splines (MARS) algorithm (Friedman, 1991). Product terms represent interactions between distinct motifs, whereas non-product terms are contributions from individual motifs. Interactions among the same motif are represented as a sum of multiple splines in that motif (Das et al, 2004). MARS is an adaptive and non-parametric method, using a greedy search strategy. The fit coefficient and the knots (Figure 1B) for each term in the model, which correspond to the weight of contribution and binding thresholds for a motif combination, are obtained by a least squares fit (see Materials and methods). Overfitting is restricted by minimizing the generalized crossvalidation (GCV) score, which assigns a penalty for the number of parameters used in the model (see Materials and methods). We have previously demonstrated in extensive simulation studies that minimization of GCV is indeed an effective means to control overfitting in spline models of gene regulation (Das et al, 2004). We also removed all motifs and motif pairs with P>0.01 to provide additional control on overfitting. We varied the maximum degree of interactions allowed in the model, and among all the prioritized matrix sets and interactions that were explored, we considered the model with maximum Δχ2 as optimal. The significance of each motif and pair was evaluated from its relative contribution to the optimal model, using an F-test (see Materials and methods). The reader may note that this is slightly different from the conventional definition of significance. The key steps in identifying significant motifs and motif pairs are summarized in Figure 1C.
The optimal model for liver had three individual motifs and five synergistic motif pairs and led to Δχ2=11.2% (Table II). Tissue-specific transcription is often achieved by cooperative action of ubiquitous and tissue-enriched factors (Locker, 2001). Our findings support this hypothesis: four out of five pairs are of this type. HNF-1 is a key regulator of hepatic transcription (Tronche et al, 1994). We find it significant and synergizing with two ubiquitous TFs, Oct-1 and HFH-3. Functional interactions of Oct-1 with HNF-1 and PPAR-DR1 have been experimentally characterized (Zhou and Yen, 1991; Kakizawa et al, 1999). In particular, Oct-1*HNF-1 pair has been shown to be liver-specific (Zhou and Yen, 1991). Cooperativity between HFH-3 and HNF-1 has also been implicated (Overdier et al, 1997). Other pairs, HFH-3*MTF-1 and Oct-1*HFH-3, are novel predictions.
Table 2.
Significant motifs and motif pairs for analysis with known vertebrate motifs as input
| Significant motifs and motif pairs | P-value | Fit coefficients |
|---|---|---|
| HNF-1 | 2.2 × 10–15 | 187.5 |
| HFH-3 | 1.3 × 10–14 | −9.8 |
| ETF | 0.002 | −2.9 |
| OCT-1*HNF-1 | 1.5 × 10–14 | 156.7 |
| OCT-1*PPAR-DR1 | 2.1 × 10–10 | 56.2 |
| HFH-3*HNF-1 | 0.001 | −120.1 |
| HFH-3*MTF-1 | 0.003 | 86.2 |
| OCT-1*HFH-3 | 2.2 × 10–15 | 30.0 |
HNF-1, PPAR-DR1 and MTF are liver-specific motifs, whereas Oct-1 and HFH-3 are ubiquitous motifs. HNF-1 is a well-established liver-specific motif. PPAR-DR1 binds several nuclear factors involved in metabolism, for example, PPAR, RXR, etc. MTF-1 is active in heavy metal metabolism and is essential for liver development. Fit coefficients are coefficients of optimal fit in the final spline model, which is reported in Supplementary note. More details on significant motif combinations are given in Supplementary Table V.
As very limited number of mammalian TF binding sites are represented in TF databases, we next included PWMs generated by the ab initio motif finder, MDscan (Liu et al, 2002; Conlon et al, 2003), along with the known matrices. The choice of MDscan was dictated by speed. Any other de novo motif finder or even a large set of candidate matrices that contains PWMs for active motifs (Smith et al, 2005) could also be used. We scanned a wide range of parameters to avoid potential bias, and obtained 1440 PWMs. We repeated the analysis as above. The reduction in variance is now further improved by ∼50% to Δχ2=16.1%, indicating improved discovery. The fitted model contains one individual motif and eight motif pairs involving nine distinct motifs (Table III), of which only HNF-1 and ARP-1 are from databases of known matrices and the rest are predicted. Among the predicted motifs, three were found to be similar to known PWMs: USF-2, FXR-IR1 and LEF-1. Matrix similarity was assessed using MatCompare (Schones et al, 2005). In all these cases, the predicted motif can explain the gene expression better than its known analog (Supplementary Table II). Clearly, the predicted motif provides a better definition of the binding site model. Although these three motifs ranked low by the original MDscan scores, we are able to detect these using adaptive splines. The other four matrices are completely novel. All eight significant pairs involve at least one such novel motif and thus are novel combinations, suggesting incompleteness of current understanding of transcriptional regulation even in well-studied mammalian systems such as liver.
Table 3.
Significant motifs and motif pairs for analysis with known and predicted matrices
| Pair ID | Significant motifs and motif pairs | P-value | Fit coefficients | Target gene count | Tissue with maximum expression | Specificity in liver | |||
|---|---|---|---|---|---|---|---|---|---|
| Model | Genome-wide | ||||||||
| No. of target genes | Percentage count | No. of target genes | Percentage count | ||||||
| — | HNF-1 | 3.0 × 10−15 | 182.8 | 10 | 1.0 | 94 | 0.6 | Liver | 2.4 × 10−15 |
| P1 | TGACCTTTG*HNF-1 | 0.0023 | 521.9 | 10 | 1.0 | 154 | 1.0 | Liver | 3.8 × 10−13 |
| P2 | TGACCTTTG*ACCCTAGACC | 0.0002 | −348.2 | 54 | 5.4 | 812 | 5.3 | Monocytes | — |
| P3 | ARP-1*ATGGAAAGA | 0.0006 | 299.2 | 17 | 1.7 | 154 | 1.0 | Liver | 6.7 × 10−6 |
| P4 | TTAACATGCA(∼FXR IR-1)*ATGGAAAGA | 0.0004 | −70.2 | 222 | 22.2 | 3416 | 22.3 | B cells | — |
| P5 | AATTGAAT(∼LEF-1)*ATGGAAAGA | 3.5 × 10−7 | −66.0 | 331 | 33.1 | 4939 | 32.3 | Monocytes | — |
| P6 | ATGGAAAGA*ACCCTAGACC | 0.0005 | 25.8 | 499 | 49.9 | 7244 | 47.3 | Monocytes | — |
| P7 | TGACCTTTG*ATGCCTGTC(∼USF-2) | 6.1 × 10−10 | 21.0 | 561 | 56.1 | 8481 | 55.4 | Monocytes | — |
| P8 | TGAATGAAT*HNF-1 | 3.0 × 10−15 | −18.6 | 989 | 98.9 | 15 212 | 99.4 | B cells | — |
Predicted matrices are represented by their consensus sequences. Matches to known PWMs are indicated in parentheses. For example, the PWM ATGCCTGTC matches USF-2. Similarity was determined using MatCompare (Schones et al, 2005). USF-2 is a ubiquitous factor, FXR is a bile acid receptor and LEF-1 is a nuclear mediator in Wnt signaling pathway. ARP-1 is involved in multiple processes including lipid metabolism. Under target gene count, model refers to the genes used to fit the spline model to microarray data (total=1000), whereas genome-wide indicates all the genes on the array whose promoters were available (total=15 309). Final spline model and the significant predicted matrices for this analysis are reported in Supplementary note.
Target genes
Are these novel motifs and combinations tissue-specific? What are their functional roles? To address these questions, we obtained the target genes of the TFs for a given motif or pair. As making the motif↔TF association is beyond the scope of our algorithm in general, we will often refer to the targets of TFs as targets of the motifs that the respective TFs bind to. As PWM scores are related to binding affinities (Berg and von Hippel, 1987), knots correspond to the activation thresholds, that is, the PWM cutoff scores (Figure 1A and B). Thus, targets are genes with PWM scores above the knot of the spline in the predicted model (Figure 1B). These constitute a set of experimentally testable hypotheses. We consider the HNF-1 motif as an example. According to the optimal model, there are 10 predicted HNF-1 targets out of 1000 genes analyzed (Table IV). We searched for corroborative evidence a posteriori. Strong HNF-1 binding was detected for four genes in the ChIP-chip assay (Odom et al, 2004). Seven targets have been experimentally validated otherwise (Table IV). Taken together, we found evidence for eight out of 10 as being direct HNF-1 targets. This is quite remarkable given that we started with a large set of matrices and microarray data from liver and made no a priori assumption in our approach apart from optimizing certain mathematical functions related to splines. Previous approaches to identifying TF targets in mammals, in contrast, are quite resource intensive, and require a combination of microarray and binding assays (Kirmizis and Farnham, 2004).
Table 4.
Target genes of HNF-1 in liver
| Gene name | Annotation | PWM score | Expression log ratio | Evidence for direct targets | ||
|---|---|---|---|---|---|---|
| Adult liver | Fetal liver | HNF-1 binding P-value | Experimental evidence (PubMed ID) | |||
| ALB | Albumin | 0.4873 | 5.07 | 4.27 | 2.3 × 10−7 | 2693890 |
| FGB | Fibrinogen, B beta polypeptide | 0.4849 | 4.37 | 4.89 | — | 8218230 |
| AFP | Alpha-fetoprotein | 0.4630 | −1.09 | 5.81 | 0.7 | 2479822 |
| CYP2E1 | Cytochrome P450, family 2, subfamily E, polypeptide 1 | 0.4622 | 5.93 | −1.89 | 8.1 × 10−9 | 7710685 |
| GC | Group-specific component (vitamin D binding protein) | 0.4592 | 4.86 | 4.49 | 0.8 | 9774468 |
| FGA | Fibrinogen, A alpha polypeptide | 0.4582 | 4.25 | 4.32 | — | 7499335 |
| GARS | Glycyl-tRNA synthetase | 0.4569 | −2.16 | −0.95 | 4.1 × 10−5 | — |
| APOH | Apolipoprotein H | 0.4533 | 5.32 | 4.77 | 5.0 × 10−8 | 14984368 |
| TXNIP | Thioredoxin interacting protein | 0.4523 | −0.96 | −2.17 | 0.1 | — |
| RPL34 | Ribosomal protein L34, transcript variant 1 | 0.4521 | −2.70 | −1.33 | 0.7 | — |
Characteristics of the predicted targets of HNF-1 among 1000 modeled genes under the given experimental condition. Expression data are log2(Eg/EgC), where Eg is the observed expression level of gene g and EgC is that of the average across all the tissues. In all cases, the mRNA levels change by at least two-fold in adult liver, and also in fetal liver, suggesting liver-specific activity. HNF-1 binding P-values were obtained from Odom et al (2004): P⩽0.001 indicates strong binding. We find evidence for eight of the 10 predicted targets as being direct HNF-1 targets: ALB, FGB, AFP, CYP2E1, GC, FGA, GARS and APOH. The detailed references for reported experimental evidence in the last column are given in Supplementary Table VI.
We used the optimal model to predict HNF-1 targets in two extra gene sets: top 2000 genes (ranked by variance across tissues) and genes upregulated by at least two-fold, genome-wide. We found previously reported experimental evidence of HNF-1 binding for 12 out of 17 predictions in the first set and 17 out of 21 in the second (Supplementary note and Supplementary Table VI). Thus, depending on which set we look at, we find existing evidence for 70–80% predictions as being direct HNF-1 targets under the experimental condition. The nine novel HNF-1 targets display strong HNF-1 binding characteristics, as follows. Their PWM scores are in the same range as the known ones. HNF-1 binds DNA as a dimer, and hence, its binding motif often contains two half-sites forming a palindrome (Locker, 2001). All novel targets have at least one half-site; seven have both. HNF-1 can, in fact, bind DNA with just one half-site available (Chung and Bresnick, 1995). Cluster Buster (Frith et al, 2003) and MATCH (Kel et al, 2003), the two popular programs that identify TF binding motifs based on sequence information only, can also find HNF-1 binding site in the promoters of some of these genes (Supplementary note and Supplementary Table VI). Hence, we think these new HNF-1 targets are biologically significant and suggest that the subnetwork defined by HNF-1 is wider than previously known (Tronche et al, 1994).
The targets for a motif combination are obtained similarly as that for a single motif. For example, for two significant motifs m1 and m2 contributing a term θ(S1−ξ1, 0) θ(S2−ξ2, 0) to the spline model, where θ's denote splines, S the PWM scores and ξ the knots (see Materials and methods), the target genes for the combination m1–m2 are those with scores S1>ξ1 and S2>ξ2 for motifs m1 and m2 respectively.
Assessment of tissue-specific roles
We next sought to determine if any of the significant motif combinations has a liver-specific role. To assess this, we obtained the targets for each active motif combination and determined the tissue where they achieve maximum transcriptional response. If this was liver, we performed tests of significance to characterize their specificity to liver (see Materials and methods). Three motif combinations, including HNF-1, were found to be significantly liver-specific (Table III and Supplementary note). We separately validated the liver-specific roles of these three combinations by examining the functional enrichment of their target genes. We searched 9133 Gene Ontology (GO) biological process terms and found 18 over-represented processes (Table V), all of which except one are known to occur in liver.
Table 5.
Enrichment of the biological process categories from Gene Ontology (GO)
| Pair ID | Motif or motif pair | GO biological process | Total no. of genes with the term | Total no. of target genes with the term | Enrichment | P-value |
|---|---|---|---|---|---|---|
| — | HNF-1 | Regulation of blood pressure | 28 | 3 | 15.2 | 0.001 |
| Regulation of body fluids | 79 | 4 | 7.2 | 0.002 | ||
| Response to pathogenic bacteria | 13 | 2 | 21.8 | 0.004 | ||
| Cytolysis | 14 | 2 | 20.2 | 0.004 | ||
| Blood coagulation | 64 | 3 | 6.6 | 0.01 | ||
| Hemostasis | 69 | 3 | 6.2 | 0.01 | ||
| P1 | TGACCTTTG*HNF-1 | Hexose metabolism | 98 | 5 | 4.7 | 0.004 |
| Monosaccharide metabolism | 101 | 5 | 4.6 | 0.005 | ||
| Muscle development | 105 | 5 | 4.4 | 0.006 | ||
| Inflammatory response | 153 | 6 | 3.6 | 0.006 | ||
| Fructose metabolism | 13 | 2 | 14.1 | 0.008 | ||
| Energy derivation by oxidation of organic compounds | 118 | 5 | 3.9 | 0.009 | ||
| Main pathways of carbohydrate metabolism | 76 | 4 | 4.8 | 0.009 | ||
| P3 | ARP-1*ATGGAAAGA | Lipid transport | 57 | 4 | 6.7 | 0.003 |
| Lipoprotein metabolism | 31 | 3 | 9.2 | 0.004 | ||
| Inflammatory response | 153 | 6 | 3.7 | 0.005 | ||
| Glycosphingolipid metabolism | 15 | 2 | 12.6 | 0.01 | ||
| Response to wounding | 235 | 7 | 2.8 | 0.01 | ||
| P2 | TGACCTTTG*ACCCTAGACC | Response to external stimulus | 556 | 48 | 1.7 | 0.0003 |
| Response to pest, pathogen or parasite | 385 | 35 | 1.8 | 0.0008 | ||
| Response to wounding | 235 | 24 | 2.0 | 0.001 |
Significant GO terms with P⩽0.01 (q⩽0.15; Storey and Tibshirani, 2003) are reported here. Enrichment (column 6) quantifies how enriched a given category is with the target genes (Zeeberg et al, 2003). P-value was calculated using a hypergeometric distribution (see Materials and methods). Parent terms with higher P-values than their child terms are not reported in this table. Terms with exactly one gene in the total and target gene sets are also not reported. Some of the P-values are close to the cutoff possibly because the terms are quite broad.
Thus, starting from expression data, we have sequentially obtained the functional motif combinations, their targets and the processes they regulate, representing the active transcriptional subnetworks (Figure 2). HNF-1 is selectively expressed in liver and is a pleiotropic regulator of liver-specific genes (Tronche et al, 1994). The statistical tests and enrichment analysis reconfirm this fact. HNF-1 is known to regulate all the processes found significant by our analysis (Table V). The two new pairs that we identified, P1 and P3, display remarkably similar liver-specific characteristics. Their targets are strongly regulated in adult liver or fetal liver or both, like the HNF-1 targets above (Supplementary Table III). Several of these genes, such as CES1, PTPRS and C4A, have been shown to have distinct pharmacological roles but their cis-regulatory mechanisms are not yet elucidated (Supplementary note) (Norris et al, 1997; Marsh et al, 2004). Our findings provide the initial hypotheses for their cis-regulation. Statistical tests demonstrate that these novel motif pairs play a strong liver-specific role. This is further supported by the functional enrichment analysis: pair P1 regulates sugar metabolism pathways, and pair P3 regulates lipid transport and metabolism (Supplementary note). Both of them are active in inflammatory response as well. Thus, these define new subnetworks active in liver that are uncovered by our approach.
Figure 2.

Schematic representation of our analysis. A snapshot of the tissue-specific transcriptional subnetworks discovered from microarray data on adult human liver under a normal condition.
We also found six motif pairs active in B cells and monocytes. Some of these are biologically significant. For example, the pair P2 that is identified as active in monocytes is also found to be involved in host defense (Table V). The mRNA sample was taken from whole liver, which receives 30% of the cardiac output and has strong hematopoietic potential (Golden-Mason and O'Farrelly, 2002). The resulting mixing effects in such mRNA samples have not been carefully considered by the existing motif analyses. To our knowledge, we are able to isolate these computationally for the first time. Furthermore, we notice that several targets achieve their maximum expression in a tissue different from the one in which the motif combination has its maximal regulatory effect (Supplementary Figure 2). This suggests that genes are coregulated across multiple tissues, as one would expect in a synexpression group (Niehrs and Pollet, 1999). An advantage of our method is that mRNA profiles from only a few conditions are necessary to reach such a conclusion.
Functional motif combinations from temporal expression profiles
We next applied our algorithm to gene expression profiles for the human cell cycle (Whitfield et al, 2002). In this set of experiments, HeLa cells were synchronized by a thymidine–nocadazole block and mRNA levels were measured at 19 time points spanning 36 h. We applied the above algorithm to each time point separately using mRNA levels from asynchronous cell cultures as reference (Whitfield et al, 2002). Our analysis revealed several responsive motifs and motif pairs across all phases of the cell cycle. We consider the 10 h time point, which is near the G1/S phase, as an example. The optimal spline model for this time point contains two individual motifs and six motif pairs arising from a total of seven distinct motifs. Among these, three are known E2F motifs from TRANSFAC, two are predicted but have strong similarity to binding motifs for AML1 and POU3F2, and the rest two are novel (Supplementary note). Combinations of E2F motifs and E2F–E2F/DP-2 are represented among the significant motif pairs. E2F and its combinations with DP proteins play a pivotal role in cell-cycle progression and regulation of G1/S-specific genes (Locker, 2001). Our findings support this fact. AML1 is a master regulator of hematopoietic cell development and is a target of chromosomal translocations responsible for aberrations in acute leukemia. It has been shown to exert strong effect on the length of G1 phase (Strom et al, 2000). We find AML1 functional at 10 h and acting synergistically with E2F. Although we did not find any direct report for this interaction, such a possibility has been indicated previously (Meyers and Hiebert, 1995). The fitted model has Δχ2=23.1%.
Our analysis confirms the activity of several important motifs at appropriate stages of cell cycle. For example, the cis-regulatory motifs for Irf, Jun, Myb, AhR:Arnt, AP-2 alpha A and Mef-2 are found to be functional at or near G1/S transition, the motif for c-Ets-1 near G2/M phase and that for ROR alpha 2 near G0/G1 phase, in accordance with previous findings (Table VI). Most of these factors have been linked to multiple types of cancer, cellular proliferation and differentiation. Lef-1 and Tcf-4, the two mediators in Wnt signaling pathway (Eastman and Grosschedl, 1999), emerge as significant at 14 h (late S phase), indicating that this pathway was potentially active at this time point. Several developmental regulators are also found to be functional at multiple time points: GATA-1 and GATA-6 at early mitotic stage, and POU3F2 and POU6F1 at G1/S and G2/M phases.
Table 6.
Individual motifs identified as significant at various phases (time points) of cell cycle
| Significant motifs | P-value | Time (h) | Phase | Experimental evidence (PubMed ID) |
|---|---|---|---|---|
| ROR alpha 2 | 4.4 × 10−5 | 22 | G0/G1 | 11241556 |
| ATF6 | 0.004 | 8 | G1/S | 10564271 |
| MEF-2 | <0.005 | 6, 14, 24, 28 | G1/S | 10322110 |
| IRF-2 | <0.01 | 22, 28, 30 | G1/S | 9417064 |
| NF-Y | 6.1 × 10−15 | 8 | G1/S | 9281303 |
| v-Myb | 0.004 | 24 | G1/S | 9111313 |
| AhR:Arnt | <0.0005 | 12, 26 | G1/S, S | 8628281 |
| AP-2 alpha | 4.6 × 10−12 | 28 | S | 9776742 |
| IRF-1 | 0.0004 | 28 | S | 1491701 |
| LEF-1 | 4.2 × 10−8 | 14 | S | 15736165 |
| Oct-1 | 5.4 × 10−16 | 28 | S | 12887926 |
| S8 | 0.009 | 28 | S | — |
| TCF-4 | 1.6 × 10−6 | 14 | S | 12408868 |
| USF2 | 0.0007 | 28 | S | — |
| v-Jun | 0.0007 | 28 | S | 12717415 |
| c-Ets-1 | 1.7 × 10−15 | 32 | G2/M | 8437861 |
| GATA-6 | 1.7 × 10−15 | 32 | G2/M | — |
| GATA-1 | 2.0 × 10−12 | 34 | M | 10216081 |
| MAZ | 3.7 × 10−6 | 2 | M | 11395515 |
| POU3F2 | <0.01 | 10, 32 | — | 14708619 |
| POU6F1 | <0.0001 | 8, 34 | — | 8900043 |
The experimental evidence for activity of a given motif, as reported in the literature, is included in the last column.
Among the significant motif pairs (Table VII), the pairs AhR–E2F, Oct1–NF-Y, Areb6–TBP and E2F–E2F/DP-1 have been previously characterized. For instance, when exposed to external toxins, Aryl hydrocarbon receptor (AhR) synergizes with retinoblastoma protein, a key regulator of E2F, to induce cell-cycle arrest (Puga et al, 2000). We detect this combination to be active at 0 h, that is, when cells were released following drug treatment. Oct-1 and NF-Y, which are significant at 6 h, coordinately regulate transcription of Gadd45 and other genes (Hirose et al, 2003; Kam et al, 2005). The cooperativity of E2F-1/DP-1–Oct1 and E2F–MEF2 has been implicated in wet-lab studies (Table VII). Functional interactions of Ik-3 with E2F and E2F-1/DP-1 are novel predictions that emerged from our study.
Table 7.
Significant motif pairs from different phases (time points) of cell cycle
| Significant pair | P-value | Time (h) | Phase | Experimental evidence (PubMed ID) |
|---|---|---|---|---|
| Oct-1*NF-Y | 8.0 × 10−9 | 6 | G1 | 14586402 |
| MEF-2*E2F-1 | 5.6 × 10−15 | 24 | G1 | 11027611 |
| TBP*AREB6 | 1.2 × 10−3 | 8 | G1/S | 14761964 |
| E2F*E2F-4/DP-2 | 1.3 × 10−15 | 10 | G1/S | 9372931 |
| E2F(M00050)*E2F(M00516) | 1.3 × 10−15 | 10 | G1/S | 15014447 |
| AML1*E2F-4/DP-2 | 1.0 × 10−10 | 10 | G1/S | — |
| AML1*E2F | 2.6 × 10−5 | 10 | G1/S | 8834231 |
| AhR*E2F | 7.3 × 10−5 | 0 | G2/M | 10644764 |
| E2F*E2F-1/DP-1 | 9.7 × 10−9 | 18 | G2/M | 7917337 |
| E2F-1/DP-1*Ik-3 | 9.7 × 10−9 | 18 | G2/M | — |
| E2F-1/DP-1*Oct-1 | 2.8 × 10−7 | 18 | G2/M | 10662552 |
| E2F*Ik-3 | 5.1 × 10−7 | 18 | G2/M | — |
The experimental evidence reported in the literature for activity of a given motif pair during cell cycle is included in the last column. Italicized Pubmed IDs indicate indirect evidence.
Among the above combinations, Oct1–NF-Y pair was determined in the computational approach based on expression coherence (EC) scores (Zhu et al, 2005b). The E2F–NF-Y pair identified in this and another computational study (Elkon et al, 2003) is marginally suboptimal in our analysis (data not shown). We also find a functional combination at 16 h, that is, near G2/M, that involves NF-Y and a motif with consensus ATTCAAA. The latter matches the reverse complement of the previously predicted CHR motif (Zhu et al, 2005b), and their PWMs are very similar (P=3.6 × 10−4) (Schones et al, 2005). Moreover, CDC25C is one of their predicted targets. Thus, this TF module is almost identical to the NF-Y–CHR module reported recently (Zhu et al, 2005b). We also detect interactions between the same regulatory motifs. YY1, GATA-6, USF and TFs with POU domain are some examples of this type (Supplementary Table VII). Of these, the last two have been identified in the EC score approach. The other active combinations uncovered by our approach involve novel motifs. Several more experimentally characterized pairs emerge as significant when only motifs from existing TF databases are used to obtain the spline fits (Supplementary Table VIII).
Some of the functional motif combinations reported here have been identified in the previous computational studies (Elkon et al, 2003; Zhu et al, 2005b), as indicated above. One of the major advantages of our approach over these methods, however, is that we can identify the appropriate time point where a given motif combination is active and enumerate its impact on gene expression levels. In addition, our computational approach has enabled identification of many more synergistic motif combinations than was achieved in the above studies. The complete list of significant motif combinations identified in our analysis is summarized in Supplementary Results.
Condition-specific gene induction by transcription factors
TFs regulate genes in a condition-specific manner, and consequently, a particular TF can activate different sets of genes under different conditions (Zhu et al, 2005a). This is brought about by changes in gene activation thresholds and accompanied by distinct cooperative partnering with other TFs. One such example is E2F, which induces separate sets of genes in G1/S and G2/M phases (Ishida et al, 2001). As the location of knots (Figure 1B), and hence the target genes, in a linear spline model is determined by the input expression profile, condition-specific gene induction can be suitably modeled in our approach. We illustrate this by focusing on targets of motif combinations E2F(TRANSFAC id: M00516)–E2F(M00050) and E2F(M00516–E2F-1/DP-1(M00736)–Ik3, which are significant at 10 h (G1/S) and 18 h (G2/M), respectively (Supplementary note). This G2/M-specific motif combination is a novel prediction of our method. Both combinations involve a common E2F motif (M00516), which partners with different motifs in the two phases. We observe that the activation threshold for this motif, as adaptively determined by the spline models, is significantly different at the two time points: the thresholds are 0.4486 and 3.4 × 10−8, leading to 98 and 21 E2F targets at 10 and 18 h, respectively (Supplementary note and Supplementary Tables IX and X). The target sets are mostly non-overlapping: 88 out of 98 G1/S targets and 11 out of 21 G2/M targets are distinct from each other. Comparison with TRED (Zhao et al, 2005), a database of manually curated E2F targets, shows that 21 G1/S and 8 G2/M targets have been validated in previous experimental studies. Many newly predicted targets are supported by other motif finding methods (Frith et al, 2003; Kel et al, 2003). Among the 77 novel G1/S targets, MATCH finds an E2F binding site for 43 (56%) genes, Cluster-Buster for 29 (38%) genes and 13 (17%) genes have a conserved E2F binding site in the mouse genome. For 13 novel G2/M targets, the respective numbers are eight (62%), one (8%) and two (15%). Of the 10 targets that are common to both phases, three are downregulated in G1/S and upregulated in G2/M phase and the other seven have exactly opposite profile. E2Fs are known to activate and repress the same gene in different phases (Locker, 2001). We suspect analogous regulatory control in these cases. In this vein, we note that all G1/S targets and 19 out of 21 G2/M targets have their maximum contribution to expression coming from an E2F motif combination (Supplementary note).
Biochemical validation of novel E2F targets
E2F is a canonical regulator of genes involved in cell-cycle progression, S-phase entry and apoptosis (Locker, 2001; Nahle et al, 2002). Additionally, it has recently become clear that it plays a key role in the G2/M phase as well (Ishida et al, 2001; Hernando et al, 2004). Indeed, Rb inactivation and deregulation of E2Fs was shown to promote genomic instability by uncoupling cell cycle progression from mitotic control (Hernando et al, 2004). Our in silico observations reaffirm a key role of E2F in the G2/M phase and expand the currently known transcriptional subnetwork controlled by E2F. CDC16 and DLG7 (also known as HURP) are among the important novel targets that we find specific to G2/M. CDC16 plays an essential role in metaphase to anaphase transition, whereas DLG7 is a recently identified cell-cycle regulator that localizes to spindle poles during mitosis. Both have been shown to play a measurable role in hepatocellular carcinomas (Yasui et al, 2002; Tsou et al, 2003), but their regulatory mechanisms are not quite understood. E2F binding sites could be detected in their promoter sequences using MATCH, but not by the other sequence-based motif finding tools (Supplementary Table X). However, as a program like MATCH does not directly use expression data, G2/M-specific activation of these targets cannot be predicted using such a method.
To determine physiologically whether E2F regulates DLG7 and CDC16, we examined their mRNA expression in NIH3T3 cells expressing a well-characterized inducible E2F-1/estrogen-receptor fusion construct (ERE2F1) (Vigo et al, 1999; Nahle et al, 2002). In this system, E2F activity can be programmatically induced simply by supplementing growth media with tamoxifen (TX), an estrogen-receptor ligand. TX triggers nuclear localization of the ERE2F1 construct where it can initiate transcription. Importantly, such inducible system allowed us to circumvent the complications encountered using constitutive E2F expression systems as unrestrained E2F-1 expression triggers apoptosis in many cell systems in a manner that prevents collection of representative experimental samples. Here, cells were infected with high-titer retroviruses harboring the E2F-1 fusion construct or a mutant lacking the transactivation domain of E2F (MERE2F1). Purely infected cell populations were selected in the appropriate antibiotic medium. Then, a time-course analysis of DLG7 or CDC16 expression was performed by QPCR using RNA extracted from ERE2F1- or MERE2F1-infected cells in the presence of TX. As shown in Figure 3A , DLG7 expression was induced approximately 4 fold upon E2F-1 activation within 8 h, whereas activation of the E2F-1 mutant fails to induce DLG7 transcription, as expected. A similar pattern of induction, albeit more modest, was obtained for CDC16 expression (Figure 3B). Of note, the fusion construct of E2F-1, ERE2F1, has been reported to transactivate at one-third the capacity of non-fusion E2F-1 constructs, presumably owing to hindrance by the ER fusion domain (Nahle et al, 2002). Therefore, we estimate a yet higher induction by endogenous E2F-1.
Figure 3.

Biochemical validation of target genes. Results of RT–PCR analysis in NIH3T3 fibroblasts expressing an inducible ERE2F1 or a transactivation-deficient mutant, MERE2F1. At time zero, medium containing 500 nM of 4-hydroxytamoxifen (OHT) was added and transcript levels of (A) DLG7 and (B) CDC16 were determined using gene-specific primers at the indicated times by RT–PCR. The respective transcript levels with 10 μg/ml of cyclohexamide (CTX) added are shown in (C) DLG7 and (D) CDC16. GAPDH was used as a standardization control. Plotted data reflect results after normalization with GAPDH.
Thus far, our analysis shows that DLG7 and CDC16 are induced by E2F. Such induction required an intact E2F transcriptional function (Figure 3A and B, MERE2F1), suggesting direct regulation of these genes by E2F. To investigate this, we induced E2F in the presence of cyclohexamide (CHX). CHX is an inhibitor of protein biosynthesis and blocks translational elongation. Hence, in the presence of CHX, de novo protein synthesis is abolished. Thus, in the event that an induction of DLG7 or CDC16 transcripts upon ERE2F activation (relocalization of pre-existing ERE2F protein) is observed, then E2F is directly regulating these genes. Consistent with this hypothesis, mRNA levels of DLG7 and CDC16 were indeed induced in the presence of both TX and CHX similar to the treatment with TX alone (Figure 3C and D, compare to A and B). Apparently, the regulation of these targets is independent of de novo protein synthesis and is the result of a direct transcriptional activation by E2F, in accordance with our computational predictions.
Discussion
In summary, we have presented an unsupervised method for learning human transcriptional subnetworks from expression data that directly accounts for the strong degeneracy of and interactions between cis-regulatory elements. The predictions are widely concordant with the experimental findings, suggesting the biological significance of this approach. We find that the linear splines provide a convenient framework to model mammalian gene regulation in the face of continuous PWM scores arising from highly degenerate TF binding sites. The average information content of the PWMs reported as significant in this paper lies in the range 0.34–1.88 (see Table I for examples). In particular, fitting single linear splines is a powerful means of prioritizing a large number of degenerate input motifs. In certain cases, one can stop at this step to identify the target genes of the active TF. The HNF-1 example discussed here is one such case. The periodic activity of the degenerate Mcm1 motif in yeast cell cycle, which was previously demonstrated using multiple spline model (Das et al, 2004), can be easily modeled by fitting a single linear spline (Supplementary Figure 3), indicating its effectiveness.
Our algorithm leads to discrete testable hypotheses for transcriptional end points of biochemical pathways active under a specific condition. As the TF binding thresholds are learnt from the expression profile, condition dependence of these end points can be naturally modeled in this framework, in contrast to clustering-based approaches. One of the difficulties in TF target determination is discriminating direct targets from indirect targets (Kirmizis and Farnham, 2004). The results here suggest that correlation with expression is a promising way to identify direct targets from expression data. In addition, they indicate significantly broader regulatory roles of master TFs like HNF-1 and E2F. A strong correlation between mitotic control and tumor progression in liver has been previously noted (Tsou et al, 2003). Intriguingly, our study suggests that part of this control may be exerted by E2F.
Our approach to target identification does not depend on arbitrary fold cutoffs, as are invoked by many comparable methods (Kirmizis and Farnham, 2004). Consequently, we can detect bona fide targets that undergo very subtle changes in expression. For example, the G1/S-specific E2F target, MCM5 (Ohtani et al, 1999), which is upregulated by only 6% at 10 h, is among the predicted targets at this time point (Supplementary Table IX). Furthermore, this technique identifies autoregulatory loops prevalent in biological networks. One of the predicted targets of the E2F motif combination at 10 h, for instance, is E2F-1 itself (Supplementary Table IX), which is a well-established example of autoregulatory loop (Chen, 1997). Such loops are difficult to infer using existing network modeling techniques (Basso et al, 2005).
Techniques based on correlation with expression have been previously proposed for yeast, but no analogous method exists, to our knowledge, for mammals. Our algorithm represents a first step in this direction. The optimal spline models for temporal expression profiles yield a Δχ2 of 21.7%, on an average. For tissue-specific data, the average is 24.4% (Supplementary Table IV). This is comparable to the percent reduction in variances achieved in lower eukaryotes (Das et al, 2004). There are several mechanisms that contribute to the mammalian gene expression, but are not captured in our models, and hence, in the above estimates of Δχ2. For instance, many genes are subject to regulation by distal long-range enhancers (Levine and Tjian, 2003). An added complexity is that the same transcriptional unit may use alternate transcription start sites (TSS) in a tissue- or developmental-specific fashion. Moreover, transcription is often coupled to other nuclear processes (Maniatis and Reed, 2002), like RNA splicing and degradation, which can potentially affect the measured mRNA levels. Finally, there are more complex modes of gene regulation, including effects of chromatin remodeling, which are not accounted for here. All of these factors need to be incorporated before we can obtain a comprehensive view of mammalian gene regulation.
Nevertheless, as this study clearly demonstrates, our approach provides a stepping stone to generate an accurate primary hypothesis of cis-regulation mediated by proximal promoters and does not require any prior system-specific knowledge. It is equally effective on expression and ChIP-chip data (Smith et al, 2005), tissue-restricted and temporal profiles. In the light of the above findings, we think this approach will help accelerate systematic understanding of regulatory network architectures in a wide range of mammalian systems and make scope for novel therapeutic interventions.
Materials and methods
Percent reduction in variance
Percent reduction in variance, Δχ2, is defined as (Bussemaker et al, 2001; Das et al, 2004)
![]()
where yg=log(Eg/EgC), rg=yg−ygp is the residual (p indicates the predicted value of y) and ȳ and r̄ are their respective means. Eg is the expression level of gene g and C refers to the control set.
Average information content of a position weight matrix
The average information content of a PWM is a measure of degeneracy of the binding sites for the corresponding TF. If we consider each PWM as a matrix of dimensions 4 × L, where rows represent the bases and columns the positions of cis-regulatory motifs, then the information content for each column (position) i is enumerated as

where pi(bi) is the probability of observing the base bi at the position i and the summation is over all possible bases at position i: A, C, G and T. The average information content of a PWM is the average of Ii taken across all the columns of the matrix. For a non-degenerate word, the average is 2, and for an exactly degenerate motif, that is, pi(bi)=0.25 for all bi's, the average is 0. For any typical mammalian PWM, the average information content interpolates between these two extremes.
Data preparation
Promoter sequences
We obtained promoter DNA sequences from CSHLmpd database (Xuan et al, 2005) (−700 and +300 nt from the TSS). For each PWM μ of width L, each L-mer in a promoter sequence was assigned a probability score S:
![]()
where pi(bi) is the probability of observing the base bi at the position i. Thus, the score S always assumes a value between 0 and 1. For a given gene g, the maximum of all S's in its promoter, Sgmax, was used as the representative score. Henceforth, we will refer to the maximum score as Sg. We used the program PATSER (Hertz and Stormo, 1999) to speed up the scoring process. log(pi(bi)) was entered as the weight for the base bi at the position i (a large negative number was used for pi(bi)=0). The scores reported by PATSER were exponentiated to obtain the scores Sg (large negative numbers converted to zero).
Expression data
Expression profile (Su et al, 2004) across 79 human tissues for a single condition each was reported for 33 689 probes representing a total of 17 543 publicly available human genes. We could obtain promoter sequences for 15 309 such genes (87% of the total) from CSHLmpd (Xuan et al, 2005). Among these, top 1000 genes by variance across all tissues were used to build the spline models. For modeling cell-cycle expression data (Whitfield et al, 2002), we used the cell-cycle-regulated genes (805 out of 874, for which promoters were available). The probability scores {Sg}, discussed above, were fitted to the logarithm of expression ratios in computing these models.
Prioritized matrix sets
We used the gap in %RIV (Supplementary Figure 1) to prepare three prioritized sets of matrices for MARS runs as follows. We first recorded the P-value of the 10th matrix below the gap, where P-value was assigned using an F-test (Das et al, 2004). This P-value was one of the cutoffs used. The other two were determined by multiplying this P-value by a factor of 10 and 0.1, respectively. For a given cutoff, all matrices with P-value lower than this cutoff were taken in the set.
MDscan
We scanned 48 parameter settings, by varying the motif width (5–10 nt) and the number of top promoter sequences, to look for candidate motifs (10, 25, 50 and 100), once each for the gene list sorted in ascending and descending order by log expression ratio, and obtained 30 PWMs for each setting using MDscan (Conlon et al, 2003), resulting in 1440 predicted PWMs. These parameter settings are very similar to those suggested by Conlon et al (2003).
Fitting a single linear spline
PWM scores across genes {Sgμ} for a given motif μ were fitted to the expression ratios {log(Eg/EgC} using the following model:
![]()
where θ(x, 0) is a linear spline: it is x, when x⩾0, and zero, otherwise. The coefficients aμ and bμ and the location of the knot ξμ were determined so as to maximize Δχ2. It is important to note that Δχ2 depends on the location of ξμ. Maximization of Δχ2 leads to an unbiased and adaptive determination of this threshold for any given PWM. We also attempted to fit the other type of linear spline, θ(ξμ−Sgμ, 0), and the spline with maximum Δχ2 was considered as the optimal choice. The latter type of spline is related to the saturation part of sigmoidal transcriptional response. The significance of the fit was enumerated using an F-test, as discussed in the context of the MARS algorithm below.
Multivariate adaptive regression splines
MARS is a non-parametric and adaptive fitting method (Friedman, 1991; Steinberg and Colla, 1999; Das et al, 2004). It builds the model using stepwise forward addition of linear splines and their products. For pairwise interactions, the fitted model has the form

where Ŝgμi=Sgμ−ξiμ or ξiμ−Sgμ, Sgμ is the PWM score for motif μ on the promoter of gene g, ξiμ is the ith knot of the motif μ and θ(x, 0) are linear splines. Egp indicates the predicted expression level of gene g. Terms and knots in the above model are selected adaptively by minimizing the residual sum of squares (RSS), ∑g(yg−ygp)2, where yg=log(Eg/EgC). This is equivalent to maximizing Δχ2 used above. We allowed up to third-order interactions in the model. The model grows until a preset maximum number of terms is reached. Terms are then deleted sequentially to obtain a set of models of various sizes. MARS controls overfitting by selecting a model that minimizes the GCV score. GCV is RSS times a factor that penalizes for model complexity:
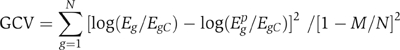
where M is the effective number of parameters, N is the total number of genes and the predicted expression level, Egp, is obtained from the fitted model above. M was obtained by 10-fold cross-validation (Friedman, 1991). The GCV-based optimization restricts the final model to a very small number of terms (Das et al, 2004). We provide additional control on overfitting by deleting motifs and motif pairs with adjusted P-values >0.01, where P-values were calculated using an F-test (Hastie et al, 2001):
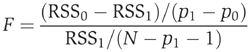
where RSS1 is the RSS of the final MARS model with p1+1 terms and RSS0 is the RSS of the model without a specific motif (or pair), which has p0+1 terms. N is the number of genes. This statistic has an F distribution with p1−p0 numerator degrees of freedom and N−p1−1 denominator degrees of freedom. The P-values were adjusted for multiple testing using the false discovery rate method as explained in an earlier work (Das et al, 2004).
As the number of input motifs and motif pairs can be very large, one needs to develop a suitable scheme to run MARS for its optimal use. When no interactions are allowed between distinct motifs (int=1), the previously developed implementation (Das et al, 2004) can be used with the prioritized set of motifs. The situation is, however, different when interactions are allowed (int>1). The previous scheme requires a prioritized set of pairs of motifs as input to MARS (Das et al, 2004). However, the pair prioritization scheme used there requires prior knowledge of whether a motif is present or absent in a given promoter, which is often not available in mammals. To address this, we used the MARS program directly to prioritize the motif pairs. A key impediment here is that when all possible interactions are allowed with a large set of motifs as input, suboptimal and typically biologically insignificant models are predicted by MARS, leading to detection of incorrect motif pairs (Das et al, 2004). To avoid this, we partitioned the prioritized set of motifs into small sets of at most k motifs (k=15 for this implementation). MARS is run on all possible pairs of such motif sets with a given interaction setting to identify prioritized set of interactions. All possible interactions are allowed for a given pair of motif sets. This process is then iterated, keeping only significant motifs and pairs from the previous iteration, until the number of significant motifs falls below 2k. The final MARS run is executed with the significant motifs identified in the above runs plus the motifs identified as significant with the int=1 setting. We used the MARS program from Salford Systems and the parameter settings for each MARS run were as described before (Das et al, 2004). More details are presented in Supplementary Methods.
Tests of significance for tissue-specific roles
We first examined if the given set of target genes has tissue-specific variation. This is identical to the question: whether a given set of subjects respond to the treatments they receive. Repeated-measures ANOVA (Glantz, 2001) is a standard way to test the significance of this hypothesis, with genes corresponding to subjects and tissues to treatments. The null hypothesis is that there is no dependence of mRNA levels on tissues for a given gene set. Because we selected genes by maximum variance across tissues, this P-value was always low (P<1.0 × 10–6).
We next obtained the tissue Tmax where the given target gene set has maximum average expression. We tested the difference in expression levels of target genes between tissue Tmax and each of the other tissues using a t-test (Glantz, 2001). A geometric mean of the calculated P-values is reported as an estimate of specificity to the tissue Tmax. We used a geometric mean of the P-values instead of the P-value of t-test to the next best tissue, because quite often regulatory TFs are selectively expressed in a few tissues instead of just one tissue.
Determination of statistical significance of GO terms
The statistical significance of a GO term was calculated using the hypergeometric distribution as follows. Consider that there are N genes on the microarray in total and m target genes for a given motif or motif combination, calculated genome-wide. Genome-wide targets were obtained using the optimal spline model discussed in the text. We performed genome-wide analysis to ensure that the test has sufficient power. If there are n genes on the array associated with a GO term and k target genes associated with the same term, then the P-value is calculated as the probability of having at least k target genes associated with this term:
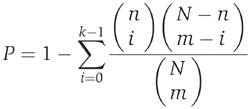
The enrichment (EN) is defined as (Zeeberg et al, 2003)
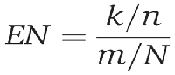
The P-values were then corrected for multiple testing using the false discovery rate method (Storey and Tibshirani, 2003). It is quantified in terms of a q-value, which provides an estimate of the fraction of false discoveries among the significant terms. For the results reported here, we used q⩽0.15.
Enumeration of characteristics of TF binding sites
We used two motif finding programs to examine the sequence-based characteristics of the predicted binding sites: MATCH (Kel et al, 2003) and Cluster-Buster (Frith et al, 2003). MATCH was run to minimize the false positive rate and Cluster-Buster was run with default settings. To determine the conservation of predicted E2F binding sites in mouse, we obtained the corresponding orthologous promoter from CSHLmpd database (Xuan et al, 2005), and examined if the relative distances of the maximum scoring E2F sites from the TSS in the two species are within 10 nt of each other. We also included the conserved E2F binding sites reported by Zhu et al (2005b).
Cells and gene transfer
NIH3T3 cells were obtained from the American Type Culture Collection (ATCC) and infected with high-titer recombinant retroviruses expressing the wild type E2F-1 (ERE2F-1), a transactivation-defective mutant of E2F-1 (pBabeHAERE2F-1(1–374)) or an empty vector control pBabeHA as described (Nahle et al, 2002). For ER constructs, E2F-1 activity was induced by the addition of 500 nM OHT (Sigma, St Louis, MO). De novo protein synthesis was inhibited by adding 10 μg/ml CHX.
Cell preparation and RNA isolation
NIH3T3 fibroblasts were harvested and total RNA was extracted using a standard Trizol (Invitrogen) procedure. Briefly, pelleted cells were resuspended in 2.0 ml Trizol and lysed by pipetting up and down. Cell suspensions were then incubated for 5 min at room temperature. Samples were centrifuged at 12 000 g for 10 min at 4°C to pellet cell debris and supernatants were transferred to fresh tubes. Chloroform (0.20 ml/ml suspension) and molecular biology grade glycogen (final concentration of 0.40 mg/ml) were added to each tube. Samples were vigorously shaken by hand for 15 s and then incubated at room temperature for 5 min. After centrifuging at 12 000 g for 15 min at 4°C, the upper aqueous phase of each tube was transferred to fresh eppendorf tubes. Isopropanol (0.50 ml/ml suspension) was added to each aqueous supernatant and incubated for 10 min. Samples were centrifuged at 12 000 g for 10 min at 4°C followed by supernatant removal. Ethanol (75%) was added to each pellet and each tube was shaken well by hand. The samples were centrifuged again at 16 100 g for 5 min at room temperature and then the supernatant was discarded. Pellets were air-dried for approximately 10 min and then rehydrated with UltraPure distilled water (GIBCO).
Real-time RT–PCR procedure
Real-time RT–PCR was performed on the SmartCycler II instrument (Cepheid) using the Superscript III Platinum SYBR Green One-Step qRT-PCR kit (Invitrogen). Each assay consisted of an RT–PCR master mix containing 12.5 μl of 2 × SYBR Green Reaction Mix, 0.5 μl of SYBR Green One-Step Enzyme Mix, 0.5 μl of each primer (final 200 nM each) and UltraPure distilled water (GIBCO) for a final volume of 25 μl and 2 μl of RNA (50 ng per assay). Negative controls contained only the RT–PCR master mix (25 ml) and 2 μl of UltraPure distilled water (GIBCO). The forward primer for DLG7 used was 5′-GTACAGCAAGGATTGGAGTCG-3′ and the reverse primer used was 5′-CTCCTTTCACAGAAGCGTGA-3′. The forward primer for CDC16 used was 5′-GACGTGGTAGTGTCTTTAGCTGAG-3′ and the reverse primer used was 5′-CTCCACAAGAGTTCCTATGTGC-3′. cDNA synthesis was performed at 50°C for 15 min followed by a 2 min incubation at 95°C to inactivate the reverse transcriptase and activate the Taq DNA polymerase. PCR amplification was performed following the 95°C incubation for 50 cycles of denaturation (15 s at 95°C), annealing (60°C for 30 s) and extension (30 s at 72°C).
Supplementary Material
Supplementary Methods
Significant Motifs and Motif Pairs for HeLa Cell-Cycle
Supplementary Figures
Supplementary Tables
Supplementary Figure 2
Supplementary Note
Acknowledgments
We thank Terri Pietka and Mike Hsieh for excellent technical assistance, X Shirley Liu for providing the updated version of MDscan, Kristian Helin for E2F-1 expression vectors and Joe W Gray, Josh Huang, Matteo Pellegrini, Nilanjana Banerjee, Nada Abumrad, Fang Zhao, Dustin Schones, Gengxin Chen, Andrew Smith, Aaron Boudreau and Zhenyu Xuan for helpful discussions. This work was supported by NIH grant HG001696 (MQZ), CSHL Association Fellowship (DD) and a grant from the Philip Morris USA External Research Program (ZN).
References
- Basso K, Margolin AA, Stolovitzky G, Klein U, Dalla-Favera R, Califano A (2005) Reverse engineering of regulatory networks in human B cells. Nat Genet 37: 382–390 [DOI] [PubMed] [Google Scholar]
- Berg OG, von Hippel PH (1987) Selection of DNA binding sites by regulatory proteins. Statistical–mechanical theory and application to operators and promoters. J Mol Biol 193: 723–750 [DOI] [PubMed] [Google Scholar]
- Bussemaker HJ, Li H, Siggia ED (2001) Regulatory element detection using correlation with expression. Nat Genet 27: 167–171 [DOI] [PubMed] [Google Scholar]
- Carey M (1998) The enhanceosome and transcriptional synergy. Cell 92: 5–8 [DOI] [PubMed] [Google Scholar]
- Chen KY (1997) Transcription factors and the down-regulation of G1/S boundary genes in human diploid fibroblasts during senescence. Front Biosci 2: d417–d426 [DOI] [PubMed] [Google Scholar]
- Chung I, Bresnick E (1995) Regulation of the constitutive expression of the human CYP1A2 gene: cis elements and their interactions with proteins. Mol Pharmacol 47: 677–685 [PubMed] [Google Scholar]
- Conlon EM, Liu XS, Lieb JD, Liu JS (2003) Integrating regulatory motif discovery and genome-wide expression analysis. Proc Natl Acad Sci USA 100: 3339–3344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das D, Banerjee N, Zhang MQ (2004) Interacting models of cooperative gene regulation. Proc Natl Acad Sci USA 101: 16234–16239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman Q, Grosschedl R (1999) Regulation of LEF-1/TCF transcription factors by Wnt and other signals. Curr Opin Cell Biol 11: 233–240 [DOI] [PubMed] [Google Scholar]
- Elkon R, Linhart C, Sharan R, Shamir R, Shiloh Y (2003) Genome-wide in silico identification of transcriptional regulators controlling the cell cycle in human cells. Genome Res 13: 773–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JH (1991) Multivariate adaptive regression splines. Ann Stat 19: 1–67 [DOI] [PubMed] [Google Scholar]
- Frith MC, Li MC, Weng Z (2003) Cluster-Buster: finding dense clusters of motifs in DNA sequences. Nucleic Acids Res 31: 3666–3668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glantz SA (2001) Primer of Biostatistics. New York, NY, USA: McGraw-Hill [Google Scholar]
- Golden-Mason L, O'Farrelly C (2002) Having it all? Stem cells, haematopoiesis and lymphopoiesis in adult human liver. Immunol Cell Biol 80: 45–51 [DOI] [PubMed] [Google Scholar]
- Hastie T, Tibshirani R, Friedman JH (2001) The Elements of Statistical Learning. New York: Springer-Verlag [Google Scholar]
- Hernando E, Nahle Z, Juan G, Diaz-Rodriguez E, Alaminos M, Hemann M, Michel L, Mittal V, Gerald W, Benezra R, Lowe SW, Cordon-Cardo C (2004) Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature 430: 797–802 [DOI] [PubMed] [Google Scholar]
- Hertz GZ, Stormo GD (1999) Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics 15: 563–577 [DOI] [PubMed] [Google Scholar]
- Hirose T, Sowa Y, Takahashi S, Saito S, Yasuda C, Shindo N, Furuichi K, Sakai T (2003) p53-independent induction of Gadd45 by histone deacetylase inhibitor: coordinate regulation by transcription factors Oct-1 and NF-Y. Oncogene 22: 7762–7773 [DOI] [PubMed] [Google Scholar]
- Ishida S, Huang E, Zuzan H, Spang R, Leone G, West M, Nevins JR (2001) Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol Cell Biol 21: 4684–4699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakizawa T, Miyamoto T, Ichikawa K, Kaneko A, Suzuki S, Hara M, Nagasawa T, Takeda T, Mori J, Kumagai M, Hashizume K (1999) Functional interaction between Oct-1 and retinoid X receptor. J Biol Chem 274: 19103–19108 [DOI] [PubMed] [Google Scholar]
- Kam KY, Jeong KH, Norwitz ER, Jorgensen EM, Kaiser UB (2005) Oct-1 and nuclear factor Y bind to the SURG-1 element to direct basal and gonadotropin-releasing hormone (GnRH)-stimulated mouse GnRH receptor gene transcription. Mol Endocrinol 19: 148–162 [DOI] [PubMed] [Google Scholar]
- Kel AE, Gossling E, Reuter I, Cheremushkin E, Kel-Margoulis OV, Wingender E (2003) MATCH: a tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res 31: 3576–3579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keles S, van der Laan M, Eisen MB (2002) Identification of regulatory elements using a feature selection method. Bioinformatics 18: 1167–1175 [DOI] [PubMed] [Google Scholar]
- Kirmizis A, Farnham PJ (2004) Genomic approaches that aid in the identification of transcription factor target genes. Exp Biol Med (Maywood) 229: 705–721 [DOI] [PubMed] [Google Scholar]
- Levine M, Tjian R (2003) Transcription regulation and animal diversity. Nature 424: 147–151 [DOI] [PubMed] [Google Scholar]
- Liu XS, Brutlag DL, Liu JS (2002) An algorithm for finding protein–DNA binding sites with applications to chromatin-immunoprecipitation microarray experiments. Nat Biotechnol 20: 835–839 [DOI] [PubMed] [Google Scholar]
- Locker J (2001) Transcription Factors. San Diego, CA, USA: Academic Press [Google Scholar]
- Maniatis T, Reed R (2002) An extensive network of coupling among gene expression machines. Nature 416: 499–506 [DOI] [PubMed] [Google Scholar]
- Marsh S, Xiao M, Yu J, Ahluwalia R, Minton M, Freimuth RR, Kwok PY, McLeod HL (2004) Pharmacogenomic assessment of carboxylesterases 1 and 2. Genomics 84: 661–668 [DOI] [PubMed] [Google Scholar]
- Meyers S, Hiebert SW (1995) Indirect and direct disruption of transcriptional regulation in cancer: E2F and AML-1. Crit Rev Eukaryot Gene Expr 5: 365–383 [DOI] [PubMed] [Google Scholar]
- Nahle Z, Polakoff J, Davuluri RV, McCurrach ME, Jacobson MD, Narita M, Zhang MQ, Lazebnik Y, Bar-Sagi D, Lowe SW (2002) Direct coupling of the cell cycle and cell death machinery by E2F. Nat Cell Biol 4: 859–864 [DOI] [PubMed] [Google Scholar]
- Niehrs C, Pollet N (1999) Synexpression groups in eukaryotes. Nature 402: 483–487 [DOI] [PubMed] [Google Scholar]
- Norris K, Norris F, Kono DH, Vestergaard H, Pedersen O, Theofilopoulos AN, Moller NP (1997) Expression of protein-tyrosine phosphatases in the major insulin target tissues. FEBS Lett 415: 243–248 [DOI] [PubMed] [Google Scholar]
- Odom DT, Zizlsperger N, Gordon DB, Bell GW, Rinaldi NJ, Murray HL, Volkert TL, Schreiber J, Rolfe PA, Gifford DK, Fraenkel E, Bell GI, Young RA (2004) Control of pancreas and liver gene expression by HNF transcription factors. Science 303: 1378–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani K, Iwanaga R, Nakamura M, Ikeda M, Yabuta N, Tsuruga H, Nojima H (1999) Cell growth-regulated expression of mammalian MCM5 and MCM6 genes mediated by the transcription factor E2F. Oncogene 18: 2299–2309 [DOI] [PubMed] [Google Scholar]
- Overdier DG, Ye H, Peterson RS, Clevidence DE, Costa RH (1997) The winged helix transcriptional activator HFH-3 is expressed in the distal tubules of embryonic and adult mouse kidney. J Biol Chem 272: 13725–13730 [DOI] [PubMed] [Google Scholar]
- Pennacchio LA, Rubin EM (2001) Genomic strategies to identify mammalian regulatory sequences. Nat Rev Genet 2: 100–109 [DOI] [PubMed] [Google Scholar]
- Puga A, Barnes SJ, Dalton TP, Chang C, Knudsen ES, Maier MA (2000) Aromatic hydrocarbon receptor interaction with the retinoblastoma protein potentiates repression of E2F-dependent transcription and cell cycle arrest. J Biol Chem 275: 2943–2950 [DOI] [PubMed] [Google Scholar]
- Schones DE, Sumazin P, Zhang MQ (2005) Similarity of position frequency matrices for transcription factor binding sites. Bioinformatics 21: 307–313 [DOI] [PubMed] [Google Scholar]
- Smith AD, Sumazin P, Das D, Zhang MQ (2005) Mining ChIP-chip data for transcription factor and cofactor binding sites. Bioinformatics 21 (Suppl 1): i403–i412 [DOI] [PubMed] [Google Scholar]
- Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, Brown PO, Botstein D, Futcher B (1998) Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell 9: 3273–3297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg D, Colla P (1999) MARS: An Introduction. San Diego, CA, USA: Salford Systems [Google Scholar]
- Storey JD, Tibshirani R (2003) Statistical significance for genomewide studies. Proc Natl Acad Sci USA 100: 9440–9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom DK, Nip J, Westendorf JJ, Linggi B, Lutterbach B, Downing JR, Lenny N, Hiebert SW (2000) Expression of the AML-1 oncogene shortens the G(1) phase of the cell cycle. J Biol Chem 275: 3438–3445 [DOI] [PubMed] [Google Scholar]
- Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, Cooke MP, Walker JR, Hogenesch JB (2004) A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci USA 101: 6062–6067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa M, Li N, Bailey TL, Church GM, De Moor B, Eskin E, Favorov AV, Frith MC, Fu Y, Kent WJ, Makeev VJ, Mironov AA, Noble WS, Pavesi G, Pesole G, Regnier M, Simonis N, Sinha S, Thijs G, van Helden J, Vandenbogaert M, Weng Z, Workman C, Ye C, Zhu Z (2005) Assessing computational tools for the discovery of transcription factor binding sites. Nat Biotechnol 23: 137–144 [DOI] [PubMed] [Google Scholar]
- Tronche F, Bach I, Chouard T, David-Wattine B, Pontoglio M, Ringeisen F, Sourdive D, Thepot D, Yaniv M (1994) Hepatocyte nuclear factor-1 (HNF1) and liver gene expression. In Liver Gene Expression, Tronche F, Yaniv M (eds) pp 155–181. Austin: RG Landes Company [Google Scholar]
- Tsou AP, Yang CW, Huang CY, Yu RC, Lee YC, Chang CW, Chen BR, Chung YF, Fann MJ, Chi CW, Chiu JH, Chou CK (2003) Identification of a novel cell cycle regulated gene, HURP, overexpressed in human hepatocellular carcinoma. Oncogene 22: 298–307 [DOI] [PubMed] [Google Scholar]
- Veitia RA (2003) A sigmoidal transcriptional response: cooperativity, synergy and dosage effects. Biol Rev Camb Philos Soc 78: 149–170 [DOI] [PubMed] [Google Scholar]
- Vigo E, Muller H, Prosperini E, Hateboer G, Cartwright P, Moroni MC, Helin K (1999) CDC25A phosphatase is a target of E2F and is required for efficient E2F-induced S phase. Mol Cell Biol 19: 6379–6395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Cherry JM, Nochomovitz Y, Jolly E, Botstein D, Li H (2005) Inference of combinatorial regulation in yeast transcriptional networks: a case study of sporulation. Proc Natl Acad Sci USA 102: 1998–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman WW, Sandelin A (2004) Applied bioinformatics for the identification of regulatory elements. Nat Rev Genet 5: 276–287 [DOI] [PubMed] [Google Scholar]
- Whitfield ML, Sherlock G, Saldanha AJ, Murray JI, Ball CA, Alexander KE, Matese JC, Perou CM, Hurt MM, Brown PO, Botstein D (2002) Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol Biol Cell 13: 1977–2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan Z, Zhao F, Wang J, Chen G, Zhang MQ (2005) Genome-wide promoter extraction and analysis in human, mouse, and rat. Genome Biol 6: R72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui K, Arii S, Zhao C, Imoto I, Ueda M, Nagai H, Emi M, Inazawa J (2002) TFDP1, CUL4A, and CDC16 identified as targets for amplification at 13q34 in hepatocellular carcinomas. Hepatology 35: 1476–1484 [DOI] [PubMed] [Google Scholar]
- Zeeberg BR, Feng W, Wang G, Wang MD, Fojo AT, Sunshine M, Narasimhan S, Kane DW, Reinhold WC, Lababidi S, Bussey KJ, Riss J, Barrett JC, Weinstein JN (2003) GoMiner: a resource for biological interpretation of genomic and proteomic data. Genome Biol 4: R28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao F, Xuan Z, Liu L, Zhang MQ (2005) TRED: a Transcriptional Regulatory Element Database and a platform for in silico gene regulation studies. Nucleic Acids Res 33: D103–D107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou DX, Yen TS (1991) The ubiquitous transcription factor Oct-1 and the liver-specific factor HNF-1 are both required to activate transcription of a hepatitis B virus promoter. Mol Cell Biol 11: 1353–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Giangrande PH, Nevins JR (2005a) Temporal control of cell cycle gene expression mediated by E2F transcription factors. Cell Cycle 4: 633–636 [DOI] [PubMed] [Google Scholar]
- Zhu Z, Shendure J, Church GM (2005b) Discovering functional transcription-factor combinations in the human cell cycle. Genome Res 15: 848–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods
Significant Motifs and Motif Pairs for HeLa Cell-Cycle
Supplementary Figures
Supplementary Tables
Supplementary Figure 2
Supplementary Note